

Abstract
BACKGROUND & AIMS
MHC class I-restricted CD8+ T cells are required for clearance of hepatitis C virus (HCV) infection. MHC class I expression is upregulated by type I and II interferons (IFNs). However, little is known about the effects of HCV infection on IFN-induced expression of MHC class I.
METHODS
We used the HCV cell culture system (HCVcc) with the genotype 2a Japanese Fulminant Hepatitis-1 strain to investigate IFN-induced expression of MHC class I and its regulatory mechanisms. HCVcc-infected Huh-7.5 cells were analyzed by flow cytometry, metabolic labeling, immunoprecipitation, and immunoblotting analyses. Protein kinase R (PKR) was knocked-down with lentiviruses that express small hairpin (sh)RNAs. The functional effects of MHC class I regulation by HCV were demonstrated in co-culture studies, using HCV-specific CD8+ T cells.
RESULTS
Although the baseline level of MHC class I was not affected by HCV infection, IFN-induced expression of MHC class I was notably attenuated in HCV-infected cells. This was associated with replicating HCV RNA, not with viral protein. HCV infection reduced IFN-induced synthesis of MHC class I protein and induced phosphorylation of PKR and eIF2α. IFN-induced MHC class I expression was restored by shRNA-mediated knockdown of PKR in HCV-infected cells. Co-culture of HCV-specific CD8+ T cells and HCV-infected cells that expressed HLA-A2 demonstrated that HCV infection reduced the effector functions of HCV-specific CD8+ T cells; these functions were restored by shRNA-mediated knockdown of PKR.
CONCLUSIONS
IFN-induced expression of MHC class I is attenuated in HCV-infected cells by activation of PKR, which reduces the effector functions of HCV-specific CD8+ T cells. This appears to be an important mechanism by which HCV circumvents antiviral adaptive immune responses.
Keywords: JFH-1, antigen presentation, immune evasion, adaptive immune response
Introduction
Hepatitis C virus (HCV) infects 170 million people worldwide and is a major cause of chronic liver disease 1. The majority of HCV-infected patients progress to chronic hepatitis, and 20% of chronically infected patients develop long-term complications such as liver cirrhosis and hepatocellular carcinoma 2.
HCV-specific CD8+ T cell responses are vital for the successful clearance of HCV infection 3–9, which has been highlighted in previous studies from HCV-infected chimpanzees 4–7 and patients with acute HCV infection 8, 9.
Major histocompatibility complex (MHC) class I molecules play a key role in the recognition of virus-infected cells by CD8+ T cells 10. When cells are infected by virus, viral peptides are processed and loaded onto MHC class I molecules. Once a stable viral peptide/MHC class I complex has been formed in the endoplasmic reticulum, it is transported to the cell surface for presentation to CD8+ T cells 10. When the T cell receptor interacts with the peptide/MHC class I complex, CD8+ T cells become activated and exert antiviral effector functions.
During viral infections, type I and III interferons (IFNs) are produced by infected cells and dendritic cells 11, 12, and type II IFN (IFN-γ) is produced by natural killer (NK) cells and T cells 13. The IFNs direct antiviral as well as immunomodulatory functions. In addition, IFNs upregulate MHC class I expression, which allows CD8+ T cells to more efficiently recognize MHC class I-upregulated, virus-infected cells.
Many viruses have developed specialized mechanisms that decrease MHC class I expression to escape CD8+ T cell responses; for example, cytomegalovirus (CMV) expresses several viral proteins that interfere with the cell surface expression of MHC class I 14. However, the effect of HCV infection on MHC class I expression is not well understood. Previous studies sought to address this by viral protein overexpression or by using HCV subgenomic replicon systems 15–17. However, these experimental models employed in those studies do not recapitulate the entire viral life cycle and do not consider the effect of cytokines, such as IFNs, those are present in the local inflammatory microenvironment during HCV infection.
In the present study, we investigated the effect of HCV infection on IFN-induced MHC class I expression using an in vitro HCV cell culture (HCVcc) system with the genotype 2a Japanese Fulminant Hepatitis-1 (JFH-1) strain 18–20, which recapitulates the complete HCV life cycle. This provided a unique opportunity to study the effect of HCV infection on MHC class I expression. Furthermore, we identified the underlying mechanism by which HCV impeded IFN-induced MHC I expression during infection, and delineated the functional significance of regulation of IFN-induced MHC class I expression by co-culture of HCV-infected cells with HCV-specific CD8+ T cells.
Materials and Methods
HCV infection and IFN treatment
The JFH-1 strain (genotype 2a) of HCVcc was produced and quantified as previously described 21. Huh-7.5 cells (provided by Apath, LLC, Brooklyn, NY) were infected with HCVcc at 0.01 to 0.1 multiplicity-of-infection (MOI), depending on the experiment. Transfection with HCV protein-encoding plasmids was performed as previously described 22. To study IFN-induced MHC class I expression, HCV-infected cells were treated with 3 ng/mL IFN-β (PeproTech, Rocky Hill, NJ), 10 ng/mL IFN-γ (PeptroTech), 100 ng/mL IFN-λ1 (R&D Systems, Minneapolis, MN) or 100 ng/mL IFN-λ2 (R&D Systems) for 24 h. Cell culture media and HCV RNA transfection are described in the Supplementary Materials and Methods section.
