

Abstract
Background:
The aim of this study was to generate in silico 3D-structure of the envelope protein of AHFV using homology modeling method to further predict its conformational epitopes and help other studies to investigate its structural features using the model.
Methods:
A 3D-structure prediction was developed for the envelope protein of Alkhumra haemorrhagic fever virus (AHFV), an emerging tick-borne flavivirus, based on a homology modeling method using M4T and Modweb servers, as the 3D-structure of the protein is not available yet. Modeled proteins were validated using Modfold 4 server and their accuracies were calculated based on their RSMDs. Having the 3D predicted model with high quality, conformational epitopes were predicted using DiscoTope 2.0.
Results:
Model generated by M4T was more acceptable than the Modweb-generated model. The global score and P-value calculated by Modfold 4 ensured that a certifiable model was generated by M4T, since its global score was almost near 1 which is the score for a high resolution X-ray crystallography structure. Furthermore, itsthe P-value was much lower than 0.001 which means that the model is completely acceptable. Having 0.46 Å rmsd, this model was shown to be highly accurate. Results from DiscoTope 2.0 showed 26 residues as epitopes, forming conformational epitopes of the modeled protein.
Conclusion:
The predicted model and epitopes for envelope protein of AHFV can be used in several therapeutic and diagnostic approaches including peptide vaccine development, structure based drug design or diagnostic kit development in order to facilitate the time consuming experimental epitope mapping process.
Keywords: 3D structure, Homology modeling, Conformational epitope, Alkhumra haemorrhagic fever virus, Envelope protein
Introduction
Alkhumra haemorrhagic fever virus (AHFV), a tick-borne flavivirus, was firstly discovered in 1995 from a butcher who had lately slaughtered a sheep in the city of Alkhumra in Makkah province. AHFV is now an emerging virus causing febrile reactions with fatal haemorrhagic and neurological manifestations (Alzahrani et al. 2010, Mohabatkar 2011, Memish et al. 2012). AHFV was misnamed as alkhurma virus for many times in scientific papers and publications but has been corrected recently by the International Committee on Taxonomy of Viruses (ICTV) (Madani 2005, King et al. 2011, Madani et al. 2011, Madani et al. 2012).
AHFV is the first tick-borne haemorrhagic virus whose whole genome has been sequenced and based on the sequence analysis, it has 89 % homology with Kyasanur Forest disease virus (KFDV) which belongs to the genus flavivirus of the flaviviridae family (Mohabatkar 2011). This genus includes nearly over 70 viruses, mostly arthropode-borne, which infect humans and animals (Pastorino et al. 2006). KFDV and its genetic variants, AHFV and seven other species including Langat, Louping ill, Omsk haemorrhagic fever, Powassan, Royal Farm, Gadgets Gully and tick-borne encephalitis are grouped in the same subclassification (Memish et al. 2010). However, AHFV among all these viruses makes a major concern in the local health authorities as it can be exported to other countries by the Muslims returning from yearly Hajj rites. Since it is suggested to be one of the most deadly flavivirus infections, with case fatality rates >30 % (Charrel et al. 2005), it can be one of our major concerns in Iran as a Muslim country.
Generally, incidence of many life-threatening viruses in humans has increased during the last two decades (Mohabatkar 2011) and there are many others which threaten to increase in the near future. These urgent situations greatly force us to accelerate our investigations and studies. Undoubtedly in vitro and in vivo studies are of major importance in discovering the therapeutic and prophylactic solutions, but the fact is that in silico studies and simulations can accelerate these experiments due to the time-saving manner they have. High-accuracy and knowledge-based in silico predictions will result in a better and faster handling of the disease being studied since it can highlights the most important parts to be studied (Mohabatkar et al. 2012).
During recent years, useful data have been computationally produced for many viruses such as human papillomavirus type 16 (HPV-16) (Mohabatkar 2007), influenza A H3N2 virus (A/Hong Kong/1/68) (Liang et al. 2012), Isfahan virus (ISFV) (Mohabatkar and Mohsenzadeh 2009) and many others parallel to that of in vivo studies, showing the complementary and necessary role of computational investigations.
