

Abstract
Clostridium difficile infection (CDI) is the principal cause of nosocomial diarrhea and pseudomembranous colitis associated with antibiotic therapy. Recent increases in the number of outbreaks attributed to highly virulent antibiotic-resistant strains underscore the importance of identifying efficacious alternatives to antibiotics to control this infection. CDI is mediated by two large exotoxins, toxins A and B. Strong humoral toxin-specific immune responses are associated with recovery and a lack of disease recurrence, whereas insufficient humoral responses are associated with recurrent CDI. Multiple approaches targeting these toxins, including intravenous immunoglobulin, neutralizing polymers, active vaccines, and, most recently, monoclonal antibodies (MAbs), have been explored, with various degrees of success. In this study, we describe the characterization of the first MAbs isolated from healthy human donors using a high-throughput B-cell cloning strategy. The MAbs were selected based on their ability to inhibit the actions of toxins A and B in vitro and because of their in vivo efficacy in a hamster challenge model. A potent 2-MAb cocktail was identified and then further potentiated by the addition of a second anti-toxin B MAb. This 3-MAb combination protected animals against mortality and also reduced the severity and duration of diarrhea associated with challenge with highly virulent strains of C. difficile toxinotypes 0 and III. This highly efficacious cocktail consists of one MAb specific to the receptor binding domain of toxin A and two MAbs specific to nonoverlapping regions of the glucosyltransferase domain of toxin B. This MAb combination offers great potential as a nonantibiotic treatment for the prevention of recurrent CDI.
INTRODUCTION
Clostridium difficile infection (CDI) is a leading cause of pseudomembranous colitis and diarrhea (C. difficile-associated diarrhea [CDAD]) causally related to a perturbation of the intestinal microbiota due to antibiotic use. Although the transmission of CDI is primarily associated with health care and long-term care facilities, C. difficile is a ubiquitous microorganism that has been found in the environment. There are documented cases of community-acquired CDI; in fact, the community-acquired C. difficile infection rates in the United States have been reported to be 7.7 cases per 100,000 person-years, of which 35% were not associated with antibiotics (1). However, the rates associated with health care and long-term care facilities are much higher, possibly due to the colocalization of a reservoir of the pathogen and a high number of susceptible individuals housed in those environments (2). As the eradication of C. difficile spores is very difficult, spore reservoirs can persist within the health care and long-term care environment for long periods (3–6). In recent years, CDI has increased in severity and incidence, and part of this increase is due to the spread of epidemic antibiotic-resistant strains (7, 8). Treatment options remain limited and even appear to be losing efficacy, as evidenced by the continued spread of the epidemic strain and increasing numbers of patients who experience relapses and recurrent disease (9).
Clostridial species are normal members of the human gut flora, usually as a small fraction of the microbiome and mostly nontoxigenic species (10). C. difficile pathogenesis in humans is associated with the disruption of the normal enteric flora and colonization with a toxigenic C. difficile strain. This is followed by overgrowth of vegetative cells and production of toxins that damage the cells of the colon through enzymatic activity of a glucosyltransferase, which glucosylates cytoskeletal regulators, such as Ras and Rac (11). Toxigenic C. difficile strains produce at least one of the two major exotoxins, toxin A or toxin B, and most produce both. Only toxigenic strains have been shown to cause intestinal inflammatory and diarrheal disease (12, 13); therefore, toxins A and B are believed to be major virulence factors of CDI, although other less-studied virulence components of the bacterium can contribute to the disease. For example, the presence of a third toxin known as binary toxin has been associated with a marked increase in disease severity and risk of death. This increase was seen in all strains carrying the gene for the binary toxin, not just the C. difficile NAP1/027 strain associated with recent virulent outbreaks (14), but it remains unclear whether the binary toxin itself causes increased virulence or if it is just a marker for virulence. Studies with isogenic toxin mutant strains implied that the binary toxin may contribute to virulence (15), and a recent report from Heinrichs (16) suggested a contribution from a binary toxin in protection against challenge with binary toxin-producing C. difficile strains in a hamster model. However, data from a phase II clinical trial showed that an antibody pair specific for toxins A and B has similar efficacy against binary toxin-negative and -positive strains (17), suggesting that antibodies against toxins A and B may be sufficient to protect against binary toxin-positive strains.
Fecal microbiota transplants, toxin binding, or neutralizing polymers, biotherapeutics to restore protective microbiota, nontoxigenic C. difficile spores, and active vaccines are some of many nonantibiotic strategies that have been attempted in the field of C. difficile study, with various degrees of success (18, 19). Additional evidence for the importance of antibodies against toxins A and B in protection from CDI is provided by clinical and preclinical studies of toxin-based vaccines and clinical studies of natural antibody responses. Sanofi Pasteur's C. difficile full-length toxoid vaccine candidate is currently being tested in phase III clinical trials. It was previously shown to be highly efficacious in preclinical studies (20) and safe and immunogenic in phase II clinical trials (21, 22). Valneva's recombinant vaccine consisting of two truncated A and B toxins has also shown a favorable safety profile and high immunogenicity in phase I. After reporting positive phase I results, Valneva is preparing for the initiation of phase II studies. Others have reported preclinical success with vaccine candidates expressed as recombinant fragments of toxins A and B (23–25). Evidence from clinical studies on naturally occurring anti-toxin antibodies suggested that the inability to mount an immune response to toxin A was associated with increased risk for recurrent CDI, whereas the development of systemic IgG to toxin A was associated with asymptomatic carriage of C. difficile (26, 27). Another study found that high serum IgG titers and the presence of neutralizing activity against toxin B correlated with clinical recovery from C. difficile-associated diarrhea without relapse (28). These findings clearly indicate that the presence and magnitude of systemic toxin-specific immunity play a major role in protection from enterotoxic and inflammatory actions of C. difficile toxins and that neither toxin A nor toxin B can be downgraded in terms of importance in novel treatments and therapeutics. Combined, these results validate the approach of targeting the two large toxins of C. difficile as a viable nonantibiotic strategy against CDI.
While the vaccine approach holds great potential for CDI prevention, the time required to mount an active immune response limits its utility for individuals with recurrent CDI and for those in need of immediate hospitalization. In contrast, a monoclonal antibody therapy offers control over timing and dose. For this reason, many have been actively working on the identification and development of toxin-specific monoclonal antibodies for treatment against recurrent CDI (29–33). Merck is currently developing a pair of monoclonal antibodies against toxins A and B. These MAbs were derived in transgenic mice and have been shown to be efficacious in a preclinical C. difficile hamster challenge model (34) and in phase II clinical trials against recurrent CDI (17). A group from Biologics PK at UCB, United Kingdom, reported on a mixture of humanized neutralizing monoclonal anti-toxin A and anti-toxin B antibodies with efficacy against a toxinotype III strain in a hamster challenge model (30). Marozsan et al. (29) reported on a pair of humanized murine toxin-specific MAbs, PA-50 and PA-41, which were shown not only to be efficacious in the hamster model but also exhibited broad neutralizing activity across multiple strains of C. difficile in vitro. Thus, the concept of protection against CDAD by MAbs targeting the two large toxins of C. difficile has been validated by multiple laboratories.
Here, we describe the characterization of a novel, efficacious, and fully human monoclonal antibody cocktail against C. difficile toxins A and B. In vitro analysis of these MAbs demonstrated that they possess potent neutralizing activity against toxins purified from the most prevalent toxinotypes in all pandemic areas, including North America, Europe, the Far East, and Latin America, namely, toxinotypes 0, III, V, VIII, and XII (35–38). Furthermore, this MAb cocktail displayed in vivo efficacy against challenge with a highly virulent toxinotype 0 strain and a binary toxin-positive toxinotype III strain in the hamster challenge model. Taken together, these data indicate that we have identified a MAb cocktail with the potential to ameliorate CDI and prevent recurrent infection.
MATERIALS AND METHODS
Initial antibody selection, expression, and purification.
The isolation of fully human candidate antibodies for this project has been carried out by using the ImmunoSpot Antibody Assay on a Chip (ISAAC) and VIVA|Screen technologies (P. Garrone, R. Abès, N. Beltraminelli, and M. Mehtali, patent application WO 2013000982 A1, January 2013) and will be detailed in a separate publication. Briefly, serum samples collected from healthy donors were screened for neutralizing activity against C. difficile toxins A and B of the VPI 10463 toxinotype 0 reference strain in the IMR-90 cell-based cytotoxicity assay. Serum samples with positive neutralizing activity were then tested in a human anti-C. difficile toxin A or toxin B IgG enzyme-linked immunosorbent assay (ELISA) for the presence of C. difficile toxin-binding activity. Purified C. difficile toxins A and B of the VPI 10463 strain were also used as capture antigens in this assay. Sera with the best neutralizing properties were further tested by Western blot assay for their capacity to recognize toxins A and B from toxinotypes III, V, VIII, XII, and XV. B cells were collected from identified individuals possessing neutralizing serum titers. After in vitro activation, isolated B cells were screened for toxin-binding and toxin-neutralizing antibody production, and single B cells producing anti-toxin A or anti-toxin B IgG were collected from microarray chips coated with toxin A or toxin B, respectively. Variable domains from MAb sequences recovered by the sequencing of individual B cells were cloned without sequence optimization into pcDNA3.1 vectors. Heavy-chain variable domains were fused to a human IgG1 constant region sequence optimized for CHO expression by DNA2.0 (Menlo Park, CA). Variable domains were fused to a human kappa constant region with sequence optimized for CHO cell expression by DNA2.0. Monoclonal antibodies were synthesized on a milligram scale by transient expression in suspension CHO cells and purified by protein A affinity chromatography before dialysis back into phosphate-buffered saline (PBS) and storage at −80°C. Recombinant MAbs were tested in an IMR-90 cell-based assay for inhibition of toxin cytotoxicity, and the most potent MAbs were selected for further characterization.
Selected MAb sequences were recloned for large-scale (>200 mg) expression in proprietary vectors. The sequences of MAbs B1 and B2 were reoptimized for CHO expression using the proprietary algorithms of GeneArt (Life Technologies, Grand Island, NY). All large-scale MAb preparations were purified using MabSelect SuRe (GE Healthcare) and stored in PBS.