Flow cytometry
The antibodies used for flow cytometry included mouse monoclonal anti-HCV core IgG1 (Clone C7-50; Thermo Scientific/Affinity BioReagents, Rockford, IL), FITC-conjugated anti-mouse IgG1 (Clone A85-1; BD Biosciences, San Jose, CA), AlexaFluor 647- or AlexaFluor 488-conjugated anti-HLA-ABC (Clone W6/32; AbD Serotec), and APC-conjugated anti-HLA-A2 (BD Biosciences). Cells were stained with ethidium monoazide (EMA) for exclusion of dead cells and then surface stained with fluorochrome-conjugated HLA-ABC or HLA-A2-specific antibodies for 30 min at 4°C. For identification of HCV-infected cells, cells were fixed and permeabilized, then stained with anti-HCV core and FITC-conjugated anti-mouse IgG1 antibodies. Multicolor flow cytometry was performed using LSR II instrument (BD Biosciences), and data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Immunoblotting and immunoprecipitation
A total of 203μg of cell lysate was loaded onto SDS–PAGE gels and analyzed by immunoblotting. The antibodies used for immunoblotting analysis included mouse monoclonal anti-HCV core IgG1 (Clone C7-50), mouse anti-HLA-ABC (Clone W6/32; BioLegend), rabbit polyclonal anti-eIF2α (Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-phospho-eIF2α (Ser51) (Cell Signaling Technology), rabbit polyclonal anti-PKR (Santa Cruz Biotechnology), and rabbit monoclonal anti-phospho-PKR (pT446) (Clone E120; Epitomics, Burlingame, CA). After overnight incubation with primary antibodies (1:1,000 dilution) at 4°C, the signal was detected using horseradish peroxidase-conjugated secondary antibodies (1:2,500 dilution; Pierce, Rockford, IL, USA) and enhanced chemiluminescence reagents (GE Healthcare/Amersham, Buckinghamshire, UK).
For immunoprecipitation of MHC class I protein, 5003μg of cell lysate was incubated overnight with anti-HLA-ABC antibody (BioLegend), subsequently with protein A agarose beads (Santa Cruz Biotechnology) for 23h. Immunoprecipitates were extracted from the beads, loaded onto SDS–PAGE gels and analyzed by immunoblotting. After overnight incubation with rabbit monoclonal anti-MHC class I (Clone EP1395Y; Epitomics) at 4°C, the signal was detected as described above. Band intensities were quantified using ImageJ software.
Metabolic labeling of MHC class I synthesis
Six hours after addition of IFN-β, cells were washed twice with PBS and incubated in methionine/cysteine-free DMEM (Sigma-Aldrich, St. Louis, MO) supplemented with 1% (v/v) dialyzed FBS (Welgene, Daegu, Korea) and L-glutamine (Sigma-Aldrich) for 1 h. The cells were then pulsed with 5003μCi of EasyTag EXPRE35S35S Protein Labeling Mix (Perkin-Elmer, Boston, MA) for 1 h and washed twice with ice-cold PBS. Cell lysates were prepared using RIPA buffer. Equal amounts of immunoprecipitates with anti-HLA-ABC (BioLegend, San Diego, CA) were loaded onto SDS-PAGE gels. 35S-labeled proteins were detected by autoradiography.
Confocal microscopy
The procedure for confocal microscopy is described in the Supplementary Materials and Methods section.
shRNA-encoding lentiviruses
Lentiviral vectors were transfected into 293TN cells (System Biosciences, Mountain View, CA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Forty-eight hours after transfection, the lentivirus particles were harvested, concentrated, and stored at −70°C until use. PKR shRNA clones are described in the Supplementary Materials and Methods section.
TaqMan real-time PCR
The procedure for TaqMan real-time PCR is described in the Supplementary Materials and Methods section.
Establishment of cell lines expressing HLA-A2 under the control of HLA-A2 promoter
The complete sequence of HLA-A*0201 with its promoter region (GenBank Accession Number GQ996941) was synthesized and cloned into the pGEM-T easy vector (Promega, Madison, WI) by Bioneer Corp (Daejeon, Korea), and designated as pGEM-T/P(HLA-A2)-(HLA-A*0201). The pcDNA3.1(+) vector was digested with BglII and EcoRI restriction enzymes to remove sequences containing the CMV promoter and part of the multiple cloning site (MCS). Subsequently, this region was replaced with P(HLA-A2)-(HLA-A*A0201) fragment, and the resultant plasmid was designated as pcDNA3.1/P(HLA-A2)-(HLA-A2). Wild-type or PKR-silenced Huh-7.5 cells were stably transfected with pcDNA3.1/P(HLA-A2)-(HLA-A2) plasmid and selected with 1 mg/mL G418 with or without 1 μg/mL puromycin.
