

Abstract
A complex interaction of genetic and environmental factors can trigger the immune-mediated mechanism responsible for type 1 diabetes mellitus (T1DM) establishment. Environmental factors may initiate and possibly sustain, accelerate, or retard damage to β-cells. The role of environmental factors in this process has been exhaustive studied and viruses are among the most probable ones, especially enteroviruses. Improvements in enterovirus detection methods and randomized studies with patient follow-up have confirmed the importance of human enterovirus in the pathogenesis of T1DM. The genetic risk of T1DM and particular innate and acquired immune responses to enterovirus infection contribute to a tolerance to T1DM-related autoantigens. However, the frequency, mechanisms, and pathways of virally induced autoimmunity and β-cell destruction in T1DM remain to be determined. It is difficult to investigate the role of enterovirus infection in T1DM because of several concomitant mechanisms by which the virus damages pancreatic β-cells, which, consequently, may lead to T1DM establishment. Advances in molecular and genomic studies may facilitate the identification of pathways at earlier stages of autoimmunity when preventive and therapeutic approaches may be more effective.
Keywords: Virus, Enterovirus, Coxsackievirus, Type 1 diabetes mellitus, Auto-immune diabetes, Pathogenesis
Core tip: A complex interaction of genetic and environmental factors can trigger the immune-mediated mechanism responsible for type 1 diabetes mellitus (T1DM) establishment. The role of environmental factors in this process has been exhaustive studied and viruses are among the most probable ones, especially enteroviruses. Improvements in enterovirus detection methods and randomized studies with patient follow-up have confirmed the importance of these viruses in the pathogenesis of T1DM. However the frequency of viruses induces autoimmunity or β-cell destruction and the mechanisms and pathways how they increment the autoimmunity in T1DM still to be determined. Here, we review these mechanisms and all evolution in enterovirus studies and T1DM. Advances in molecular and genomic studies may facilitate the identification of pathways at earlier stages of autoimmunity when preventive and therapeutic approaches may be more effective.
INTRODUCTION
Type 1 diabetes mellitus (T1DM) is a chronic endocrine disorder that is caused by the progressive destruction of pancreatic β-cells, which results in insulin deficiency. A complex interaction between genetic and environmental factors may trigger this immune-mediated mechanism[1]. The most important T1DM susceptibility genes are located in the HLA-DR and DQ loci[2]. However, T1DM is not induced by genetic susceptibility alone, and environmental factors may initiate and possibly sustain, accelerate, or retard the damage to β-cells[3,4]. The role of environmental factors in the development of T1DM has been suggested because of the seasonal variation in the incidence of T1DM[5] and the conspicuous variation in the incidence of T1DM between different countries[6,7]. Immigrants often acquire a level of risk for developing T1DM that is typical for their new home country[8]. In addition, the incidence of T1DM has rapidly increased during the last decade[9-11] despite the increased prevalence of protector genes for T1DM and a concomitant decrease in high-risk genes[12,13]. Changes in the environment and how individuals respond to these variations have been indicated as being responsible for this increase in T1DM.
The following environmental factors have been suspected to contribute to the development of T1DM: dietary factors, such as cow’s milk proteins[14,15], vitamin D deficiency[16,17] and gluten[18]; pancreatic toxins[19,20], such as streptozotocin and nitrites; psychological factors[21]; and viral infection factors[22]. Viruses are among the most probable environmental factors in the development of T1DM, including rubella virus[23], rotavirus[24], mumps virus, cytomegalovirus and enteroviruses[25-27]. Recent studies using different approaches have suggested that the most promising candidates for viral triggers with clinically significant associations with T1DM development are enteroviruses[28-31].
However, it has been difficult to establish viruses as the inducers of T1DM. First, the link between infections and autoimmunity is multifactorial[32]. Several infections may act together or in an appropriate temporal sequence to trigger clinical autoimmunity. Furthermore, the particular virus that is involved in triggering T1DM may be hard to detect systemically or in the target organ after the initiation of the autoimmune response[33]. Second, the long duration of time between the possible triggering effect and the onset of the clinical symptoms of diabetes makes it difficult to establish a direct relationship. Third, T1DM patients and healthy individuals undergo multiple viral infections during their lifetime, and several of these viruses may even protect individuals from autoimmune disease[34,35]. Fourth, the “fertile field hypothesis” suggests that viral infections render tissue a “fertile ground” for autoaggressive lymphocytes to invade and expand, which leads to T1DM[36,37]. Therefore, the activation of the immune system may have a role in the pathogenesis of this disease[38].
In this review, the potential mechanisms of enterovirus infections in the establishment of T1DM will be discussed.