Recombinant C. difficile toxin fragment preparation.
Segments of the genes for toxins A and B were cloned by PCR from C. difficile DNA of strain VPI 10463 (Table 1), according to the boundaries set by Kink and Williams (39).
TABLE 1.
C. difficile toxin A and toxin B recombinant fragments used for domain mapping
| Toxin | Domain | Amino acids | Fragment name |
|---|---|---|---|
| A | C terminus of GST, N terminus of proteasea | 300–660 | TcdA300–660 |
| C terminus of protease, N terminus of translocation domain | 660–1100 | TcdA660–1100 | |
| B | GTD | 10–520 | TcdB10–520 |
| Protease | 510–1110 | TcdB510–1110 | |
| Translocation domain of N terminus | 1110–1530 | TcdB1110–1530 | |
| Translocation domain of C terminus | 1530–1750 | TcdB1530–1750 | |
| CTD | 1750–2360 | TcdB1750–2360 |
GST, glutathione S-transferase.
A methionine start codon was added to the N terminus, and a 6×His tag followed by a stop codon was added to the C terminus of each construct. The resulting PCR products were ligated into the multiple-cloning site of plasmid pET24+. The constructs were transformed into Escherichia coli strain BL21(DE3) and induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG).
The TcdA300–660 and TcdA660–1100 constructs (TcdA, C. difficile toxin A) expressed but were insoluble and were purified by denaturing chromatography, while constructs TcdB10–520 through TcdB1750–2360 were at least partly soluble and were purified by nondenaturing chromatography. Soluble constructs were grown to liter scale in Luria-Bertani (LB) medium at 37°C. Cells were pelleted by centrifugation and lysed by microfluidization (Microfluidics Corp., Newton, MA) in 50 mM NaHPO4, 300 mM NaCl, and 20 mM imidazole (pH 8.0). Insoluble material was removed by centrifugation, and the cleared lysate was loaded onto a nickel nitrilotriacetic acid (Ni-NTA) column (Qiagen). The column was washed with 50 mM NaHPO4, 300 mM NaCl, and 20 mM imidazole (pH 8.0) and eluted with 50 mM NaHPO4, 300 mM NaCl, and 250 mM imidazole (pH 8.0). Insoluble constructs were grown and harvested as for soluble ones, but the cell pellet was resuspended in 8 M urea, 100 mM NaHPO4, and 10 mM Tris-HCl (pH 8.0) before microfluidization. Insoluble material was removed by centrifugation, and the cleared lysate was loaded onto a Ni-NTA column and washed with 8 M urea, 100 mM NaHPO4, and 10 mM Tris-HCl (pH 6.3). The column was eluted with 8 M urea, 100 mM NaHPO4, and 10 mM Tris-HCl (pH 4.5), and protein-containing fractions were dialyzed with multiple changes against 50 mM NaHPO4, 300 mM NaCl, and 250 mM imidazole (pH 8.0).
C-terminal domains.
A QuickExtract DNA extraction kit (Epicentre) was used to isolate genomic DNA from 1-ml samples of cultures of six C. difficile strains representing different toxinotypes (0, III, V, VIII, XII, and XV). The following primers were designed to amplify the last 900 amino acids (amino acids 1811 to 2710 in the VPI 10463 reference strain sequence), or 2,700 bp of the toxin A of the toxinotype 0 C-terminal domain (CTD): FP (5′-CACCATGGGATTTAAAATAATAGATAATAAAACTTATTAC-3′) and RP (5′-GCCATATATCCCAGGGGC-3′).
Amplification was performed using Pfx50 DNA polymerase and a standard touchdown PCR protocol (40). A band of the correct size (∼2,700 bp for toxinotypes 0, III, V, XII, and XV) was purified by excision from an agarose gel, followed by gel extraction. The PCR product was directionally cloned into the expression plasmid pET101-D-Topo using a ligation-independent cloning strategy, as per the manufacturer's instructions (Champion pET directional TOPO expression kit; Invitrogen).
Directionality and sequence were confirmed by DNA sequencing using the forward and reverse cloning primers. Recombinant expression of these proteins yielded a protein with the following sequence: Met-GFKIIDNKTYY-(toxinotype-specific toxin A amino acids 1823 to 2704)-APGIYG-KGELNSKLEGKPIPNPLLGLDSTRTGHHHHHH.
For toxin B, DNA samples were isolated from the same six C. difficile strains. The following primers were designed to amplify the last 615 amino acids, excluding the final 6 amino acids of the toxin B CTD (amino acids 1752 to 2360) or 1,827 bp of the toxin B toxinotype 0 CTD: FP (5′-CGGATCCGAATTCATTCTTATGTCAACTAGTGAAGAAAATAAGG-3′) and RP (5′-GTGGTGGTGCTCGAGAGCTGTATCAGGATCAAAATAATAC-3′).
Amplification was performed using TaKaRa LA Taq DNA polymerase and a standard touchdown PCR protocol. The annealing temperature was decreased from 60°C to 45°C over 15 cycles, followed by 20 cycles at 45°C. A band of the correct size (about 1,827 bp for all toxinotypes) was purified by excision of the band from an agarose gel, followed by gel extraction. The PCR product was cloned into the pET24A+ expression vector by restriction digested with XhoI and EcoRI, followed by ligation with T4 DNA ligase. Directionality and sequence were confirmed by DNA sequencing using the forward and reverse cloning primers. Recombinant expression of these proteins yields a protein of the following sequence: Met-ASMTGGQQMGRGSEFIL-MSTSEENK-(toxinotype-specific toxin B amino acids 1760 to 2352)-YYFDPDTA-LE-(6×His tag). All native toxin B CTDs end in PDTAQLVISE, and thus, all 8 constructs have a 6-amino acid deletion of native sequence at the C terminus prior to the addition of the His tag.
All cloned CTDs were expressed as soluble full-length His-tagged proteins by expression in the E. coli strain BL21 Star (DE3) using the IPTG-free Overnight Express autoinduction system 1, as per the manufacturer's instructions (Novagen). Proteins were purified under native conditions by bind-and-elute affinity chromatography on Ni-NTA resin, followed by anion exchange in negative (pass-through) purification mode.
Glucosyltransferase domain.
The glucosyltransferase domain contains the first 546 amino acids of the VPI 10463 strain toxin A sequence in pET28a. The gene was completely synthetic, with a sequence optimized for expression in E. coli. A 6His tag was added to the N terminus.
Partial purification of C. difficile toxins from the small-scale culture filtrates.
Clinical C. difficile strains representative of toxinotypes 0, III, V, VIII, XII, and XV were grown anaerobically at a 250-ml scale. The representative strain of toxinotype 0 was the C. difficile reference strain VPI 10463 (ATCC 43255). The representative toxinotype III strain C. difficile CDC2005099 was a hypervirulent NAP1/027 strain isolated from an outbreak in Montreal, Canada, in 2005. The representative toxinotype V strain was C. difficile CDC2004255. The representative toxinotype VIII strain was C. difficile CDC2005195. The representative toxinotype XII strain was C. difficile CDC2004097. The representative toxinotype XV strain was C. difficile CDC2004012.
The supernatants were recovered by tangential flow filtration through a 0.2-μm pore membrane and adjusted to 0.4 M ammonium sulfate using a 3.7 M stock solution. The supernatant was loaded on a 1-ml phenyl Sepharose fast-slow (FF) (hi-sub) column (GE Healthcare), and the column was washed with buffer A (45 mM Tris-HCl, 45 mM NaCl, 0.4 M NH4SO4, 1 mM dithiothreitol [DTT], 0.2 mM EDTA [pH 7.5]). The crude toxins were eluted using a 200-ml gradient to buffer B (50 mM Tris-HCl, 50 mM NaCl, 1 mM DTT, 0.2 mM EDTA [pH 7.5]). Fractions containing toxins were identified by SDS-PAGE. The fractions were stored in SDS-PAGE loading buffer to prevent autoproteolysis prior to Western blot analysis.
Western immunoblotting analysis.
Western immunoblotting was used for both mapping the binding sites of MAbs to domains of the toxins and for determining which toxinotypes were bound by a given MAb. The mapping analysis used the recombinant proteins, and the toxinotype analysis used partially purified toxins isolated from strains of various toxinotypes.
Recombinant toxin fragments or partially purified toxins (about 20 ng) were analyzed using SDS-PAGE on a NuPAGE 4 to 12% polyacrylamide gel run at 200 V using SeeBlue Plus2 standards (Invitrogen). The proteins in the gel were transferred to a nitrocellulose membrane in 6 min using the Invitrogen iBlot gel blotting system. The blot was blocked with PBST (10 mM sodium phosphate, 2 mM potassium phosphate, 2.7 mM potassium chloride, 137 mM sodium chloride, 0.1% Tween 20) containing 5% nonfat dry milk (NFDM) for 1 h at room temperature. The blot was probed with the antibody of interest diluted 1:5,000 in 2.5% NFDM-PBST for 1 h at room temperature and then washed 3 times for 5 min each with PBST. The blot was incubated with goat anti-human alkaline phosphatase conjugate (Sigma) (1:6,600 dilution in 2.5% NFDM-PBST) for 1 h at room temperature. The blot was washed 3 times for 5 min each with PBST and developed with one 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium (BCIP/NBT) tablet (Sigma) in 10 ml of water. Development was stopped by putting the blot in deionized water.
Antibody affinity measurements in Bio-Layer interferometry.
The binding affinities of all MAbs were determined using Bio-Layer interferometry on a fortéBIO::Octet Red96 at 30°C. The technique involves the real-time measurement of the thickness of a protein layer on the end of a sensor. Sensors coated with immobilized protein A were first wet for 10 min in kinetics buffer (PBS [pH 7.4] containing 0.002% Tween 20 and 0.1 mg/ml bovine serum albumin [BSA]) and then dipped for 300 s into a 10 μg/ml MAb solution to capture the antibody on the end of the sensor. The stability of the protein A-MAb complex was then monitored for 300 s. Next, the antibody-captured sensors were dipped into four solutions containing C. difficile toxin A or toxin B at concentrations ranging from 20 to 0.7 μg/ml. After 400 s, the sensors were moved into kinetics buffer for 900 s to allow the bound toxin to dissociate. During all these steps, the samples were agitated at 1,000 rpm. The association and dissociation data at different toxin concentrations were fit into a single set of association and dissociation constants. The entire set of binding data for a given MAb and a given ligand was fit using least-squares minimization to standard 1:1 binding and zero-order dissociation equations. There was no background subtraction or weighting of one curve over another.