MHC class I stabilization assay
Huh-7.5/P(HLA-A2)-(HLA-A2) cells were infected with HCV at 0.1 MOI. After 3 days of infection, the cells were pulsed with excess amounts (10 μg/mL) of HLA-A2-restricted HCV genotype 1a NS31073 peptide (CINGVCWTV) or dimethyl sulfoxide (DMSO) in the presence or absence of 3 ng/mL IFN-β. Twenty-four hours later, cells were analyzed for surface expression levels of HLA-A2 using flow cytometry.
HCV-specific CD8+ T cell lines
Whole blood was obtained from HLA-A2-positive patients with acute HCV infection (genotype 1a) with approval of the local institutional review boards and patient consent. Peripheral blood mononuclear cells (PBMCs) were isolated, and CD8+ T cells were negatively selected using magnetic beads (Miltenyi Biotec, Auburn, CA). HCV-specific CD8+ T cells were subsequently selected with PE-conjugated HLA-A2-NS31073 (CINGVCWTV) or HLA-A2-core35 (YLLPRRGPRL) pentamers (ProImmune, Oxford, UK) and anti-PE microbeads (Miltenyi Biotec). Selected cells were expanded for 3 weeks in 48-well plates with irradiated autologous feeder cells in RPMI 1640 (5% FBS) with 40 ng/mL anti-CD3, 200 U/mL interleukin (IL)-2, 10 ng/mL IL-7 and 100 ng/mL IL-15.
Co-culture experiment
shControl-transduced or shPKR#4-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) cells were infected with HCV for 3 days and treated with 3 ng/mL IFN-β for 24 h. HLA-A2 expression was determined by flow cytometry. The cells were pulsed with HLA-A2-restricted HCV NS31073 peptide (CINGVCWTV) for 1 h, then co-cultured with the HCV NS31073-specific CD8+ T cell line for 12 h at an effector:target ratio of 5:1. Considering the frequency of HCV-specific T cells in the cell line (~50%) and the infection rate of target cells (~90%), the final effector:infected Huh-7.5 cell ratio was calculated to be 2.78:1 and the effector:uninfected Huh-7.5 cell ratio was 2.78:0.1. In another set of experiments, target cells were co-cultured with the HCV core35-specific CD8+ T cell line without peptide pulsing. The effector functions of HCV-specific CD8+ T cells were assessed by intracellular staining for IFN-γ and TNF-α.
Statistical analysis
Statistical analysis is described in the Supplementary Materials and Methods section.
Results
IFN-induced MHC class I expression is attenuated by HCV infection
To address whether HCV infection has a significant effect on MHC class I expression in human hepatoma cells, we used an in vitro JFH-1 genotype 2a HCVcc system in Huh-7.5 cells (Figure 1A). HCV infection tended to decrease the baseline level of MHC class I expression; however, the difference was not statistically significant (Figure 1B). Next, we studied the effect of HCV infection on type I or II IFN-induced MHC class I expression. Huh-7.5 cells were infected with HCV and 3 days later, type I or II IFN was added for 24 h. As expected, type I or II IFN upregulated MHC class I expression in uninfected cells; however, IFN-induced MHC class I expression was remarkably attenuated in HCV-infected cells (Figure 1C). This was confirmed at various time points after HCV infection (days 3–7) (data not shown). We further confirmed our finding by HCV core and MHC class I co-staining. In an HCV-inoculated culture, HCV-infected cells were identified by HCV core staining (Figure 1D), and IFN-induced MHC class I expression was compared between HCV-infected cells and uninfected cells. As shown in Figure 1E, IFN-induced MHC class I expression was attenuated in HCV-infected cells compared with uninfected cells. In HCV-infected cells, type III IFN-induced MHC class I expression was also attenuated (Supplementary Figure 1A and B). This diminished induction of MHC class I expression in response to IFNs in HCV-infected Huh-7.5 hepatoma cells was also confirmed by immunofluorescence analysis (Figure 1F). Taken together, these data demonstrate that IFN-induced MHC class I expression is attenuated by HCV infection.
Figure 1. IFN-induced MHC class I expression is attenuated by HCV infection.

(A–C) Huh-7.5 cells were infected with HCVcc at 0.1 MOI and cultured for 3 days, with an infection rate of 85–95%. (A) The infection rate was determined with anti-HCV core immunostaining and flow cytometry. (B) Surface expression of MHC class I was evaluated in HCV-infected or uninfected Huh-7.5 cells by flow cytometry, and mean fluorescence intensity (MFI) was compared between HCV-infected and uninfected cells (n = 3). F, fluorescence intensity of stained sample; F0, fluorescence intensity of isotype control. ns, not significant. (C) HCV-infected or uninfected Huh-7.5 cells were treated with 3 ng/mL IFN-β or 10 ng/mL IFN-γ or untreated (control). After 24 h, cells were stained for MHC class I expression and analyzed by flow cytometry. Data from five independent experiments are presented as a bar graph (mean + SEM). ***, P < 0.001; ns, not significant. (D–F) For this experiment, Huh-7.5 cells were inoculated with HCVcc at 0.01 MOI and cultured for 3 days, with an infection rate of 30–45%. (D) Co-staining for HCV core and MHC class I was performed in an HCV-inoculated culture with gating on HCV core-positive (blue) and HCV core-negative (red) populations, respectively. (E) HCV-inoculated cultures were treated with 3 ng/mL IFN-β or 10 ng/mL IFN-γ or were left untreated (control). HCV core and MHC class I are shown for HCV-infected cells (blue) and uninfected cells (red). Data from five independent experiments are presented as a bar graph (mean + SEM). **, P < 0.01; ***, P < 0.001; ns, not significant. (F) Immunofluorescence microscopy of HCV core (red) and MHC class I (green) expression after 24 h of IFN-β treatment. The HCV-infected cell displayed lower intensity of MHC class I staining compared to the adjacent uninfected cell. The nuclei were stained by DAPI (blue).