A comprehensive in silico study on medically important structural properties of protein E of AHFV was done by Mohabatkar 2011, including the prediction of its secondary structure, glycosylation sites, tetraspan membrane protein of hair cell stereocilia (TMH), disulfide connectivity, subcellular localization, evolutionary distance and T-cell epitope prediction, but was not including conformational epitope prediction while lacking a good 3D model (Mohabatkar 2011). It should be noted that, protein E plays a central role in the biology of flaviviruses. It is the dominant antigen in eliciting neutralizing antibodies and plays a main role in inducing immunologic responses in the infected host (Wu et al. 2003).
The aim of this study was to generate in silico 3D-structure of the envelope protein of AHFV using homology modeling method to further predict its conformational epitopes and help other studies to investigate its structural features using the model. However, better models can be generated in the future due to the fact that bioinformatics servers and tools are progressing rapidly.
Materials and Methods
Retrieval of target amino acid sequence
The amino acid sequence of the envelope protein of AHFV was obtained from the protein database of NCBI (accession number NP_775470.1). After ensuring that there is no 3D-structure of the protein in Protein Data Bank (PDB) (http://www.rcsb.org/pdb/), the present work was held.
Homology modeling and quality assessment
Modeling was done using two homology modeling programs, M4T (Fernandez-Fuentes et al. 2007) and Modweb (http://salilab.org/modweb) which are both automated web-servers. Since unfavorable distortions are exerted on the side chains of amino acids during homology modeling process, energy minimization was performed on both models to move them to their optimal condition where the protein is in its most stable state. Here energy minimization process was performed using GROMOS96 force-Field implementation in Swiss PdbViewer software (version 4.0.1) (Kaplan et al. 2001). Refined models were then evaluated via quality assessment methods. The most common method is root-mean-square deviation (RMSD) metric which shows the mean distance between the corresponding atoms in the two structures (Maiorov 1994). RMSDs of the modeled structures from the selected template were calculated using rmsd calculator tool in Swiss-PdbViewer (version 4.0.1) based on the backbone atoms (N, Cα and C). The other method to evaluate the generated protein models used here was based on the use of ModFold4 web-based server. The server allows user to make quantitative judgments about the credibility of models using different quantitative parameters like global score and P-value (McGuffin et al. 2013).
Finally, the overall stereochemical property of the more accurate model was further assessed by Ramachandran plot analysis using Rampage server. (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php).
Conformational epitope prediction
The best selected model was subjected to predict its possible conformational epitopes with DiscoTope 2.0 server (Haste Andersen et al. 2006), which uses a combination of amino acid statistics, spatial information, and surface exposure. The server is trained on a compiled data set of discontinuous epitopes from 76 X-ray structures of antibody-antigen protein complexes (Haste Andersen et al. 2006) and is one of the most known and reliable epitope prediction servers. According to the server constructors study, DiscoTope detects 15.5 % of residues located in discontinuous epitopes with a specificity of 95 %. This level of specificity is higher than several similar servers (Haste Andersen et al. 2006).
Results
Homology modeling, quality assessment and streochemical quality evaluation
Homology modeling was performed using two programs: M4T and Modweb which are servers for automated comparative protein structure modeling that calculate their models based on the best available template structures from PDB without the need of manually identifying a template and doing pairwise alignment. However, results of the alignment and the choice of template can be observed in the servers’ output (Eswar et al. 2003). Here, the template by which the two modeling programs generated their models was the crystal structure of chain A of envelope glycoprotein from tick-borne encephalitis virus (PDB: 1SVB_A) at 2 Å resolution (Rey et al. 1995). Alignment results performed automatically by the server revealed that the selected template has almost 82 % identity with the query sequence.
Although the accuracy of the generated model is highly dependent on its sequence identity with the template sequence, by having the results of RMSD metric and Modfold4 server the quality of the model can be reliably assessed. The results of RMSD and Modfold4 for the final energy-minimized models are provided in Table 1. During energy minimization process which was perfomed for 5 runs, the final calculated energy of M4T and Modweb generated models were reduced to −15335.445 KJ/mol and −14327.418 KJ/mol respectively. Before energy minimization process, these values were −4808.651 KJ/mol and −3147.817 KJ/mol for M4T and Modweb generated models respectively.
Table 1.
Quality assessment of models using different parameters
| Modeling program | confidence | P-value | Global model quality score | RMSD (Å) |
|---|---|---|---|---|
| M4T | Certifiable | 2.92E−4 | 0.7730 | 0.46 |
| ModWeb | Medium | 1.399E−2 | 0.4408 | 3.45 |
In case of RMSD, M4T generated model showed 0.46 Å rmsd with the template and is therefore more accurate than the model generated by Modweb with 3.45 Å rmsd.