Antibody binding epitope mapping in PepSet ELISA.
C. difficile toxin A PubMed (Swiss-Prot: GenBank accession no. P16154.2), reference no. 2109310, nucleotide sequence (genomic DNA) of the strain ATCC 4325/VPI 10463 and C. difficile toxin B PubMed (Swiss-Prot: GenBank accession no. P18177.3), reference no. 2374729, nucleotide sequence (genomic DNA) of the strain ATCC 4325/VPI 10463 were used as the reference sequences to design the PepSet library of biotinylated linear peptides that cover the N-terminal (glucosyltransferase) and C-terminal (receptor binding) domains of both toxins A and B. The N-terminal peptides were 15 amino acids in length and consisted of overlapping 10-amino acid sequences and a moving window of 5 amino acids. The C-terminal peptides that covered the oligopeptide repeats were designed as nonoverlapping peptides, and most were 20 amino acids in length. The C-terminal peptides that covered the gaps between the oligopeptide repeats were 15 amino acids long and consisted of overlapping 10-amino acid sequences and a moving window of 5 amino acids. All biotin-SGSG-peptides were synthesized by Mimotopes (Minneapolis, MN). To measure MAb binding to peptides, Nunc MaxiSorp 96-well plates were coated with 100 μl of 5 μg/ml streptavidin in a carb-bicarbonate coating buffer solution. The plates were washed with PBS with 0.01% Tween 20 (PBST) and blocked with 3% BSA. The plates were washed again with PBST prior to the addition of 100 μl per well of 100 ng/ml biotinylated C. difficile peptides. Plates were allowed to incubate at 25°C for 60 min. The plates were washed again prior to the addition of MAbs at 5 μg/ml, which were then incubated at 25°C for 60 min. Unbound antibody was removed by additional washing with PBST, and MAb binding was detected using horseradish peroxidase (HRP)-conjugated goat anti-human antibody. HRP-conjugated antibody was incubated on plates for 1 h at 25°C. Plates were then washed with PBST. TMB (3,3′,5,5′-tetramethylbenzidine) substrate was added to each well and incubated for 10 min at 25°C. The colorimetric reaction was stopped by adding acidic stop solution, and the plates were read at a wavelength of 450 nm at 25°C. As all human MAbs tested bound to peptides VTGWRIINNKKYYFNPNNAI and QNRFLHLLGKIYYFGNNSKA (data not shown), this was considered a nonspecific interaction; therefore, these sequences were not reported as specific binding sites for any MAb.
Vero cell-based toxin neutralization assay.
Vero cells (African green monkey kidney epithelial cells) were seeded at 2.5 × 104 cells/well with 5% heat-inactivated fetal bovine serum (FBS) in a 96-well tissue culture plates and incubated at 37°C for 20 h. Solutions of the toxins were prepared in Vero cell complete medium (Eagle's minimum essential medium [EMEM] plus 5% heat-inactivated FBS) and used at a final concentration of 4× the 50% maximum cytopathic concentration (MC50), unless otherwise indicated. MC50 was defined as the lowest concentration of toxin inducing ≥50% maximum cytopathic response. Purified toxinotype 0 toxins were produced in-house from the reference strain VPI 10463 (ATCC 43255), as per the Sanofi Pasteur manufacturing process. One MC50 dose was 1.95 pM for toxin A and 0.016 pM for toxin B. To assess antibody toxin neutralizing activity, 2-fold dilutions of the MAbs were prepared in Vero cell medium and added to a 96-well plate. An equal volume of 8 MC50 C. difficile toxin A or toxin B solution and individual dilutions of the antibody solutions were combined in a new 96-well plate and incubated at 37°C with 5% CO2 and humidity for 1 h. There were at least four orders of magnitude molar excess of MAbs over toxin, even at the lowest concentration of antibody assessed. After 1 h, complete Vero cell medium was removed from 96-well plates containing the Vero cell monolayer, and 100 μl of antibody-toxin mixture was added to the wells. The plates were incubated for 72 h at 37°C supplied with 5% CO2 and humidity.
After 72 h of incubation, the cells were washed twice with minimum essential medium (MEM) (Invitrogen) that did not contain phenol red, l-glutamine, or FBS. Next, 100 μl of the MEM and 10 μl of alamarBlue (Invitrogen) were added to each well. The plates were gently mixed and incubated at 37°C for 4 h before reading fluorescence at 560 to 590 nm with a cutoff at 590 nm. Resazurin, the active ingredient of alamarBlue, is a nontoxic cell-permeable blue compound. As only living cells are able to reduce resazurin to a red fluorescent compound, the viable cell number is directly proportional to red fluorescence. The fluorescence results were plotted over antibody concentration. The NT50, which was defined as the lowest concentration of antibody that resulted in ≥50% neutralization of cytotoxicity, was calculated for each antibody using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). The controls for each assay were treatment with toxin A or B alone and treatment with medium alone. The calculation of maximum completeness of protection was done as follows: (average MAb fluorescence at upper asymptote − average fluorescence of medium-only control)/(average fluorescence of toxin-only control − average fluorescence of medium-only control) × 100.
The breadth of antibody protection against various toxinotypes was assessed using additional native purified toxins (TGC Biomics, Bingen, Germany). MC50 values for the toxins of each individual toxinotype were identified in ≥2 separate experiments. The MC50 values ranged from 0.9 to 1.4 pM for toxin A and 0.08 to 0.26 pM for toxin B. Each individual MAb was tested in at least three separate experiments. Intra-assay precision was ±20%.
Transepithelial electrical resistance T84 cell-based toxin neutralization assay.
The transepithelial electrical resistance (TEER) assay uses a polarized monolayer of the human colonic adenocarcinoma cell line T84 and was designed to mimic the human colon in vitro. The assay measures changes in the electrical resistance across the monolayer of T84 cells postexposure to purified C. difficile toxin A or toxin B.
In order to induce the polarization of T84 human colonic carcinoma-derived cells (ATCC CCL-248), the cells were seeded into 0.4-μm polyester transwell plates (Costar) at a seeding density of 3.6 × 105 cells/cm2. The cells were maintained at 37°C with 5% CO2 in 10% heat-inactivated FBS in Dulbecco's modified Eagle medium (DMEM)-F12 culture medium for 10 to 12 days until stable transepithelial resistance was achieved. Transepithelial resistance was measured using a Millipore Millicell ERS-2 V-Ohm meter. Medium was replaced in both the upper and lower compartments daily from day 6 and on the day of assay.
For potency testing of MAbs, either toxin A or toxin B was combined with antibody at a 1:1 ratio by volume and incubated at 37°C with gentle rocking for 30 min before being added to polarized T84 cells. For toxin A TEER assays, toxin-only or toxin-antibody mixtures were added to the upper compartment of the transwell, exposing only the apical surface of T84 cells to toxin. A final concentration of 0.6 nM toxin A purified from toxinotype 0 was used as the challenge dose. This dose was equivalent to 6 times the challenge dose required to produce a loss of transepithelial electrical resistance of 50% (6 TEER50). The apical surface of T84 was previously shown to be less sensitive to toxin B than the basolateral surface (41); therefore, toxin B TEER assays were performed by adding toxin B or toxin-antibody combinations to the lower compartment, exposing the basolateral surface to toxin. A final concentration of 0.3 nM toxin B purified from toxinotype 0 was used for the challenge dose and was equivalent to 5 TEER50. For testing of anti-toxin B MAbs in “stress tests,” a 20 TEER50 concentration of 1.1 nM toxin B was used. The controls consisted of at least one well per plate of toxin challenged without antibody and one well containing medium only. Medium was removed from the appropriate compartment, and the toxin-antibody mixture was added to the well. After preparation of the sample, transepithelial resistance was measured immediately (T0) before sample addition and then after 2.5 to 6 h (T150 − T360) of incubation at 37°C and 5% CO2.
Percent TEER loss was calculated for each sample using the equation [(T0 − T150)/T0] × 100% − %TEER loss in negative well. The percent protection for antibody was calculated for each treatment using the equation: (% TEER loss in toxin-only challenge) − (% TEER loss in antibody-neutralized toxin challenge). NT50 was defined as the lowest concentration of antibody conferring ≥50% protection. The percent completeness of protection represents the proportion of toxin-induced damage that was prevented by the highest concentration of MAb. Antibody concentrations were increased until no further protection was observed.
For testing antibodies against toxins from multiple toxinotypes, the same procedure was used as described for potency testing; however, TEER50 toxin concentrations varied by toxinotype and were adjusted to maintain 6 TEER50 toxin A challenge and 5 TEER50 toxin B challenge. The amount of toxin A used for challenge ranged from 0.5 to 1 nM and for toxin B from 0.3 to 4 nM. Each individual MAb was tested in at least three separate experiments. Intra-assay precision was ±20%.
ELISA for testing toxin A and B concentrations in C. difficile culture filtrates.
The ELISA for culture filtrates was performed as previously described (42). Briefly, ELISA plates (Corning) were coated with polyclonal goat anti-toxin IgG and then blocked with 2.5% NFDM in PBS. Purified toxin A or toxin B and clarified filtrates were serially diluted and incubated with antibody. Plates were washed, and then bound toxin was detected using polyclonal rabbit anti-toxin IgG. The plates were washed again, and anti-rabbit IgG conjugated to alkaline phosphate (SouthernBiotech) was added. The plates were washed again, and disodium p-nitrophenyl phosphate (pNPP) was added to generate a colorimetric signal. The plates were read in a plate reader at an absorbance of 405 nm. A 4-parameter fit standard curve was constructed based on a purified toxin-only signal and used to determine the amount of toxin present in the filtrate.
Animals.