Replicating HCV RNA, not viral protein, is responsible for suppression of IFN-induced MHC class I expression
We repeated the experiment using HCV RNA transfection instead of HCVcc infection to determine if replicating HCV RNA was sufficient to suppress IFN-induced MHC class I expression. Huh-7.5 cells were transfected with full-genome JFH-1 HCV RNA. In HCV RNA-transfected culture, transfected cells were identified by HCV core staining, and IFN-induced MHC class I expression was compared between HCV RNA-transfected cells and untransfected cells. IFN-induced MHC class I expression was attenuated in HCV RNA-transfected cells compared with untransfected cells (Figure 2A). When Huh-7.5 cells were transfected with the replication-defective JFH-1/GND mutant RNA 19, IFN-induced MHC class I expression was not attenuated (data not shown). Next, we studied the effect of each viral protein on IFN-induced MHC class I expression by transfecting Huh-7.5 cells with HCV protein-encoding plasmids. However, IFN-induced MHC class I expression was not affected by overexpression of any HCV protein (Figure 2B and C). Transfection was also performed with pairs of HCV gene plasmids in 28 combinations. In addition, transfection was performed separately with all structural protein genes, all non-structural protein genes and all HCV genes. However, none of these transfections affected IFN-induced MHC class I expression (Supplementary Figure 2A and B). Collectively, these data indicate that replicating HCV RNA, not viral protein, is responsible for the diminished expression of IFN-induced MHC class I.
Figure 2. Replicating HCV RNA, not viral protein per se, leads to attenuation of IFN-induced MHC class I expression.

(A) Huh-7.5 cells were transfected with replication-competent JFH-1 HCV RNA and 2 days later, 3 ng/mL IFN-β or 10 ng/mL IFN-γ was added for 24 h. The expression level of MHC class I in transfected (blue) or untransfected (red) cells was evaluated by flow cytometry after co-staining for HCV core and MHC class I. Data from three independent experiments are presented as a bar graph (mean + SEM). ***, P < 0.001; ns, not significant. (B and C) Huh-7.5 cells were transfected with HCV protein-encoding plasmids, followed by treatment with 3 ng/mL IFN-β for 24 h. The expression level of MHC class I was analyzed by flow cytometry (B), and presented as MFI (C). F, fluorescence intensity of stained samples; F0, fluorescence intensity of isotype control.
HCV infection attenuates IFN-induced synthesis of MHC class I protein
We then evaluated the underlying mechanism responsible for the attenuation of IFN-induced MHC class I expression during HCV infection. First, the mRNA expression level of hla-a, a representative MHC class I gene, was evaluated by real-time quantitative PCR. Unexpectedly, IFN-induced MHC class I mRNA expression was elevated rather than suppressed in HCV-infected cells (Figure 3A), suggesting that HCV infection regulates IFN-induced MHC class I expression at the translational or post-translational level.
Figure 3. HCV infection attenuates IFN-induced synthesis of MHC class I protein.
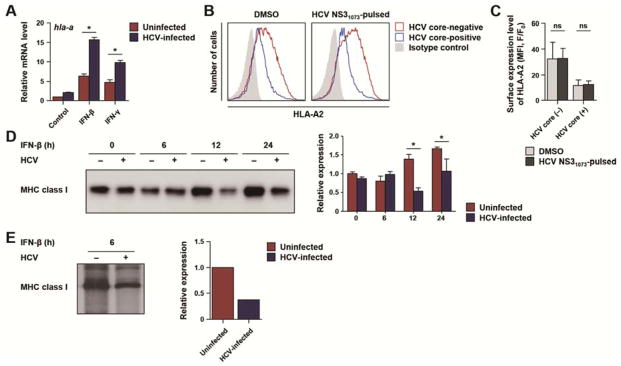
(A) Huh-7.5 cells were infected with HCVcc at 0.1 MOI and cultured for 3 days, with an infection rate of 85–95%. HCV-infected or uninfected cells were treated with 3 ng/mL IFN-β or 10 ng/mL IFN-γ for 24 h, and the mRNA level of hla-a, a representative MHC class I gene, was determined by real-time quantitative PCR and normalized to β-actin mRNA. Data represent mean + SEM of four independent experiments. *, P < 0.05. (B and C) MHC class I stabilization assay. Three days after inoculating HCV to Huh-7.5/P(HLA-A2)-(HLA-A2) cells at 0.01 MOI (yielding an infection rate of 30–45%), 10 μg/mL HLA-A2-restricted HCV NS31073 peptide was pulsed in the presence of IFN-β for 24 h. The expression level of HLA-A2 in HCV-infected (blue) or uninfected (red) cells was analyzed by flow cytometry after co-staining for HCV core and HLA-A2 (B). HLA-A2 expression was compared between peptide-pulsed cells and dimethyl sulfoxide (DMSO)-treated control cells (C). Data represent mean + SEM of three independent experiments. ns, not significant. (D and E) Huh-7.5 cells were infected with HCVcc at 0.1 MOI and cultured for 3 days, with an infection rate of 85–95%. (D) HCV-infected or uninfected Huh-7.5 cells were treated with 3 ng/mL IFN-β. Total cell lysates were subjected to immunoprecipitation of MHC class I protein. MHC class I immunoprecitates were visualized by immunoblotting. The density of the immunoblot bands from three independent experiments are presented as a bar graph (mean + SEM). *, P < 0.05. (E) HCV-infected or uninfected cells were treated with 3 ng/mL IFN-β for 6 h, followed by 35S-Met/Cys metabolic labeling for 1 h. Equal amounts of protein extracts were immunoprecipitated for MHC class I and analyzed by SDS-PAGE and autoradiography. The density of the immunoblot bands was quantified and presented in the graph on the right.