ENTEROVIRUSES
The Enterovirus genus of the Picornaviridae family consists of small, non-enveloped, positive, single-strand RNA viruses, including polio viruses (PVs), Coxsackie viruses A and B (CVA and CVB), echoviruses (EVs) and new enteroviruses. Three different PVs (types 1-3) and more than 60 non-polio enteroviruses cause disease in humans. These human enteroviruses (HEVs) include 23 CVAs (types 1-24; type 23 does not exist), 6 CVBs (types 1-6), 28 EVs (types 1-33; types 10, 22, 23 and 28 do not exist) and 4 other enteroviruses (EV 68-71)[39].
Enterovirus infections are transmitted from person to person by fecal-oral and, less commonly, respiratory routes, which indicates that these infections usually begin in the gastrointestinal or respiratory mucosa. After replicating in the mucosa, the virus spreads through the lymphatic system into the circulation after a brief viremic phase at secondary replication sites, which determines the types of symptoms[40].
Most enterovirus infections are asymptomatic or produce subclinical or mild symptoms, such as nonspecific febrile disease, muscle pain, sore throat, gastrointestinal distress, headache and abdominal discomfort. However, a wide variety of symptoms that affect various organs may occur, such as hand, foot and mouth disease; acute hemorrhagic conjunctivitis; aseptic meningitis; myocarditis; severe neonatal sepsis-like disease; and acute flaccid paralysis[41].
Independent of location and symptom intensity, viral replication is continuous in the lymphatic tissue. The incubation period varies from 2-30 d, and enteroviruses can be detected in various types of biological specimens from humans. Enteroviruses may be detected most readily in stools for up to 3-4 wk but rarely more than 2-3 mo. Serologic diagnoses in the acute phase are difficult because most cases are asymptomatic[42].
Systemic enterovirus infection may lead to viral dissemination to other target organs, and enterovirus RNA and protein may be detected in intestinal, heart or pancreatic tissues by reverse transcription-polymerase chain reaction (RT-PCR), immunohistochemistry or in situ hybridization[43].
ASSOCIATION BETWEEN ENTEROVIRUSES AND T1DM: A HISTORICAL OVERVIEW
This are convincing experimental results for the role of EV infection in T1DM development using mouse models[44-46], and some mechanisms of beta cell damage have been proposed based on experiments with non-obese diabetic (NOD) mice[47].
In humans, enterovirus infection has been suspected to be involved in the pathogenesis of T1DM since the late 1960s, when Gamble et al[5] described a seasonal variation in the incidence of T1DM following enterovirus infection[48] and demonstrated that the frequency of neutralizing antibodies against the CVB4 serotype was increased in newly diagnosed T1DM patients[49]. A CVB4 virus was subsequently isolated from the pancreas of a child who died from diabetic ketoacidosis, and this virus strain caused diabetes in a susceptible mouse strain[50]. However, subsequent studies failed to replicate this result.
In many serological studies, enterovirus antibodies have been more prevalent in diabetic patients than in healthy children[51]. However, critics have questioned these data because control patients were not matched for HLA risk alleles, and the detection methods for enterovirus cannot differentiate between HEV types. Several reports have not presented the same outcome, and the role of enterovirus infection in the development of T1DM has remained controversial[52].
Oikarinen et al[53] analyzed the role of past exposure to different CVB serotypes by measuring neutralizing antibodies specifically against each of the six serotypes in 249 children who were newly diagnosed with T1DM and in 249 control subjects from five European countries. Antibodies against CVB1 were more frequently detected in diabetic children than in the control group. This study suggests that coxsackie virus B may include a diabetogenic virus group and indicates that CVB1 may be a member of this group. However, because of the cross-sectional study design, the findings do not support a causality link between enterovirus infection and T1DM. Nevertheless, the same virus type has recently been observed to increase the risk of T1DM in the prospective Diabetes Prediction and Prevention (DIPP) study as a potential initiator of the β-cell-damaging process[54].
Much attention has been paid to the possible immunological cross-reactivity that is induced by a homology sequence in the 2C non-structural CVB protein and a principal diabetes autoantigen glutamic acid decarboxylase (GAD65), which share a common amino acid sequence[55,56]. GAD65 is an important target antigen in the pathogenic process of diabetes. In mice, the insulitis establishment coincides with GAD65 specific reactivity, and tolerance induction to GAD65 can prevent the disease[57,58]. Humoral and cellular responses have been detected against GAD65 before the onset of clinical diabetes[59], and auto antibodies are positive several years before diagnosis[60]. The importance of this homology in T1DM pathogenesis is supported by data showing that T cells that respond to this sequence are present both in NOD mice and T1DM patients[61,62]. This mechanism will be discussed below.