Using Modfold 4, the global model quality score is calculated and from this feature a P-value is calculated which represents the probability that a model is incorrect. The global model quality scores range between 0 and 1 and scores less than 0.2 indicates an incorrectly modeled proteins while scores greater than 0.4 are models with higher quality and indicate that the models are highly similar to the native structures. The other output parameter of the server is P-value. In general, P-values more than 0.05, represent low-quality models. When P-value reaches 0.01–0.05, it can be concluded that the model quality is medium. High quality models show 0.001–0.1 P-values and if having absolutely certifiable models, the P-values of less than 0.001 are expected. Here both the global model quality score and P-value are acceptable especially for the model generated by M4T, where P-value is less than 0.001 and the global model quality score is near 1, at about 0.77. The P-value and global model quality of 2.92E−4 and 0.773, respectively, shows that the model scores are both in the range required for a high quality model. Therefore the model generated by M4T was then visualized by PyMol (Fig. 1).
Fig. 1.

3D predicted model visualized by Pymol
The evaluation of M4T generated model for its stereochemical quality was performed by Ramachandran plot analysis using Rampage server and the output plot is shown in Fig. 2. It showed 98 % of residues in favored region, 1.5 % in allowed region and just one residue (0.3 %) in outlier region which highly indicates a good streochemical quality of the predicted model. The detailed result from Rampage is given in Table 2.
Fig. 2.
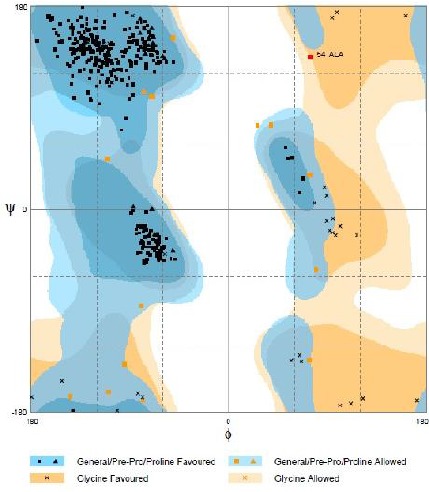
Ramachandran plot for the model
Table 2.
Ramachandran plot analaysis results using Rampage
| Analysis result | Residue | Position | (Φ, Ψ) | % |
|---|---|---|---|---|
| Thr | 56 | (−106.51, −157.54) | ||
| Leu | 202 | (−81.16, −79.63) | ||
| Leu | 223 | (−34.43, 122.46) | ||
| In allowed region | Ala | 249 | (43.99, −123.34) | 1.5 |
| Asn | 351 | (82.85, −42.90) | ||
| Asn | 361 | (21.20, 69.10) | ||
| In outlier region | Ala | 54 | (90.29, 127.61) | 0.3 |
| In favored region | All other residues | All other positions | - | 98.2 |
B-cell conformational epitope prediction
Having an acceptable model, the prediction of envelope protein of AHFV was undertaken using DiscoTope 2.0. The input is the PDB file format of the model. Disco Tope 2.0 utilizes calculation of surface accessibility -estimated in terms of contact numbers- and a novel epitope propensity amino acid score. The final scores are calculated by combining the propensity scores of residues in spatial proximity and the contact numbers. In the most recent version of DiscoTope which is version 2.0 used in this paper, novel definition of the spatial neighborhood is used to sum propensity scores and half-sphere exposure as a surface measure (Kringelum et al. 2012a). Results from DiscoTope 2.0 showed 26 residues out of the whole 395 total residues of the model as epitopes (Table 3). These residues can then interact as the consequence of protein folding and form conformational epitopes.
Table 3.