For cell surface expression of MHC class I, peptides need to be produced by the intracellular antigen processing machinery and bind to MHC class I. Therefore we investigated whether a defective supply of MHC-binding peptides accounts for the diminished expression of IFN-induced MHC class I using a standard MHC class I stabilization assay. In this assay, MHC class I expression on the cell surface was examined after addition of excess amounts of an MHC-binding peptide. We performed MHC class I stabilization assays using human leukocyte antigen (HLA)-A2-restricted HCV NS31073 peptide and the Huh-7.5/P(HLA-A2)-(HLA-A2) cell line, which stably expresses HLA-A2 under the control of the HLA-A2 promoter. We found that excess amounts of HCV NS31073 peptide did not increase IFN-induced MHC class I expression on the cell surface in uninfected and HCV-infected cells (Figure 3B and C), suggesting that attenuated expression of IFN-induced MHC class I is not attributed to defective antigen processing and/or peptide loading. Next, the amount of total cellular MHC class I protein was analyzed by immunoprecipitation and immunoblotting. IFN-induced MHC class I expression was diminished not only on the cell surface but also in total cell lysates of HCV-infected cells (Figure 3D).
Finally, we studied the synthesis rate of MHC class I protein. HCV-infected Huh-7.5 cells were treated with IFN-β for 6 h followed by 1 h of metabolic labeling with 35S-Met/Cys, and the cell lysates were analyzed by MHC class I immunoprecipitation and autoradiography to quantify the synthesis of MHC class I protein. IFN-β-induced synthesis of MHC class I protein was reduced in HCV-infected cells compared with uninfected cells (Figure 3E), demonstrating that HCV inhibits the translation of IFN-induced MHC class I protein. Taken together, these results indicate HCV infection attenuates IFN-induced MHC class I expression at the translational level rather than at the transcriptional or post-translational level.
Activation of the PKR pathway by HCV infection is responsible for attenuated MHC class I expression
Protein kinase R (PKR) and its substrate eIF2α are regulators of translation initiation. A previous study reported that the PKR-eIF2α pathway is activated in HCV-infected cells resulting in suppressed translation of antiviral IFN-stimulated genes (ISG) 23. We confirmed that HCV infection resulted in phosphorylation of PKR and eIF2α (Figure 4A). We therefore studied the role of PKR in the regulation of IFN-induced MHC class I expression by silencing PKR expression. Five clones of PKR-silenced Huh-7.5 cells were generated by transduction with five different clones of PKR shRNA-lentivirus. Huh-7.5 cells with complete (shPKR#4) or partial (shPKR#1) silencing of PKR expression were confirmed by immunoblotting analysis (Figure 4B). IFN-induced MHC class I expression was examined after HCV infection of these cells. Whereas IFN-β-induced MHC class I expression was attenuated in HCV-infected shControl-transduced Huh-7.5 cells, it was not affected in HCV-infected Huh-7.5 cells (shPKR#4) with complete silencing of PKR (Figure 4C and D). In Huh-7.5 cells (shPKR#1) with partial silencing of PKR, IFN-β-induced MHC class I expression was partially restored (Figure 4C and D). These data illustrate a critical role for the PKR-eIF2α pathway in suppressing IFN-induced synthesis of MHC class I protein in HCV-infected cells.
Figure 4. HCV activates the PKR-eIF2α pathway, and PKR-silencing restores MHC class I expression.