Female Golden Syrian hamsters (Mesocricetus auratus) (Charles River, Wilmington, MA, or Harlan, Indianapolis, IN) weighing 70 to 90 g were used for the challenge studies. All animals were housed individually to prevent the transmission of infection. All procedures involving animals were conducted under protocols approved by the Sanofi Pasteur Institutional Animal Care and Use Committee.
Golden Syrian hamster challenge model.
To assess MAb efficacy, groups of 5 to 10 hamsters were intraperitoneally (i.p.) injected with MAb on four consecutive days beginning 3 days prior to the administration of C. difficile spore challenge. On the last day of antibody administration, hamsters were challenged intragastrically (i.g.) with the 100% lethal dose (LD100) CFU of spores of the specified C. difficile strain via a feeding needle. Clindamycin (1 mg per animal) was administered i.p. 24 h prior to spore challenge. When MAbs were assessed in combinations of two, the total dose of anti-toxin A or anti-toxin B antibody equaled 50 mg/kg of body weight/dose or 6 mg/kg/dose. Accordingly, treatment with a single anti-toxin A MAb and 2 anti-toxin B MAbs at 50 mg/kg/dose meant that each animal received 50 mg/kg/dose of anti-toxin A MAb and 25 mg/kg/dose of each anti-toxin B MAb. The concentrations for each MAb used individually or in combination of two or three are specified in the graphs (see Fig. 2 to 7). To control for the lethality of the bacterial challenge dose, each study included a group of hamsters treated with PBS. The postchallenge animals were observed at least twice a day for morbidity and mortality. Animals were assigned individual illness scores of 0, no disease; 1, loose feces; 2, wet tail and perianal region; or 3, wet perianal region, belly, and hind paws. Diarrheal disease was reported as a group mean score. The endpoints of the study were weight loss of >30%, lethargy, and nonresponsiveness or death. The studies were terminated when all surviving animals were considered healthy for at least two consecutive days.
FIG 2.

Survival (A) and protection against illness (B) in hamsters treated with 50-mg/kg/dose of A2+B2, A2+B1, A2+B4, or A2+B6 MAb combinations following challenge with clinical C. difficile strain 630. X, death. The numbers in parentheses are the doses in milligrams per kilogram of body weight.
FIG 7.
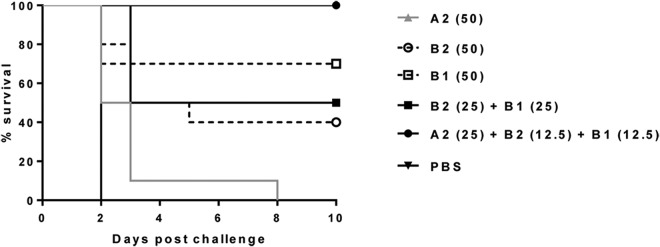
Survival of hamsters treated with total 50 mg/kg/dose of individual or combinations A2, B2, and B1 MAbs following challenge with C. difficile strain 630. The numbers in parentheses are the doses in milligrams per kilogram of body weight.
C. difficile spore preparation for challenge.
For spore preparation, C. difficile was grown for 24 h in thioglycolate broth (BD BBL). Bacterial cultures were then inoculated on anaerobic Columbia blood agar plates (BD BBL) and incubated at 37°C until confluent (3 to 4 days). After reaching confluence, the plates were incubated for an additional 3 days to induce spore formation (approximately 7 days duration). Spores were harvested into PBS without Ca or Mg, washed once, and then heated to 56°C for 10 min to kill vegetative cells. The spore suspensions were centrifuged at 500 × g for 30 min and resuspended in 20% glycerol in PBS. The spore preparations were then frozen at below −65°C for long-term storage. Viable spore counts (CFU/ml) were assessed using C. difficile selective agar (CDSA) (BD Company) plates. The spore stock was thawed at 37°C, and serial 10-fold dilutions were prepared in water. The dilutions were plated in triplicate onto prereduced CDSA agar plates. The plates were incubated under anaerobic conditions at 37°C for ≥48 h, at which time colony counts were used to determine the CFU per milliliter.
Statistical analyses.
Statistical analysis was done using the GraphPad Prism software (version 6.01; GraphPad Software, CA). A comparison of the survival data was done using the log rank test.
RESULTS
Initial screening and down-selection of individual toxin A and toxin B monoclonal antibodies using in vitro assays.
While serum from 3,000 healthy individuals without a history of CDAD was screened, only 8 individuals were identified as having both toxin-neutralizing activity and toxin binding to multiple toxinotype activities. After B cells were collected from these 8 broadly neutralizing donors, variable IgG domains from single B cells producing toxin-specific IgGs were sequenced, cloned, and used to synthesize recombinant MAbs. Monoclonal antibodies were prescreened in an IMR-90 cell-based assay for inhibition of toxin cytotoxicity, and the top 39 unique MAbs were selected for further evaluation. All of these MAbs were assessed in vitro for their activity against native toxinotype 0 toxins using the Vero cell-based cytotoxicity and T84 cell-based TEER assays. The Vero cytotoxicity assay, one of the most commonly used in the C. difficile field, was employed to evaluate the ability of the anti-toxin A and anti-toxin B MAbs to prevent C. difficile toxin-mediated cytopathic effect. The TEER assay was developed for an assessment of the ability of MAb candidates to neutralize the enterotoxicity of both toxins and potentially ameliorate diarrhea. Because both in vitro assays assessed toxin-neutralizing activity, which is considered to be the key activity for potency in vivo, we did not disqualify any MAbs from further evaluation based on a lack of performance in a single functional assay. Average potency and completeness of protection measurements from both assays for the 11 best MAbs are shown in Table 2.
TABLE 2.
Neutralizing titer and completeness of protection of selected toxin-specific MAbs in the Vero cell-based and TEER neutralization assaysa
| MAb | NT50 (pM) |
Maximum % completeness |
||
|---|---|---|---|---|
| Vero | TEER | Vero | TEER | |
| A1 | 64 | <1,300 | 90 | 80 |
| A2 | 33 | 700 | 96 | 75 |
| A3 | 1,130 | 1,300 | 90 | 80 |
| A4 | 2,700 | 4,700 | 90 | 65 |
| A5 | 2,000 | 7,300 | 75 | 60 |
| B1 | 33 | 270 | 90 | 100 |
| B2 | 33 | 70 | 70 | 100 |
| B3 | 100 | 600 | 100 | 100 |
| B4 | ND | 130 | 0 | 100 |
| B5 | 270 | 190 | 95 | 65 |
| B6 | ND | 100 | 0 | 95 |
| B4+B6 | 70 | NT | 100 | NT |
Intra-assay precision for both methods was ±20%. The data are the average of ≥3 experiments. ND, tested but not detected; NT, not tested.
In the Vero cell-based cytotoxicity assay, the anti-toxin A MAb candidate A2 demonstrated the best potency and completeness of protection in comparison with the other anti-toxin A MAbs tested. Out of all anti-toxin B MAbs tested, the B1 and B2 MAbs appeared to have the best potency in the Vero cell assay. Individually, anti-toxin B MAbs B4 and B6 showed no activity in the Vero cell assay. However, when mixed together to the same final concentration, the resulting mixture displayed high potency and complete protection, suggesting synergy between these MAbs.
The anti-toxin A MAbs had higher NT50s in the TEER assay than those in the Vero cell assay but retained the same relative order. Because the A2 candidate showed far superior potency and completeness of protection in both cell-based assays in comparison with the other anti-toxin A MAbs, it was the only MAb selected out of all anti-toxin A MAb candidates for further evaluation. Unlike the situation with the anti-toxin A MAbs, there was no single anti-toxin B antibody that was superior in both in vitro assays; therefore, we selected a variety of anti-toxin B MAbs that performed well in at least one cell-based assay for further evaluation. For example, while individual B4 and B6 displayed no detectable activity in the Vero cell assay, both were selected for further evaluation, as they were among the most potent individual anti-toxin B MAbs in TEER.
Binding affinity of MAb candidates.
Binding to C. difficile toxin A or toxin B with a dissociation constant (Kd) of ≤500 pM was one criterion used to prioritize candidate MAbs. The affinities of selected MAbs were determined using the fortéBIO Octet, and the results are shown in Table 3. The off-rate constant (Koff) error of the Octet Red96 defines a lower limit of quantitation of about 10 pM for an antibody, with an on-rate constant (Kon) of 4 × 105 M−1 s−1. Thus, the dissociation constants of MAbs A2, B6 (Table 3), A1, and A4 (data not shown) were all below the limit of quantitation. The rest of the 39 MAbs had Kd values between 48 and 1,930 pM (data not shown).
TABLE 3.
Binding affinity of selected toxin-specific MAbs, as measured by Bio-Layer interferometry
| MAb | Kd (M) | Kon (M−1 s−1) | Kon error | Koff (s−1) | Koff error |
|---|---|---|---|---|---|
| A2 | <LOQa | 3.56 × 105 | 2.16 × 103 | <LOQ | 3.16 × 10−6 |
| B1 | 4.77 × 10−11 | 5.85 × 105 | 4.34 × 103 | 2.79 × 10−5 | 3.73 × 10−6 |
| B2 | 7.97 × 10−11 | 4.20 × 105 | 3.18 × 103 | 3.35 × 10−5 | 3.92 × 10−6 |
| B3 | 4.87 × 10−10 | 1.93 × 105 | 1.45 × 103 | 9.40 × 10−5 | 3.73 × 10−6 |
| B4 | 1.21 × 10−10 | 5.31 × 105 | 4.46 × 103 | 6.44 × 10−5 | 4.26 × 10−6 |
| B5 | 2.02 × 10−10 | 4.77 × 105 | 2.15 × 103 | 9.62 × 10−5 | 2.35 × 10−6 |
| B6 | <LOQ | 7.30 × 105 | 4.61 × 103 | <LOQ | 3.13 × 10−6 |
LOQ, limit of quantitation.
Identification of binding epitopes recognized by MAb candidates.
We used several techniques to define the binding epitopes of the various candidate MAbs. Identification of the binding domain was important for many reasons, not the least of which was the potential for using multiple antibodies specific for disparate domains in the final MAb cocktail.
Domain mapping by Western blotting.