Predicted conformational epitiope residues by DiscoTope 2
| Epitope residue | Residue position | Discotope score |
|---|---|---|
| Glu | 51 | −2.368 |
| Asn | 52 | −0.729 |
| Ala | 54 | −3.180 |
| Asn | 154 | −3.107 |
| Asp | 178 | −3.487 |
| Glu | 277 | −3.678 |
| Gly | 278 | −0.777 |
| Ser | 279 | −1.855 |
| Met | 302 | −3.086 |
| Thr | 303 | −0.787 |
| Ser | 335 | −3.399 |
| Lys | 336 | −3.567 |
| Ala | 346 | −3.463 |
| His | 347 | −3.001 |
| Gly | 348 | −1.157 |
| Glu | 349 | −2.459 |
| Pro | 350 | −3.312 |
| Asn | 351 | −3.003 |
| Thr | 366 | −1.287 |
| Thr | 367 | −1.642 |
| Pro | 378 | −0.886 |
| Gly | 379 | −1.292 |
| Asp | 380 | −1.495 |
| Gln | 391 | −3.275 |
| Phe | 393 | −1.356 |
| Lys | 395 | −1.260 |
Discussion
Interaction between B-cell receptors or soluble antibodies and protein antigens is one of the major mechanisms of immune system to overcome infectious pathogens. Identification of the epitopes which antigens can bind to antibodies is undoubtedly essential in many biomedical applications including vaccine design, immunotherapeutics studies and also for developing and designing of diagnostic kits. Experimental epitope mapping is difficult and expensive in term of time and cost, making in silico methods a good complementary accelerant (Kringelum et al. 2012a).
Although most B-cell epitopes are conformational, computational methods and studies often concentrate on mapping linear epitopes (Yasser and Honavar 2010). In recent years interests are increasing for mapping conformational epitopes and this assertion can be confirmed by the fact that different tools potential for predicting conformational epitopes are increasingly developing, these are including Ellipro (Ponomarenko et al. 2008), PEPITO (Sweredoski and Baldi 2008), SEPPA (Sun et al. 2009), EPSVR (Liang et al. 2010), DiscoTope 1.0 and 2.0 and many others. However due to the computational complexity and lack of sufficient knowledge about antibody-antigen complex structures, there are still a limited number of high accuracy web-based servers and tools.
Linear B-cell and T-cell epitopes of envelope protein of AHFV have already been predicted and both have essential roles in the immune response to pathogens (Mohabatkar 2011).
In this paper, predicting the conformational epitope of the envelope protein of AHFV using a structure-based prediction method was held after generating a certifiable 3D model. There are sequence-based prediction methods available for conformational epitope mapping which does not require the 3D structure of the protein being studied.
However, the structure-based prediction can be more accurate if equipping with strong algorithms and therefore, the latter was used in the present study.
Although we can more conveniently gain information about protein 3D structures in silico, X-ray crystallography and NMR are undoubtedly the first and best choices to get such information. Homology modeling of proteins is an alternative way when there is no information about their experimentally-prepared 3D structures. The critical step in homology modeling is the identification of a 3D template chosen by virtue of having the highest sequence identity with the target sequence, if indeed any are available. Sometimes there is no suitable template structure available or there are templates with medium-low sequence identities (less than 50–70 % identity). When there are template structures available but with low sequence identity to our target protein sequence, it is not recommended to perform homology modeling.
Here, the selected template structure (1SVB) showed 82 % sequence identity to our target sequence. Having 82 % sequence identity, it was ensured that homology modeling is an appropriate method to predict the 3D structure of our target protein. Furthermore, model accuracy was described based on its RMSD with the template structure and also by Modfold4. According to related literatures and to provide a frame of reference for RMSD values, up to 0.5 Å rmsd occurs in independent determinations of relatively the same protein, meaning that this range of RMSD value for any modeled protein shows high modeling accuracy (Chothia and Lesk 1986). RMSDs of M4T and Modweb generated models were 0.46 and 3.45 Å respectively and it is obvious that M4T-generated model is more accurate. According to previous studies of homology modeling cases, highly accurate models were reported to have 0.4–1.5 Å rmsd (Lindin et al. 2013, Zhu et al. 2014). Thus, our predicted protein structure has excellent backbone RMSD and can be referred to as highly accurate.
It is important to note that, there is a defect in the use of RMSD as a quality assessment method: RMSD metric is sensitive to outlier regions created by poor modeling of individual loop regions in a structure that its core is accurately modeled and for this reason there are cases when as many as one in ten highly accurate homology models (60%–95% identity), have an RMSD value of more than 5 Å vs. the empirical structure (Guex et al. 2009). Due to this defect, Modfold4 quality assessment server was also used along with RMSD-based accuracy assessment. Using the scores given by Modfold4, the similarity between the template and target structures which are identical in amino acid sequences but different in tertiary structures can be described. Both models were regarded as accurate due to the results of Modfold4, however, again these scores were better for the M4T generated model (0.7730 and 2.92E-4 for Global model quality score and P-value, respectively). Using this server, the model is ranked by global model quality score, and a P-value is calculated for it, which relates to the likelihood that the global model is incorrect (McGuffin et al. 2013). Our M4T-generated model has gained a high global model quality score along with a very low P-value, meaning that the model is highly certifiable. Ramachandran plot analysis also verified this fact that the model is accurate.