(A) Huh-7.5 cells were infected with HCVcc at 0.1 MOI and cultured for 3 days, with an infection rate of 85–95%. HCV-infected or uninfected cells were treated with 3 ng/mL IFN-β or 10 ng/mL IFN-γ for 24 h, and cell lysates were analyzed by immunoblotting. (B) PKR-silenced stable cell lines were established by PKR shRNA-lentivirus transduction. Complete (shPKR#4) or partial (shPKR#1) silencing of PKR expression was confirmed by immunoblotting. The density of the immunoblot bands was quantified and presented in the graph on the bottom. (C and D) PKR-silenced or unsilenced Huh-7.5 cells were inoculated with HCV at 0.01 MOI, cultured for 3 days (yielding an infection rate of 30–45%) and treated with 3 ng/mL IFN-β. The expression level of MHC class I in HCV-infected (blue) or uninfected (red) cells was analyzed by co-staining for HCV core and MHC class I and flow cytometry (C). Expression of MHC class I was compared between HCV core-positive and -negative cells (D). Data represent mean + SEM of three independent experiments. **, P < 0.01; ***, P < 0.001; ns, not significant.
Attenuated expression of MHC class I by HCV infection leads to suppressed effector functions of HCV-specific CD8+ T-cells
To demonstrate the functional significance of MHC class I regulation in HCV infection, we co-cultured an HCV-specific CD8+ T cell line with HCV-infected HLA-A2-expressing Huh-7.5 cells. PKR-silenced or unsilenced Huh-7.5/P(HLA-A2)-(HLA-A2) cells were infected with HCV for 3 days and treated with IFN-β for 24 h. As expected, cell surface expression of HLA-A2 was attenuated by HCV infection and restored by PKR-silencing (Figure 5A). These cells were pulsed with HLA-A2-restricted NS31073 peptide (CINGVCWTV), then co-cultured with an NS31073-specific CD8+ T cell line, which was derived from an HLA-A2-positive patient with acute HCV infection (genotype 1a) (Supplementary Figure 3A). This cell line did not cross-recognize the genotype 2a JFH-1 sequence that we used to infect Huh-7.5 cells. Co-culture with HCV-uninfected, peptide-pulsed cells resulted in production of IFN-γ and TNF-α from CD8+ T cells. However, co-culture with HCV-infected, peptide-pulsed cells resulted in reduced production of IFN-γ and TNF-α from CD8+ T cells. Production of IFN-γ and TNF-α was reinstated in CD8+ T cells when they were co-cultured with HCV-infected, peptide-pulsed cells without PKR expression (Figure 5B and C).
Figure 5. Effector functions of HCV-specific CD8+ T cells relate to MHC class I expression.

(A) Cells were infected with HCVcc at 0.1 MOI and cultured for 3 days, with an infection rate of 90%. Uninfected, shControl-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) (left), HCV-infected, shControl-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) (middle), or HCV-infected, shPKR#4-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) (right) cells were treated with 3 ng/mL IFN-β for 24 h, and HLA-A2 expression was determined by flow cytometry. (B-E) Uninfected, shControl-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) (left), HCV-infected, shControl-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) (middle), or HCV-infected, shPKR#4-transduced Huh-7.5/P(HLA-A2)-(HLA-A2) (right) cells were treated with 3 ng/mL IFN-β for 24 h, pulsed with (B and C) or without (D and E) 10 μg/mL CINGVCWTV NS31073 peptide and co-cultured for 12 h with NS31073-specific CD8+ T cells (B and C) or core35-specific CD8+ T cells (D and E) at an effector:target ratio of 5:1. IFN-γ and TNF-α production of HCV-specific CD8+ T cells was assessed by intracellular cytokine staining and flow cytometry. Quadrant statistics are shown (B and D), and the percentages of IFN-γ+ or TNF-α+ T cells are presented as bar graphs (C and E).
Next, we examined CD8+ T cell recognition of endogenously processed epitope peptides. PKR-silenced or unsilenced Huh-7.5/P(HLA-A2)-(HLA-A2) cells were infected with HCV and treated with IFN-β, then co-cultured with a core35-specific CD8+ T cell line, which was derived from another HLA-A2-positive patient with acute HCV infection (genotype 1a) (Supplementary Figure 3B). Notably, this cell line cross-recognized the conserved core35 peptide of genotype 2a JFH-1. Whereas CD8+ T cells did not produce IFN-γ and TNF-α when co-cultured with uninfected cells, a small percentage of T cells (3.3%) produced IFN-γ and/or TNF-α when co-cultured with HCV-infected cells (Figure 5D and E). Importantly, co-culture with PKR-silenced, HCV-infected cells increased the frequency of IFN-γ and/or TNF-α-producing CD8+ T cells to 6.9% (Figure 5D and E). This increase was not attributed to a higher infection rate in PKR-silenced cells. In fact, HCV infection rate in PKR-silenced cells was not higher than that in shControl-transduced cells after HCV infection and IFN-β treatment (Supplementary Figure 4). Collectively, these data demonstrate that the effector functions of HCV-specific CD8+ T cells are dictated by the expression level of MHC class I on the cell surface of HCV-infected cells, and that HCV suppresses IFN-induced MHC class I expression and evades recognition by HCV-specific CD8+ T cells.