Western immunoblotting using recombinant fragments of toxins A and B (Table 1) was used to determine the domain specificity of selected MAbs. Three out of all the anti-toxin B MAbs tested, B1, B2 (Fig. 1A), and B3 (data not shown) bound to the toxin B N-terminal glucosyltransferase domain (GTD), and one, MAb B4, bound to the central translocation domain of toxin B (Fig. 1A). Anti-toxin A MAb A2 (data not shown) and anti-toxin B MAb B6 bound to the CTD fragments of their respective toxins. As MAb B6 did not bind to any protein in a Western format (not shown), it was probed as a dot blot, using nondenatured CTD fragments expressed and purified for each of the six available toxinotypes (Fig. 1B).
FIG 1.

Binding epitopes of B1, B2, B4, and B6 anti-toxin B MAbs. (A) Immunoblot of cloned fragments using candidate MAbs. (B) Dot blot of cloned CTDs of various toxinotypes with MAb B6. MWM, molecular weight marker.
Localization of linear epitopes by PepSet ELISA.
The PepSet ELISA was developed to identify linear epitopes recognized by the selected anti-C. difficile toxin MAbs. Anti-toxin A MAb A2 showed specific binding to six peptides in the CTD: VTGWQTINGKKYYFDINTGA, VTGWQTIDGKKYYFNLNTAE, ATGWQTIDGKKYYFNLNTAE, ATGWQTIDGKKYYFNTNTFI, VTGWQTINGKKYYFNTNTSI, and VTGWQTINGKVYYFMPDTAM. These peptides contained the consensus sequence TGWQTI (underlined), suggesting that this is potentially the linear epitope for the MAb A2.
Anti-toxin B MAbs B2 and B6 did not bind any linear epitopes in the peptide library; however, their functional activity in other in vitro assays suggests that these MAbs may recognize conformational rather than linear epitopes. The candidate MAb B1 bound three peptides: TDICIDTYKKSGRNK , DTYKKSGRNKALKKF, and SGRNKALKKFKEYLV. These three peptides contain the consensus sequence SGRNK (underlined), which lies in a 4-helix bundle near the N terminus of the glucosyltransferase domain. These data suggest that N-terminal binding MAbs B1 and B2 recognize nonoverlapping epitopes.
Assessment of breadth of protection of MAb candidates.
All initial characterization experiments were performed using toxinotype 0 toxins. However, there are multiple genetic variants of the toxin genes, and C. difficile strains are classified into different toxinotype variants based on the heterogeneity in toxins A and B (35). Therefore, we included an assessment of toxin neutralization across strains of various toxinotypes as a criterion in our selection of MAb candidates.
Immunoblotting experiments were performed with partially purified toxins from culture supernatants to determine the breadth of MAb binding. The anti-toxin A MAb candidate A2 recognized all five toxinotypes in the panel. While all six anti-toxin B MAbs recognized toxin B of toxinotypes 0 and III, only B3 and B6 recognized all 6 toxinotypes (data not shown).
The breadth of protection was also tested in functional assays by assessing the neutralizing activity of MAbs against purified full-length native toxins of C. difficile strains of clinically important toxinotypes. As toxins of different toxinotypes varied in their potencies, the MC50 values for all the different toxins were established in both assays. The anti-toxin B MAbs were assessed against C. difficile strains of toxinotypes 0, III, V, VIII, and X, whereas the anti-toxin A MAb candidate A2 was assessed against toxinotypes 0, III, and V, as toxinotypes VIII and X do not express full-length toxin A protein. We tested the neutralizing activity of antibodies against equal toxin potency challenge rather than at the same toxin concentrations to meaningfully compare MAb cross-neutralization against multiple toxinotypes. In both assays, candidate A2 displayed potent neutralizing activity and complete protection against toxins A of all three toxinotypes tested (Tables 4 and 5). Anti-toxin B MAb candidates B1 and B2 displayed activity against toxin B of all five toxinotypes tested (Tables 4 and 5). Taken together, these data indicate that we had identified MAbs with broad neutralizing activity.
TABLE 4.
Neutralizing titer and completeness of protection of selected toxin-specific MAbs in Vero cell-based assay against C. difficile toxins from strains of various toxinotypesa
| Measure by toxinotype | MAb tested |
||
|---|---|---|---|
| Anti-A (A2) | Anti-B |
||
| B2 | B1 | ||
| 0 | |||
| NT50 (pM) | <67 | <33 | <33 |
| Maximum % completeness | 98 | 78 | 95 |
| III | |||
| NT50 (pM) | 53 | <33 | <33 |
| Maximum % completeness | 97 | 90 | 89 |
| V | |||
| NT50 (pM) | <33 | 33 | 67 |
| Maximum % completeness | 99 | 80 | 76 |
| VIII | |||
| NT50 (pM) | <33 | <33 | |
| Maximum % completeness | 96 | 95 | |
| X | |||
| NT50 (pM) | <33 | <33 | |
| Maximum % completeness | 94 | 96 | |
The data are the average of ≥3 experiments.
TABLE 5.
Neutralizing titer and completeness of protection of selected toxin-specific MAbs in TEER assay against C. difficile toxins from strains of various toxinotypesa
| Measure by toxinotype | MAb tested |
||
|---|---|---|---|
| Anti-A (A2) | Anti-B |
||
| B2 | B1 | ||
| 0 | |||
| NT50 (pM) | 467 | 200 | 447 |
| Maximum % completeness | 94 | 85 | 84 |
| III | |||
| NT50 (pM) | 933 | 353 | 627 |
| Maximum % completeness | 87 | 91 | 81 |
| V | |||
| NT50 (pM) | 333 | 400 | 467 |
| Maximum % completeness | 100 | 80 | 87 |
| VIII | |||
| NT50 (pM) | 267 | 533 | |
| Maximum % completeness | 74 | 74 | |
| X | |||
| NT50 (pM) | 807 | 1,533 | |
| Maximum % completeness | 93 | 97 | |
The data are the average of ≥3 experiments.
Efficacy of selected anti-toxin A and anti-toxin B MAb candidate combinations in passive protection in the hamster C. difficile challenge model.
The best performers in each in vitro assay, namely, anti-toxin A MAb A2 and anti-toxin B MAbs B1, B2, B4, and B6, were chosen for evaluation in the in vivo hamster challenge model. These MAbs were also shown to bind a unique epitope and to exhibit neutralizing activity against a wide breadth of clinically important toxinotypes. Four MAb pairs were created by combining the anti-toxin A MAb A2 with one of the four anti-toxin B MAbs, B1, B2, B4, or B6. These combinations were tested at 50 mg/kg/dose for each individual MAb. This dose was chosen, as it was the highest dose used for the preclinical testing of anti-toxin specific MAbs, which subsequently have shown efficacy in a phase II clinical trial at 10 mg/kg (17, 34).
The primary challenge hamster model and dosing schedule were adapted from the same preclinical studies, described in Babcock et al. (34). First, the MAb combinations were tested against challenge with strain 630, a toxinotype 0 clinical isolate. While all control animals in the PBS-treated group succumbed to challenge by day 2, the MAb pairs showed strong protection against both death and diarrheal disease. Three of the toxin A/B MAb combinations (A2 paired with B1, B2, or B4) protected 100% of the animals, while one pair, A2 and B6 (A2+B6), conferred protection to 60% of animals (Fig. 2A). Not surprisingly, the pair A2+B6 was also the least efficacious in its protection against illness. It took 10 days for the surviving animals treated with this combination to be free of all disease symptoms, whereas the other MAb-treated groups exhibited very minor, if any, disease symptoms, which were fully resolved by day 6. By day 10, diarrhea symptoms were fully resolved in all surviving animals (Fig. 2B). These data suggest that the A2+B6 combination was the least efficacious out of the four combinations tested.
To allow for further discrimination between the three top anti-toxin B MAb candidates, the MAb combinations were next tested at a lower dose of 6 mg/kg. At this dose level, only the A2+B2 pair was fully protective, whereas A2+B1 and A2+B4 protected only 40% of animals (Fig. 3A). Moreover, treatment with the A2+B2 combination led to only mild disease, with a shorter period of 6 days to full resolution, whereas treatment with either the A2+B1 or A2+B4 combination allowed for moderate disease, with a longer resolution of 9 to11 days (Fig. 3B). A similar result was observed when weight loss was measured as an additional endpoint (data not shown).
FIG 3.

Survival (A) and protection against illness (B) in hamsters treated with 6-mg/kg/dose of A2+B1, A2+B2, or A2+B4 MAb combinations following challenge with clinical C. difficile strain 630. X, death. The numbers in parentheses are the doses in milligrams per kilogram of body weight.
Next, the most efficacious A2+B2 MAb pair was tested against hypervirulent C. difficile strains VPI 10463 and 13695#7 of toxinotypes 0 and III, respectively. Strain 13695#7 is a binary toxin-positive strain of ribotype 027. The levels of toxin A and B production by the reference VPI 10463 and clinical epidemic 13695#7 strains were assessed by ELISA in culture filtrates after 72 h of culture and compared with the levels of toxins produced in clinical strain 630. Strain VPI 10463 produced 40 to 247 times as much toxin (8,567 and 3,698 ng/ml of toxins A and B, respectively), and clinical isolate 13695#7 produced 7 to 60 times as much toxin (1,577 and 868 ng/ml of toxins A and B, respectively) as strain 630 (216 and 15 ng/ml of toxins A and B, respectively).
When tested against the high-toxin-producing strain VPI 10463, treatment with 50 mg/kg of the MAb pair A2+B2 was unable to protect from death but showed limited prolongation of life, with hamsters surviving until day 6 compared to survival until day 2 in the negative-control groups. Less protection was observed in the 6-mg/kg/dose group, as all animals reached an endpoint by day 3 (Fig. 4). As expected, the negative-control groups treated with PBS or IVIG (purified fraction of human IgG for intravenous injection, Gamunex/26N9971) showed no protection, as all animals succumbed to challenge and reached an endpoint by day 2. Similarly, when tested against clinical isolate 13695#7, the MAb pair A2+B2 showed no protection for either the 50-mg/kg or 6-mg/kg dose, as all animals succumbed to challenge and reached an endpoint by day 4 (Fig. 5). One out of 10 (10%) animals in the PBS-treated control group survived lethal challenge with toxinotype III clinical isolate 13695#7. The reason for this survival is unknown; however, it could be due to a lack of colonization. It was shown by other researchers (43) that intragastric inoculation of hamsters with C. difficile spores of toxinotype III strains did not lead to 100% colonization, whereas strain 630 of toxinotype 0 colonized 100% of animals. Once colonized, hamsters demonstrated 100% mortality. Similarly, in current study, we observed 100% mortality in the unprotected groups with strains 630 and VPI 10463 but not with 13695#7 of toxinotype III.