Here, the choice of epitope prediction server was relied on a study showed that DiscoTope 2.0 server has the highest predictive performance among some other similar servers (Kringelum et al. 2012b). Results from DiscoTope 2.0 showed 26 residues as conformational epitopes. According to Ramachandran plot analysis, alanine 54 which was one of the predicted epitope residues, was shown to be in outlier region of Ramachandran plot. Therefore, it seems to be unacceptable to name this residue as an epitope compartment. However, it should be also noted that it is not bizzare when a residue or more adopt a disallowed stereochemical angle. It can be explained by the fact that they do so to maintain the stability of the whole protein structure. Due to this reason, alanine can adopt disallowed conformations to stabilize the whole protein tertiary structure with high rate of occurrence (Pal and Chakrabarti 2002). Thus, this residue can be also accepted as an epitope compartment regardless of being in the outlier region of Ramachandran plot.
Conclusion
The predicted model for envelope protein of AHFV is of an acceptable quality. Since no therapeutic or prophylactic vaccine is available for AHFLV, such data on its potential conformational epitopes might be helpful for scientists in future drug and vaccine development.
Acknowledgements
Support of this study by University of Isfahan is highly acknowledged. The authors declare that there is no conflict of interests.
References
- 1. Alzahrani AG, Al Shaiban HM, Al Mazroa MA, Al-Hayani O, Macneil A, Rollin PE, Memish ZA. ( 2010) Alkhurma hemorrhagic fever in humans, Najran, Saudi Arabia. Emerg Infect Dis. 16: 1882– 1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Charrel RN, Zaki AM, Fakeeh M, Yousef AI, de Chesse R, Attoui H, de Lamballerie X. ( 2005) Low diversity of Alkhurma hemorrhagic fever virus, Saudi Arabia, 1994–1999. Emerg Infect Dis. 11: 683– 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chothia C, Lesk A M. ( 1986) The relation between the divergence of sequence and structure in proteins. EMBO J. 5: 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B. ( 2003) Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31: 3375– 3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandez-Fuentes N, Madrid-Aliste CJ, Rai BK, Fajardo JE, Fiser A. ( 2007) M4T: a comparative protein structure modeling server. Nucleic Acids Res. 35: 363– 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guex N, Peitsch MC, Schwede T. ( 2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis. 30: 162– 173. [DOI] [PubMed] [Google Scholar]
- 7. Haste Andersen P, Nielsen M, Lund O. ( 2006) Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 15: 2558– 2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaplan W, Littlejohn TG. ( 2001) Swiss-PDB viewer (deep view). Brief Bioinform. 2: 195– 197. [DOI] [PubMed] [Google Scholar]
- 9. King AM, Adams MJ, Lefkowitz EJ, Carstens EB. ( 2011) Virus taxonomy: IXth report of the International Committee on Taxonomy of Viruses. Vol 9 Elsevier. [Google Scholar]
- 10. Kringelum JV, Lundegaard C, Lund O, Nielsen M. ( 2012) Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol. 8: e1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liang L, Huang P, Wen M, Ni H, Tan S, Zhang Y, Chen Q. ( 2012) Epitope peptides of influenza H3N2 virus neuraminidase gene designed by immunoinformatics. Acta Biochim Biophys Sinica. 44: 113– 118. [DOI] [PubMed] [Google Scholar]
- 12. Liang S, Zheng D, Standley D, Yao B, Zacharias M, Zhang C. ( 2010) EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC bioinformatics. 11: 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Madani TA. ( 2005) Alkhumra virus infection, a new viral hemorrhagic fever in Saudi Arabia. J Infect. 51: 91– 97. [DOI] [PubMed] [Google Scholar]