Discussion
The current study demonstrated that HCV decreases MHC class I expression and evades recognition by HCV-specific CD8+ T cell responses. Only a few studies have investigated the effect of HCV on MHC class I expression with conflicting results. In a study by Moradpour et al. 15, the expression of MHC class I was not affected by overexpression of HCV proteins whereas another study showed upregulation of MHC class I expression by the HCV core 17. On the other hand, Tardif et al. 16 demonstrated downregulation of MHC class I expression using an HCV RNA subgenomic replicon system. These inconsistent results are likely due to differences in the experimental models used in each study. To the best of our knowledge, ours is the first study to evaluate the effect of HCV infection on MHC class I expression using the HCVcc system, in which HCV proteins are expressed at physiological levels, though it was studied in a hepatoma cell line, Huh-7.5. Another unique aspect of our study is that we examined the effect of HCV infection on MHC class I expression also in the presence of exogenous IFNs. During viral infections, type I and III IFNs are produced by infected cells and dendritic cells 11, 12, and type II IFN (IFN-γ) is produced by NK cells and T cells 13. We demonstrated that HCV infection significantly attenuates IFN-induced MHC class I expression (Figure 1C and E).
Several HCV proteins inhibit host responses when overexpressed in vitro 22, 24–27. The HCV core inhibits STAT1 activation 24, the NS3/4A protease blocks IRF-3 25, and the E2 and NS5A proteins inhibit kinase activity of PKR 26, 27. However, we found that viral proteins per se do not play a role in the attenuation of IFN-induced MHC class I expression, because overexpression of each viral protein did not affect IFN-induced MHC class I expression (Figure 2B and C). Instead, replicating HCV RNA is responsible for the suppression of IFN-induced MHC class I expression, as demonstrated by the repressed induction of MHC class I by IFNs in the presence of replication-competent JFH-1 RNA (Figure 2A), but not in the presence of replication-defective JFH-1/GND mutant RNA.
In the present study, we showed that HCV infection induced phosphorylation of PKR and eIF2α (Figure 4A), and that IFN-induced MHC class I expression was restored by PKR-silencing in HCV-infected cells (Figure 4C and D). A similar role of PKR was previously reported for diminished ISG induction in HCV-infected cells 23. In that report, Garaigorta and Chisari found that translation of ISG protein is suppressed in HCVcc-infected cells as a result of PKR and eIF2α phosphorylation 23. Their findings are consistent with our results on the molecular mechanisms, which demonstrated that HCV-induced activation of PKR and eIF2α is responsible for the attenuated induction of IFN-stimulated protein expression in HCV-infected cells. Garaigorta and Chisari studied attenuated ISG induction by HCV infection in that study; thus, the significance of their findings was limited to the innate antiviral responses 23. In contrast, we studied attenuation of IFN-induced MHC class I expression by HCV infection; thus, we extended the significance of the PKR-eIF2α pathway to viral immune evasion from CD8+ T cell responses. In fact, the effector functions of HCV-specific CD8+ T cells were dictated by the expression level of MHC class I. This was shown in two different types of co-culture experiments (Figure 5B and D). First, genotype 1a NS31073 (CINGVCWTV)-specific CD8+ T cells were used, and HLA-A2+ target cells were pulsed with CINGVCWTV peptide, because CINGVCWTV-specific CD8+ T cells were reported not to recognize JFH-1 (genotype 2a) NS31073 (TISGVLWTV) 28. Second, we investigated CD8+ T cell recognition of endogenously processed epitope peptides in the JFH-1 HCV infection system. In this experiment, we used core35 (YLLPRRGPRL)-specific CD8+ T cells from another genotype 1a patient. YLLPRRGPRL-specific CD8+ T cells recognized endogenously processed core35 peptide in JFH-1-infected cells because the epitope sequence is conserved. Although fewer cytokine-producing CD8+ T cells were stimulated by endogenously processed peptide than by externally loaded peptide, both experiments demonstrated a close relation between the effector function of HCV-specific CD8+ T cells and the expression level of MHC class I on HCV-infected cells. Collectively, these data imply that specific inhibition of PKR may lead to recovery of MHC class I expression in HCV-infected cells, enhancement of HCV-specific CD8+ T cell responses and possibly clearance of HCV in patients.
If HCV attenuates IFN-induced MHC class I expression, the role of NK cells needs to be considered. NK cells contribute to antiviral innate immune responses by recognizing virus-infected cells that lack MHC class I or overexpress ligands for activating receptors 29. Whereas diminished expression of IFN-induced MHC class I protein in HCV-infected cells may facilitate immune evasion from HCV-specific CD8+ T cells, it might lead to recognition of HCV-infected cells by NK cells. However, HCV is known to evade the NK cell responses in several ways. The HCV envelope protein directly suppresses NK cell activation 30, 31 and HCV-infected cells induce downregulation of NKG2D on NK cells 32. Thus, NK cell activation may be avoided.
In summary, we utilized an in vitro HCVcc system to delineate the impact of HCV on the expression of MHC class I, while considering the effect of IFNs. We demonstrate that IFN-induced MHC class I expression is attenuated by HCV infection at the translational level through the PKR-eIF2α pathway, leading to reduction of the antiviral effector functions of HCV-specific CD8+ T cells. Moreover, we show that PKR-silencing in HCV-infected cells restores the diminished expression of IFN-induced MHC class I in a dose-dependent manner, and accordingly recovers HCV-specific CD8+ T cell responses. Our findings illustrate one mechanism by which HCV may evade adaptive antiviral immune responses.