FIG 4.
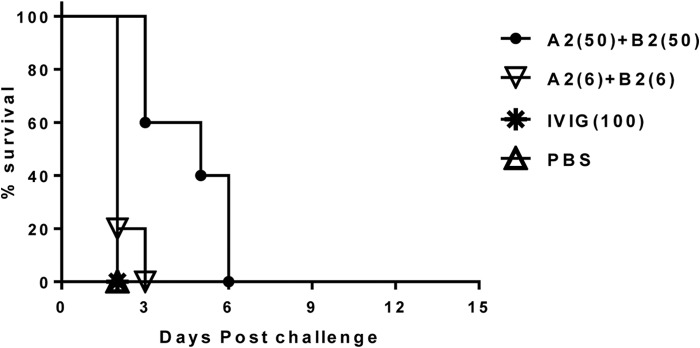
Survival in hamsters treated with the A2+B2 MAb combination postchallenge with C. difficile strain VPI 10463. The numbers in parentheses are the doses in milligrams per kilogram of body weight.
FIG 5.
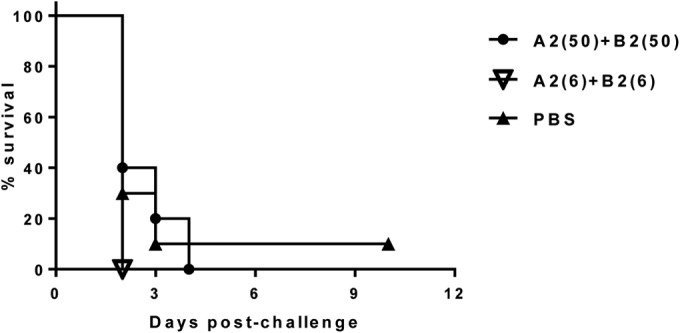
Survival in hamsters treated with the A2+B2 MAb combination postchallenge with C. difficile clinical isolate 13695#7. The numbers in parentheses are the doses in milligrams per kilogram of body weight.
Identification of synergistic anti-toxin B MAb cocktails in functional assays in vitro.
While our best MAb pair, A2+B2, protected hamsters against challenge with the lower-toxin-producing 630 strain, it offered little to no protection against challenge with the higher-toxin-producing strains 13695#7 and VPI 10463. As none of the individual anti-B MAbs had shown superior activity in both in vitro screening assays, we hypothesized that failure to protect against 13695#7 and VPI 10463 challenges could be due to weakness in our individual anti-B antibodies and that combining anti-B MAbs with nonoverlapping epitopes of toxin B might reveal synergy and enhanced potency in vitro, and, more importantly, in vivo.
Our initial characterization and down-selection data were very suggestive of this possibility, as the MAbs B6 and B4 displayed virtually no activity in the Vero cell cytotoxicity assay when assessed individually, but when combined, they were very potent (Table 2). As some of the MAbs were already displaying very high activity when assessed individually in both the Vero cell cytotoxicity and TEER assays to assess combinations of MAbs, we had to develop a more stringent stress test version of the in vitro assay using elevated toxin challenge doses to screen MAb combinations. To differentiate between combinations of MAbs in the Vero cell cytotoxicity assay, we assessed neutralizing activity against increasing toxin B concentrations of 12×, 36×, 108×, and 324× the MC50. In order to stress test the MAb combinations in the TEER assay, we increased the toxin concentration to 20 TEER50 and extended the exposure time up to 6 h. In both stress assays, the highest toxin concentrations reached a low-nanogram per milliliter level, which was comparable with the fecal toxin levels associated with a CDI diagnosis based on commercially available C. difficile toxin ELISA kits. The stress tests were performed with native toxins of toxinotypes 0 (Tables 6 and 7) and III (data not shown), with similar results.
TABLE 6.
Neutralizing activity of combinations of anti-toxin B MAbs against increasing concentrations of C. difficile toxin B in the Vero cell assaya
| MAb combination | Results for MC50 times: |
|||||||
|---|---|---|---|---|---|---|---|---|
| 12 |
36 |
108 |
324 |
|||||
| NT50 (pM) | Maximum % completeness | NT50 (pM) | Maximum % completeness | NT50 (pM) | Maximum % completeness | NT50 (pM) | Maximum % completeness | |
| B2+B1 | <67 | 90 | 107 | 91 | >133,333 | 86 | >133,333 | 14 |
| B2+B4 | 1,667 | 69 | >133,333 | 7 | >133,333 | 11 | >133,333 | 0 |
| B2+B6 | 107 | 84 | 700 | 93 | 4,433 | 90 | >133,333 | 37 |
The data are the average of ≥3 experiments.
TABLE 7.
Neutralizing activity of combinations of anti-toxin B MAbs against elevated concentration of C. difficile toxin B (20 TEER50) in the TEER assaya
| MAb combination | Results at time (h): |
|||||
|---|---|---|---|---|---|---|
| 2.5 |
4 |
6 |
||||
| NT50 (pM) | Maximum % completeness | NT50 (pM) | Maximum % completeness | NT50 (pM) | Maximum % completeness | |
| B2+B1 | 1,067 | 100 | 1,867 | 83 | 3,333 | 50 |
| B2+B4 | 520 | 90 | 733 | 62 | >3,333 | 22 |
| B2+B6 | 953 | 100 | 1,867 | 85 | 3,333 | 58 |
The data are the average of ≥3 experiments.
We chose to assess the anti-toxin B MAb pairs B1+B2, B2+B6, and B2+B4. These combinations were assembled based on a desire to improve on epitope coverage across toxin B, as B2 and B1 recognize nonoverlapping epitopes within the N-terminal domain, whereas B6 and B4 recognize epitopes in the CTD and translocation domain of toxin B, respectively. At the lower concentrations of toxin (12× the MC50) in the Vero cell neutralization assay, it was not possible to differentiate the three MAb combinations; however, at higher toxin concentrations, the combinations could be differentiated. Increasing the toxin B concentration made it apparent that B2+B1 and B2+B6 were superior to a B2+B4 combination. The ranking of B2+B1 and B2+B6 was not clear, as B2+B1 had greater activity, as measured by the NT50 at lower toxin concentrations (12× and 36× the MC50, respectively), but the B2+B6 pair maintained activity at a higher toxin concentration (108× the MC50) while B2+B4 did not (Table 6). As we were not certain which property is more important or more relevant in terms of efficacy in vivo, we were unable to differentiate B2+B1 from B2+B6. However, it was clear that both pairs were superior to B2+B4.
In the TEER assay after 2.5 h of exposure to the toxin, there was a minimal difference between the MAb combinations. At later time points (4 and 6 h), the superiority of the B2+B1 and B2+B6 combinations over the B2+B4 combination became apparent. Similar to the results seen in the Vero cell assay, it was not possible to differentiate B2+B1 from B2+B6 (Table 7). These data suggest a synergistic impact of combining MAbs, as the B2+B1 and B2+B6 combinations had greatly increased potency in both in vitro assays over what was seen with equimolar amounts of single antibodies.
Efficacy of one anti-toxin A and a 2-anti-toxin B MAb candidate cocktail in passive protection in a hamster challenge model.
Based on our in vitro data, suggesting a synergistic impact of combining selected anti-toxin B MAbs against different epitopes, we decided to assess if bringing an additional anti-toxin B MAb to the MAb cocktails would improve the in vivo efficacy against high-toxin-producing strains. We assessed three MAb combinations in the hamster challenge model: A2+B2+B1, A2+B2+B4, and A2+B2+B6. All three combinations conferred protection against the binary toxin-positive strain 13695#7. The group treated with a lower dose of A2+B2+B4 had 80% survival, whereas all other MAb combination-treated groups showed complete protection (Fig. 6A). Moreover, only the group treated with a lower dose of A2+B2+B4 showed diarrheal symptoms up to day 5, whereas all other combinations were completely protective against disease (Fig. 6B). These data demonstrate that protection against both morbidity and mortality caused by a binary toxin-producing strain is possible with toxin A- and toxin B-specific antibodies only. The same three MAb combinations were also tested against the high-toxin-producing binary toxin-negative strain VPI 10463, with the same outcome (data not shown).
FIG 6.

Survival (A) and protection against illness (B) in three-MAb-cocktail-treated hamsters postchallenge with C. difficile strain 13695#7. The numbers in parentheses are the doses in milligrams per kilogram of body weight.
Thus, in the hamster challenge model, the A2+B2+B1 and A2+B2+B6 combinations were more efficacious than A2+B2+B4. These results were in agreement with the in vitro stress test, the ranking of which showed that the B2+B4 combination was the weakest out of the three cocktails tested (Tables 7 and 8). The A2+B2+B1 combination was more efficacious in vivo than A2+B2+B6 when assessed at the lower 6-mg/kg dose level; however, due to the number of animals used for these studies, this difference did not reach statistical significance.
Efficacy of individual anti-toxin A or anti-toxin B MAb candidates in passive protection in hamster model.
To understand the contribution of each individual anti-toxin A and anti-toxin B MAb to protection, MAbs were tested alone and in combination at a total IgG dose level of 50 mg/kg/dose/animal against C. difficile challenge with strain 630. The less virulent C. difficile 630 strain was chosen over the hypervirulent 13695#7 strain for an in vivo efficacy assessment of individual MAbs, because no protection was shown even for the highest dose of 50 mg/kg of anti-toxin A plus anti-toxin B MAb combination tested against the 13695#7 strain (Fig. 5).