- 14. Madani TA, Azhar EI, Abuelzein el TM, Kao M, Al-Bar HM, Abu-Araki H, Niedrig M, Ksiazek TG. ( 2011) Alkhumra (Alkhurma) virus outbreak in Najran, Saudi Arabia: epidemiological, clinical, and laboratory characteristics. J Infect. 62: 67– 76. [DOI] [PubMed] [Google Scholar]
- 15. Madani TA, Azhar EI, Abuelzein E-T, Kao M, Al-Bar HM, Niedrig M, Ksiazek TG. ( 2012) Alkhumra, not Alkhurma, is the correct name of the new hemorrhagic fever flavivirus identified in Saudi Arabia. Intervirology. 55: 259– 260. [DOI] [PubMed] [Google Scholar]
- 16. Maiorov VN, Crippen GM. ( 1994) Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J Mol Biol. 235: 625– 634. [DOI] [PubMed] [Google Scholar]
- 17. McGuffin LJ, Buenavista MT, Roche DB. ( 2013) The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic Acids Res. 41: W368– W372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Memish ZA, Charrel RN, Zaki AM, Fagbo SF. ( 2010) Alkhurma haemorrhagic fever--a viral haemorrhagic disease unique to the Arabian Peninsula. Int J Antimicrob Agents. 36( 1): 53– 57. [DOI] [PubMed] [Google Scholar]
- 19. Memish ZA, Fagbo SF, Assiri AM, Rollin P, Zaki AM, Charrel R, Mores C, MacNeil A. ( 2012) Alkhurma viral hemorrhagic fever virus: proposed guidelines for detection, prevention, and control in Saudi Arabia. PLoS Negl Trop Dis. 6: e1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mohabatkar H. ( 2007) Prediction of epitopes and structural properties of Iranian HPV-16 E6 by bioinformatics methods. Asian Pac J Cancer Prev. 8: 602– 606. [PubMed] [Google Scholar]
- 21. Mohabatkar H, Mohsenzadeh S. ( 2009) Bioinformatics Comparison of G Protein of Isfahan Virus with the Same Proteins of Two Other Closely Related Viruses of the Genus Vesiculovirus. Protein Pept Lett. 16: 1017– 1023. [DOI] [PubMed] [Google Scholar]
- 22. Mohabatkar H. ( 2011) Computer-based comparison of structural features of envelope protein of Alkhurma hemorrhagic fever virus with the homologous proteins of two closest viruses. Protein Pept Lett. 18: 559– 567. [DOI] [PubMed] [Google Scholar]
- 23. Mohabatkar H, Keyhanfar M, Behbahani M. ( 2012) Protein bioinformatics applied to virology. Curr Protein Pept Sci. 13: 547– 559. [DOI] [PubMed] [Google Scholar]
- 24. Pal D, Chakrabarti P. ( 2002) On residues in the disallowed region of the Ramachandran map. Biopolymers. 63: 195– 206. [DOI] [PubMed] [Google Scholar]
- 25. Pastorino BA, Peyrefitte CN, Grandadam M, Thill MC, Tolou HJ, Bessaud M. ( 2006) Mutagenesis analysis of the NS2B determinants of the Alkhurma virus NS2BNS3 protease activation. J Gen Virol. 87: 3279– 3283. [DOI] [PubMed] [Google Scholar]
- 26. Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ( 2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC bioinformatics. 9: 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. ( 1995) The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature. 1995 May 25 375( 6529): 291– 298. [DOI] [PubMed] [Google Scholar]
- 28. Sun J, Wu D, Xu T, Wang X, Xu X, Tao L, Li Y, Cao Z-W. ( 2009) SEPPA: a computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res. 37: W612– W616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sweredoski MJ, Baldi P. ( 2008) PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics. 24: 1459– 1460 [DOI] [PubMed] [Google Scholar]
- 30. Wu KP, Wu CW, Tsao YP, Kuo TW, Lou YC, Lin CW, Cheng JW. ( 2003) Structural Basis of a Flavivirus Recognized by Its Neutralizing Antibody solution structure of the domain iii of the japanese encephalitis virus envelope protein. J Biol Chem. 278: 46007– 46013. [DOI] [PubMed] [Google Scholar]
- 31. Yasser E-M, Honavar V. ( 2010) Recent advances in B-cell epitope prediction methods. Immunome Res. 6: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu K, Day T, Warshaviak D, Murrett C, Friesner R, Pearlman D. ( 2014) Antibody Structure Determination Using a Combin'ation of Homology Modeling, Energy-Based Refinement and Loop Prediction. Proteins: Struct, Funct, Bioinf. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]