In more recent studies, RT-PCR has been used to detect CVB-specific RNA in the sera of newly diagnosed T1DM patients[63]. Yeung et al[64] conducted a useful systematic review and meta-analysis of observational molecular studies on the detection of enterovirus in T1DM patients. Observational case-control studies measured enterovirus RNA or viral protein in the blood, stool or tissue of prediabetic and diabetic patients by molecular methods. The 24 selected papers and two abstracts demonstrated a clinically significant association between enterovirus infection and autoimmunity/T1DM (odds ratios ranging from 5.5 to 17.4).
Human studies on the relationship between enterovirus and T1DM have been retrospective and based on the detection of virus infections in newly diagnosed T1DM patients. However, this study design does not allow for the evaluation of possible causal associations. To overcome this issue, several prospective studies have been performed to assess the role of CVB and other HEV infections in the induction and acceleration of T1DM and islet autoimmunity. These studies include the Childhood Diabetes in Finland (DiMe) study[65,66], the DIPP study[67,68], and the Trial to Reduce T1DM in the Genetically at Risk (TRIGR)[69], which were conducted in Finland; the BABYDIAB[70] and Babydiet[71] studies in Germany; the Diabetes and Autoimmunity Study in the Young (DAISY)[72,73] in Colorado, United States; and the Environmental Triggers of Type 1 Diabetes (MIDIA)[74] in Norway. These studies included children with an increased risk of T1DM, which was defined as a first-degree family history of T1DM, HLA susceptibility genes or both. The sampling frequency and the method of enterovirus detection varied between these studies (Table 1).
Table 1.
Longitudinal studies evaluating the association between enterovirus infection and autoimmunity/type 1 diabetes mellitus
| Study | Enterovirus infection and autoimmunity/T1DM | Cases/controls | Infection diagnose method | End point | Ref. |
| DiMe | + | 22/110 | Antibody assays | Diabetes | [65] |
| DiMe | + | 49/105 | EV RNA in serum | Diabetes | [66] |
| DIPP | + | 21/104 | Antibody assays; EV RNA in serum and stool-RT-PCR | Autoimmunity | [67] |
| DIPP | + | 41/196 | Antibody assays; EV RNA in serum-RT-PCR | Autoimmunity | [68] |
| TRIGR | + | 19/84 | Antibody assays; EV RNA in serum-RT-PCR | Autoimmunity | [69] |
| BABYDIAB | - | 28/51 | Antibody assays | Autoimmunity | [70] |
| Babydiet | - | 22/82 | EV RNA in stool | Autoimmunity | [71] |
| DAISY | - | 26/39 | EV RNA in serum, saliva and rectal swab-RT-PCR | Autoimmunity | [72] |
| DAISY | + | 50/90 | EV RNA in serum | Diabetes | [73] |
| MIDIA | - | 27/53 | EV RNA in stool-RT-PCR | Autoimmunity | [74] |
EV RNA: Enterovirus RNA; RT-PCR: Reverse transcription-polymerase chain reaction; T1DM: Type 1 diabetes mellitus; DiMe: Diuretics In the Management of Essential Hypertension; DIPP: Department of industrial policy and promotion; TRIGR: Trial to Reduce IDDM in the Genetically at Risk; DAISY: Diabetes and Autoimmunity Study in the Young.
A positive association between EV infections and a rapid progression from autoimmunity to clinical T1DM was observed both in the DiMe study as well as in the DAISY follow-up study (human longitudinal studies). However, there was no agreement in the studies’ conclusions between EV infection and islet autoimmunity development.
The results of these prospective studies may be controversial due to heterogeneity in the study design, the small number of patients in each study and the low sensitivity of the methods used to detect enterovirus infection. Another important confounding factor is the frequency of sampling because EV RNA can rarely be found continuously in stool samples for more than 3 mo, and it is found for a shorter time in serum samples[75]. The studies that indicated a positive association between enterovirus infection and T1DM used smaller sampling intervals and a wider panel of enterovirus assays than the studies that indicated no association. Similarly, most enterovirus infections are asymptomatic, and a negative result for the virus at diagnosis does not mean that its contribution is meaningless.
The prevalence of EV infections varies in populations, and independent of this, the vast majority of people infected will not develop autoimmunity or T1DM, as illustrated by Sarmineto[76]. This study showed that in Cubans that were exposed to an echovirus epidemic, a large number of patients seroconverted to islet autoantibody positivity, but T1DM prevalence has not increased. It remains to be determined how often enteroviruses induce β cell damage, autoimmunity development and clinical diabetes.