Treatment with MAb A2 alone prolonged survival compared to that in the PBS-treated group, but all animals eventually succumbed to challenge and reached an endpoint by day 8. In contrast, treatment with individual or a combination of two anti-toxin B MAbs allowed the survival of 40% (B2), 50% (B2+B1), and 70% (B1) of the animals. As expected, the combination of three MAbs against both toxin A and B, A2+B2+B1, protected 100% of the animals (Fig. 7). The difference in survival in the single MAb A2-treated versus the A2+B2+B1-treated group was significant (P < 0.0001). There was also a statistically significant difference in the survival of the single-MAb B2-treated group (P = 0.0041) or the MAb combination B2+B1-treated group (P = 0.0118) and the 3-MAb combination-treated group. The difference in the survival of the 3-MAb-treated group versus the group treated with B1 alone was close to being significant (P = 0.0671). There was no statistically significant difference in survival between the three groups treated with the anti-toxin B MAbs. While treatment against a single toxin can offer some protection against mortality, these data clearly indicate that combination treatment with both anti-toxin A and anti-toxin B offers a significant therapeutic benefit.
DISCUSSION
In humans, strong humoral toxin-specific immune responses elicited by natural C. difficile infection are associated with recovery and lack of disease recurrence, whereas insufficient humoral responses are associated with recurrent CDI (26–28). The principal role of circulating toxin-neutralizing antibody in immunity against disease has also been clearly demonstrated in animal models (20, 34). Accordingly, therapeutic approaches for CDI that target the two major virulence factors of the C. difficile bacterium, toxins A and B, remain in high demand. In this study, we identified and characterized efficacious fully human MAbs against C. difficile toxins A and B, with unique binding epitopes and broad neutralizing activity against clinically prevalent worldwide toxinotypes 0, III, V, and VIII. This work is the first example of efficacious anti-toxin antibodies isolated from human donors, which were assumed to be exposed to native holotoxins via natural infection. By concentrating on healthy donors and by using a functional assay to select B-cell candidates for sequencing, we were able to identify MAbs of high potential value.
We performed our initial screening using toxins purified from toxinotype 0 VPI 10463, because a substantial majority of strains worldwide are reported to belong to toxinotype 0 (35). However, taking into consideration that the prevalence of variant toxinotypes in the human population is increasing (35), for further selection, we used recombinant toxin fragments or partially purified toxins of C. difficile strains of prevalent toxinotypes to focus on donors whose sera recognized multiple toxinotypes, allowing for the subsequent identification of antibodies with breadth.
Based on the initial screening, 39 C. difficile toxin-neutralizing MAbs were isolated from 8 healthy individuals. These MAbs were prioritized based on (i) binding to C. difficile toxin A or toxin B, (ii) broad neutralization of toxin A or toxin B in the Vero cell-based cytotoxicity assay, and (iii) the ability to prevent toxin A- or toxin B-induced loss of transepithelial electrical resistance in the T84 cell monolayer. By using three distinct in vitro functional assays to rank candidates, including one assay using polarized human colon epithelial cells, we hoped to identify candidate MAbs that possessed in vitro activity that would translate into clinically relevant activity in vivo.
Epitope mapping was performed using peptides and toxin fragments to define the binding domains of the candidate MAbs. Using the toxin fragments depicted in Table 3, as defined by Kink and Williams (39), we determined that the MAb A2 binds to the CTD of toxin A and the MAb B6 binds to the CTD of toxin B. Anti-toxin B MAbs B1 and B2 bind to the GTD, and MAb B4 binds to the translocation domain. Only MAbs A2 and B1 recognized linear peptide epitopes, suggesting that B2, B4, and B6 bind to conformational epitopes within their identified binding domains.
There are longstanding and unanswered questions about the specific neutralizing epitopes within each toxin. Neutralizing antibodies to the CTD or receptor binding domain (RBD) of both toxins have long been implicated in efficacy (39). Supporting the essential role of the RBD is a recent study in which hamsters immunized with a fusion protein containing RBD of toxins A and B developed neutralizing antibodies and were protected against C. difficile spore challenge (24). In agreement with these studies, a pair of anti-toxin A and anti-toxin B RBD-specific monoclonal antibodies developed by Merck was found to be efficacious in a hamster model (34) and in humans against recurrent CDI when tested in a phase II clinical trial (17). However, in our study, we have confirmed the effectiveness of antibodies binding outside the RBD of toxin B, as neither B1 nor B2 MAbs bound to epitopes within the RBD; yet, they were effective in protecting hamsters against challenge both alone and in combination with an anti-toxin A MAb. Furthermore, we found that expansion of the toxin B epitope coverage by adding a second anti-toxin B MAb with nonoverlapping specificity resulted in an improvement in potency, as measured in the in vitro and in vivo models. The high percentage of potent antibodies we found were against the N terminus of toxin B, and examples elsewhere in the literature (29) suggest that while MAbs against the C terminus of toxin B can be efficacious, the N terminus of toxin B also plays a critical role in virulence.
When assessed in vitro, the toxin-neutralizing ability of the combinations of B2+B1 and B2+B6 was far greater than what was seen with equimolar total amounts of any of the single antibodies. This impact of combining anti-toxin B MAbs was also seen in vivo; when paired with MAb A2 at the 6-mg/kg/dose, the B1+B2, B1+B4, and B1+B6 pairs were able to offer protection against challenge with strains VPI 10463 and 13695#7, while treatment at 50 mg/kg with the A2+B2 pair provided hamsters virtually no protection against challenge with those same strains. Taken together, these data suggest that combining certain anti-toxin B MAbs offers a powerful synergistic benefit. The impact of a second antibody against the same toxin has been noted before (33), but the mechanism for this interaction remains unclear.
There is an ongoing discussion focused on the relative roles of toxins A and B in the context of C. difficile-associated disease. Data from various animal studies have demonstrated a leading role of anti-toxin A antibody in protection (23, 34). In contrast, a recent study using the infant gnotobiotic pig model showed that treatment with Merck's anti-toxin B antibody alone or in combination with the anti-toxin A antibody led to reduced gastrointestinal (GI) inflammation and full protection against systemic CDI following C. difficile challenge with hypervirulent epidemic strain NAP1/027/BI. However, when anti-toxin A antibody alone was used, animals experienced exacerbated GI inflammation and a greater fatality rate than those with the placebo-treated control (44). This anti-toxin A-mediated exacerbation has not been reported in rodent models. In Lyras et al. (45), the authors constructed isogenic tcdA and tcdB mutant strains of C. difficile to analyze the relative contributions of each toxin to morbidity and mortality in the hamster challenge model. While hamsters challenged with the toxin A mutants were as likely to die as those infected with the wild-type strain, animals challenged with the toxin B mutant were much more likely to survive challenge. These data suggest that toxin B plays a more essential role in C. difficile virulence than toxin A. A recent phase II study of hospitalized patients with CDI analyzing the effectiveness of treatment with Merck's neutralizing monoclonal antibody against C. difficile toxin A also demonstrated the importance of antibodies to toxin B, as lower serum concentrations of anti-toxin B antibody were associated with and predictive of CDI recurrence (46). In light of these recent findings, our data showing that 40 to 70% of hamsters survived lethal C. difficile challenge after treatment with anti-toxin B antibodies (either B1, B2, or B1+B2) while there were no survivors in the group administered the anti-toxin A MAb were not surprising. While it is clear that treatment against both toxins A and B is the most effective treatment, our data also add to the continuing body of work demonstrating the essential role that toxin B plays in C. difficile virulence.
In summary, we have identified and characterized the first fully human anti-C. difficile toxin MAbs isolated from human donors. These MAbs bind to unique broadly neutralizing epitopes against the C. difficile toxinotypes prevalent in pandemic areas, such as the Americas, Europe, and Asia. Individually, each MAb provided limited protection; the combination of MAbs A2 and B2 provided full protection against challenge with clinical strain 630. Moreover, the addition of a third MAb, B1, to the cocktail of MAbs A2 and B2 exhibited markedly improved potency against the more virulent strains VPI 10463 and 13695#7, with 13695#7 also secreting binary toxin. Thus, in this study, we demonstrated the possibility of protection with the C. difficile toxin A- and toxin B-specific antibodies against both morbidity and mortality caused by a binary toxin-producing strain. A bispecific approach may be considered a cost-saving option for manufacturing the two anti-toxin B candidates. These fully human MAbs are attractive candidates for further evaluation as a therapeutic option against C. difficile-associated disease.
ACKNOWLEDGMENTS
This paper is dedicated to the memory of Majid Mehtali, former CSO of Vivalis, from his collaborators and colleagues for his outstanding contribution to this work.
We thank Svetlana Stegalkina, Robert Sabharwal, and Chris Rogers for technical assistance and critical reagents. We also thank Elizabeth Masterjohn, R. Bridge Hunter, and Jennifer M. Totman for providing critical reagents. We also thank Charlotte Houston for providing cells for all of our in vitro studies.