Supplementary Material
Acknowledgments
Grant support: This research was supported by the National Research Foundation of Korea, funded by the Ministry of Science, ICT & Future Planning of Korea (2010-0030075 and 2012-M3C1A1-048860). This work was partly supported by the Korea Advanced Institute of Science and Technology Future Systems Healthcare Project from the Ministry of Science, ICT & Future Planning of Korea. This study was also supported by the Yonsei Liver Blood Bank (YLBB), in part by a grant from Sanofi-Aventis Korea. This research was supported in part by the Intramural Research Program of the NIH, The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- MHC
major histocompatibility complex
- HCV
hepatitis C virus
- IFN
interferon
- HCVcc
cell culture-derived hepatitis C virus
- PKR
protein kinase R
- NK
natural killer
- CMV
cytomegalovirus
- JFH-1
Japanese Fulminant Hepatitis-1
- HLA
human leukocyte antigen
- MOI
multiplicity-of-infection
- shRNA
short-hairpin RNA
- mRNA
messenger RNA
- cDNA
complementary DNA
- Ig
immunoglobulin
- MFI
mean fluorescence intensity
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Footnotes
Disclosures: None to be disclosed.
Author contribution:
WK: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; statistical analysis
PSS, SHP, SY: acquisition of data; analysis and interpretation of data
DYC, JKK: study concept and design; acquisition of data
SK: acquisition of data; drafting of the manuscript
KHH: study concept and design; obtained funding; study supervision
BR, YJC: study concept and design; analysis and interpretation of data; study supervision
ECS: study concept and design; analysis and interpretation of data; drafting of the manuscript; obtained funding; study supervision
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36:S35–46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 3.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–52. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 4.Cooper S, Erickson AL, Adams EJ, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10:439–49. doi: 10.1016/s1074-7613(00)80044-8. [DOI] [PubMed] [Google Scholar]
- 5.Thimme R, Bukh J, Spangenberg HC, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A. 2002;99:15661–8. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shoukry NH, Grakoui A, Houghton M, et al. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J Exp Med. 2003;197:1645–55. doi: 10.1084/jem.20030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin EC, Park SH, Nascimbeni M, et al. The Frequency of CD127+ HCV-specific T cells but not the Expression of Exhaustion Markers Predict the Outcome of Acute Hepatitis C Virus Infection. J Virol. 2013 doi: 10.1128/JVI.03122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lechner F, Wong DK, Dunbar PR, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thimme R, Oldach D, Chang KM, et al. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. Philadelphia: Elsevier; 2012. [Google Scholar]
- 11.Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy DE, Marie IJ, Durbin JE. Induction and function of type I and III interferon in response to viral infection. Curr Opin Virol. 2011;1:476–86. doi: 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biron CA. Expansion, maintenance, and memory in NK and T cells during viral infections: responding to pressures for defense and regulation. Plos Pathog. 2010;6:e1000816. doi: 10.1371/journal.ppat.1000816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 15.Moradpour D, Grabscheid B, Kammer AR, et al. Expression of hepatitis C virus proteins does not interfere with major histocompatibility complex class I processing and presentation in vitro. Hepatology. 2001;33:1282–7. doi: 10.1053/jhep.2001.23793. [DOI] [PubMed] [Google Scholar]
- 16.Tardif KD, Siddiqui A. Cell surface expression of major histocompatibility complex class I molecules is reduced in hepatitis C virus subgenomic replicon-expressing cells. J Virol. 2003;77:11644–50. doi: 10.1128/JVI.77.21.11644-11650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herzer K, Falk CS, Encke J, et al. Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J Virol. 2003;77:8299–309. doi: 10.1128/JVI.77.15.8299-8309.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 19.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang W, Shin EC. Colorimetric focus-forming assay with automated focus counting by image analysis for quantification of infectious hepatitis C virions. PLoS One. 2012;7:e43960. doi: 10.1371/journal.pone.0043960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park J, Kang W, Ryu SW, et al. Hepatitis C virus infection enhances TNFalpha-induced cell death via suppression of NF-kappaB. Hepatology. 2012;56:831–40. doi: 10.1002/hep.25726. [DOI] [PubMed] [Google Scholar]
- 23.Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe. 2009;6:513–22. doi: 10.1016/j.chom.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bode JG, Ludwig S, Ehrhardt C, et al. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17:488–90. doi: 10.1096/fj.02-0664fje. [DOI] [PubMed] [Google Scholar]
- 25.Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–8. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 26.Taylor DR, Shi ST, Romano PR, et al. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–10. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 27.Gale MJ, Jr, Korth MJ, Tang NM, et al. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–27. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 28.Fytili P, Dalekos GN, Schlaphoff V, et al. Cross-genotype-reactivity of the immunodominant HCV CD8 T-cell epitope NS3-1073. Vaccine. 2008;26:3818–3826. doi: 10.1016/j.vaccine.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 29.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 30.Crotta S, Stilla A, Wack A, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195:35–41. doi: 10.1084/jem.20011124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195:43–9. doi: 10.1084/jem.20011145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoon JC, Lim JB, Park JH, et al. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J Virol. 2011;85:12557–69. doi: 10.1128/JVI.00838-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.