REFERENCES
- 1.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 2.Le Monnier A, Zahar JR, Barbut F. 2014. Update on Clostridium difficile infections. Med Mal Infect 44:354–365. doi: 10.1016/j.medmal.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 3.Dawson LF, Valiente E, Donahue EH, Birchenough G, Wren BW. 2011. Hypervirulent Clostridium difficile PCR-ribotypes exhibit resistance to widely used disinfectants. PLoS One 6:e25754. doi: 10.1371/journal.pone.0025754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerding DN, Muto CA, Owens RC Jr. 2008. Measures to control and prevent Clostridium difficile infection. Clin Infect Dis 46(Suppl 1):S43–S49. doi: 10.1086/521861. [DOI] [PubMed] [Google Scholar]
- 5.Lawley TD, Clare S, Deakin LJ, Goulding D, Yen JL, Raisen C, Brandt C, Lovell J, Cooke F, Clark TG, Dougan G. 2010. Use of purified Clostridium difficile spores to facilitate evaluation of health care disinfection regimens. Appl Environ Microbiol 76:6895–6900. doi: 10.1128/AEM.00718-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, Wren BW, Fairweather NF, Dougan G, Lawley TD. 2012. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun 80:2704–2711. doi: 10.1128/IAI.00147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuijper EJ, Coignard B, Tüll P, ESCMID Study Group for Clostridium difficile; EU Member States, European Centre for Disease Prevention and Control. 2006. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect 12(Suppl 6):S2–S18. doi: 10.1111/j.1469-0691.2006.01580.x. [DOI] [PubMed] [Google Scholar]
- 8.Miller M, Gravel D, Mulvey M, Taylor G, Boyd D, Simor A, Gardam M, McGeer A, Hutchinson J, Moore D, Kelly S. 2010. Health care-associated Clostridium difficile infection in Canada: patient age and infecting strain type are highly predictive of severe outcome and mortality. Clin Infect Dis 15:194–201. doi: 10.1086/649213. [DOI] [PubMed] [Google Scholar]
- 9.Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, Pepin J, Wilcox MH, Society for Healthcare Epidemiology of America, Infectious Diseases Society of America. 2010. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 31:431–455. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 10.Peterfreund GL, Vandivier LE, Sinha R, Marozsan AJ, Olson WC, Zhu J, Bushman FD. 2012. Succession in the gut microbiome following antibiotic and antibody therapies for Clostridium difficile. PLoS One 7:e46966. doi: 10.1371/journal.pone.0046966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonna I, Welsby PD. 2005. Pathogenesis and treatment of Clostridium difficile infection. Postgrad Med J 81:367–369. doi: 10.1136/pgmj.2004.028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–714. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 14.Bacci S, Mølbak K, Kjeldsen MK, Olsen KE. 2011. Binary toxin and death after Clostridium difficile infection. Emerg Infect Dis 17:976–982. doi: 10.3201/eid/1706.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuehne SA, Collery MM, Kelly ML, Cartman ST, Cockayne A, Minton NP. 2014. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis 209:83–86. doi: 10.1093/infdis/jit426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinrichs JH. 2012. Design, production and pre-clinical evaluation of a novel toxin-based vaccine for the prevention of Clostridium difficile disease, p 30 International Clostridium difficile Symposium, 20 to 22 September 2012, Bled, Slovenia. [Google Scholar]
- 17.Lowy I, Molrine DC, Leav BA, Blair BM, Baxter R, Gerding DN, Nichol G, Thomas WD Jr, Leney M, Sloan S, Hay CA, Ambrosino DM. 2010. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med 362:197–205. doi: 10.1056/NEJMoa0907635. [DOI] [PubMed] [Google Scholar]
- 18.Gerding DN, Johnson S. 2010. Management of Clostridium difficile infection: thinking inside and outside the box. Clin Infect Dis 51:1306–1313. doi: 10.1086/657116. [DOI] [PubMed] [Google Scholar]
- 19.Gerding DN. 2012. Clostridium difficile infection prevention: biotherapeutics, immunologics, and vaccines. Discov Med 13:75–83. [PubMed] [Google Scholar]
- 20.Anosova NG, Brown AM, Li L, Liu N, Cole LE, Zhang J, Mehta H, Kleanthous H. 2013. Systemic antibody responses induced by a two-component Clostridium difficile toxoid vaccine protect against C. difficile-associated disease in hamsters. J Med Microbiol 62(Pt 9):1394–1404. doi: 10.1099/jmm.0.056796-0. [DOI] [PubMed] [Google Scholar]
- 21.Greenberg RN, Marbury T, Foglia G, Warny M. 2012. Phase I dose finding studies of an adjuvanted Clostridium difficile toxoid vaccine. Vaccine 30:2245–2249. doi: 10.1016/j.vaccine.2012.01.065. [DOI] [PubMed] [Google Scholar]
- 22.Foglia G, Shah S, Luxemburger C, Pietrobon PJ. 2012. Clostridium difficile: development of a novel candidate vaccine. Vaccine 30:4307–4309. doi: 10.1016/j.vaccine.2012.01.056. [DOI] [PubMed] [Google Scholar]
- 23.Permpoonpattana P, Hong HA, Phetcharaburanin J, Huang JM, Cook J, Fairweather NF, Cutting SM. 2011. Immunization with Bacillus spores expressing toxin A peptide repeats protects against infection with Clostridium difficile strains producing toxins A and B. Infect Immun 79:2295–2302. doi: 10.1128/IAI.00130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian JH, Fuhrmann SR, Kluepfel-Stahl S, Carman RJ, Ellingsworth L, Flyer DC. 2012. A novel fusion protein containing the receptor binding domains of C. difficile toxin A and toxin B elicits protective immunity against lethal toxin and spore challenge in preclinical efficacy models. Vaccine 30:4249–4258. doi: 10.1016/j.vaccine.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 25.Karczewski J, Zorman J, Wang S, Miezeiewski M, Xie J, Soring K, Petrescu I, Rogers I, Thiriot DS, Cook JC, Chamberlin M, Xoconostle RF, Nahas DD, Joyce JG, Bodmer JL, Heinrichs JH, Secore S. 2014. Development of a recombinant toxin fragment vaccine for Clostridium difficile infection. Vaccine 32:2812–2818. doi: 10.1016/j.vaccine.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 26.Kyne L, Warny M, Quamar A, Kelly CP. 2000. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med 342:390–397. doi: 10.1056/NEJM200002103420604. [DOI] [PubMed] [Google Scholar]
- 27.Kyne L, Warny M, Quamar A, Kelly CP. 2001. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357:189–193. doi: 10.1016/S0140-6736(00)03592-3. [DOI] [PubMed] [Google Scholar]
- 28.Aronsson B, Granström M, Möllby R, Nord CE. 1985. Serum antibody response to Clostridium difficile toxins in patients with Clostridium difficile diarrhoea. Infection 13:97–101. doi: 10.1007/BF01642866. [DOI] [PubMed] [Google Scholar]
- 29.Marozsan AJ, Ma D, Nagashima KA, Kennedy BJ, Kang YK, Arrigale RR, Donovan GP, Magargal WW, Maddon PJ, Olson WC. 2012. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J Infect Dis 206:706–713. doi: 10.1093/infdis/jis416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies NL, Compson JE, MacKenzie B, O'Dowd VL, Oxbrow AK, Heads JT, Turner A, Sarkar K, Dugdale SL, Jairaj M, Christodoulou L, Knight DE, Cross AS, Hervé KJ, Tyson KL, Hailu H, Doyle CB, Ellis M, Kriek M, Cox M, Page MJ, Moore AR, Lightwood DJ, Humphreys DP. 2013. A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin Vaccine Immunol 20:377–390. doi: 10.1128/CVI.00625-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussack G, Arbabi-Ghahroudi M, van Faassen H, Songer JG, Ng KK, MacKenzie R, Tanha J. 2011. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. Biol Chem 286:8961–8976. doi: 10.1074/jbc.M110.198754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Jin K, Xiao Y, Cheng Y, Huang Z, Wang S, Lu S. 2013. Potent monoclonal antibodies against Clostridium difficile toxin A elicited by DNA immunization. Hum Vaccin Immunother 9:2157–2164. doi: 10.4161/hv.25656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demarest SJ, Hariharan M, Elia M, Salbato J, Jin P, Bird C, Short JM, Kimmel BE, Dudley M, Woodnutt G, Hansen G. 2010. Neutralization of Clostridium difficile toxin A using antibody combinations. MAbs 2:190–198. doi: 10.4161/mabs.2.2.11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babcock GJ, Broering TJ, Hernandez HJ, Mandell RB, Donahue K, Boatright N, Stack AM, Lowy I, Graziano R, Molrine D, Ambrosino DM, Thomas WD Jr. 2006. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun 74:6339–6347. doi: 10.1128/IAI.00982-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rupnik M. 2008. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol Rev 32:541–555. doi: 10.1111/j.1574-6976.2008.00110.x. [DOI] [PubMed] [Google Scholar]
- 36.Cheknis AK, Sambol SP, Davidson DM, Nagaro KJ, Mancini MC, Hidalgo-Arroyo GA, Brazier JS, Johnson S, Gerding DN. 2009. Distribution of Clostridium difficile strains from a North American, European and Australian trial of treatment for C. difficile infections: 2005–2007. Anaerobe 15:230–233. doi: 10.1016/j.anaerobe.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Stabler RA, Dawson LF, Valiente E, Cairns MD, Martin MJ, Donahue EH, Riley TV, Songer JG, Kuijper EJ, Dingle KE, Wren BW. 2012. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS One 7:e31559. doi: 10.1371/journal.pone.0031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balassiano IT, Yates EA, Domingues RM, Ferreira EO. 2012. Clostridium difficile: a problem of concern in developed countries and still a mystery in Latin America. J Med Microbiol 61(Pt 2):169–179. doi: 10.1099/jmm.0.037077-0. [DOI] [PubMed] [Google Scholar]
- 39.Kink JA, Williams JA. 1998. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect Immun 66:2018–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korbie DJ, Mattick JS. 2008. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat Protoc 3:1452–1456. doi: 10.1038/nprot.2008.133. [DOI] [PubMed] [Google Scholar]
- 41.Stubbe H, Berdoz J, Kraehenbuhl J, Corthesy B. 2000. Polymeric IgA is superior to monomeric IgA and IgG carrying the same variable domain in preventing Clostridium difficile toxin A damaging of T84 monolayers. J Immunol 164:1952–1960. doi: 10.4049/jimmunol.164.4.1952. [DOI] [PubMed] [Google Scholar]
- 42.Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. 2005. Toxin production by and emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 366:1079–1084. [DOI] [PubMed] [Google Scholar]
- 43.Razaq N, Sambol S, Nagaro K, Zukowski W, Cheknis A, Johnson S, Gerding DN. 2007. Infection of hamsters with historical and epidemic BI types of Clostridium difficile. J Infect Dis 196:1813. doi: 10.1086/523106. [DOI] [PubMed] [Google Scholar]
- 44.Steele J, Mukherjee J, Parry N, Tzipori S. 2013. Antibody against toxin TcdB, but not toxin TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis 207:323–330. doi: 10.1093/infdis/jis669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, Poon R, Adams V, Vedantam G, Johnson S, Gerding DN, Rood JI. 2009. Toxin B is essential for virulence of Clostridium difficile. Nature 458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leav BA, Blair B, Leney M, Knauber M, Reilly C, Lowy I, Gerding DN, Kelly CP, Katchar K, Baxter R, Ambrosino D, Molrine D. 2010. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28:965–969. doi: 10.1016/j.vaccine.2009.10.144. [DOI] [PubMed] [Google Scholar]