ENTEROVIRUSES AND THE DEVELOPMENT OF T1DM
Hygiene hypothesis
The hygiene hypothesis was first proposed by Strachan[77] to explain the increasing rates of asthma in highly developed countries, suggesting that contacts with a high number of infections early in life could properly modulate the adaptive immune system, and the significant changes in human living standards and the improvement of sanitary conditions meant that people had less exposure to infection, favoring an impaired immune response to environmental triggers[35,78]. This concept may be applied to many autoimmune diseases, but it does not explain all of these diseases, as there is a complex interplay between environmental exposure, the host, and other confounding variants[78-80].
Exposure to HEV, which is typically transmitted through a fecal-oral pathway, becomes less common as individual age, and infection with HEV later in life could result in an unbalanced immune response. In other words, where enterovirus infections are frequent, children develop an efficient immune response to these viruses, and when they are exposed in the future, the effects are not exacerbated or harmful. This may explain the rising worldwide incidence of T1DM over the last decade, mainly in developed societies where enterovirus infections are less prevalent[81-83].
The age when individuals are first exposed to enteroviruses may be critical in determining how the virus interacts with the host immune system (as recently demonstrated in a NOD mouse model[84]) and ultimately, whether T1DM develops. Enterovirus infections during the first year of life have been correlated with protection from the onset of T1DM[85]. A study of enterovirus-specific cellular immunity in Estonian and Finnish children at 9 mo of age found that enterovirus infections were inversely correlated with T1DM risk[86]. Estonian children who were immunized with live-attenuated PV at early ages had stronger T cell responses to CVB4 and PV type 1 compared with Finnish children who were immunized with inactivated PV. This stronger T cell immunity led to higher cross-reactivity with other enterovirus serotypes. Despite the higher incidence of enterovirus infections in Estonia, the incidence of T1DM is five times lower than that in Finland[87]. In addition, the frequency of T1DM is higher in the firstborns of multiplex families than in younger children, which could be explained by a lower exposure of firstborns than siblings to infections[88]. Therefore, viral infections during childhood may protect individuals from developing T1DM or may delay disease onset.
Implication of HEV in T1DM
CVB infections may have two different roles in the etiology of T1DM in humans: protective or triggering[89]. These proposed models are based on observations that CVBs do not replicate productively in healthy, non-inflamed pancreatic islets from NOD mice[90]. In individuals who are genetically predisposed to T1DM but not to insulitis, CVB infection may induce a protective Treg population that prevents the development of pathogenic autoimmune islet-specific T cells, thereby reducing the risk of autoimmune T1DM. However, in the absence of a protective Treg population, the extent of insulitis tends to increase with the depletion of β-cells and results in an elevated risk of developing autoimmune diabetes. The likelihood of developing T1DM requires a basic condition of genetic predisposal with the presence of anti-islet autoimmunity due to a particular virus strain and infectious dose in addition to extensive insulitis at the time of infection. A significant number of β-cells must be destroyed before the adaptive immune system may be activated. Therefore, depending on the host environment during infection, the virus may either induce protection against autoimmune T1DM onset or, with significant insulitis, may induce T1DM development (Figure 1)[89].
Figure 1.
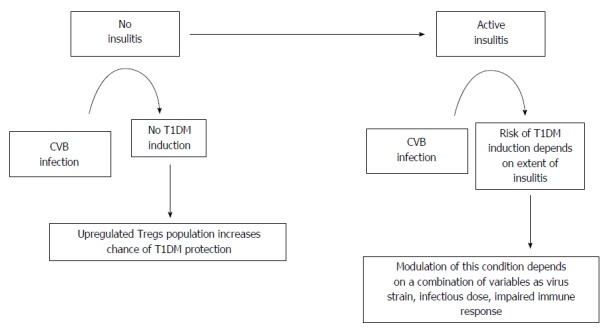
Enterovirus infection pathways on type 1 diabetes mellitus pathogenesis (adapted from Tracy et al[47]). T1DM: Type 1 diabetes mellitus; CVB: Coxsackie viruses.
Autoimmune islet inflammation may facilitate productive virus replication and induce β-cell damage[47].
Roles of enteroviruses in β-cell injury mechanisms that lead to the development of T1DM
Several hypotheses have been proposed to explain how enteroviruses affect T1DM (Figure 2)[28,40,89]. The mechanisms differ for each enterovirus; however, their coexistence is supported[33]. Enteroviruses have a strong pancreotropism[91]; human islets express coxsackie virus receptor[92], and beta cells are susceptible to enteroviruses in vitro[93]. During enterovirus infection, pancreatic islet cells may exhibit cytolysis, which exposes previously hidden self-components[94,95]. Dotta et al[96] detected enterovirus in three of six pancreatic tissue samples from patients with T1DM. Additionally, Coxsackie viruses lead to direct pancreatic injury. Elshebani et al[97] demonstrated that enteroviruses that were isolated from patients who were newly diagnosed with T1DM infected induced the destruction of human pancreatic islets in vitro.
Figure 2.
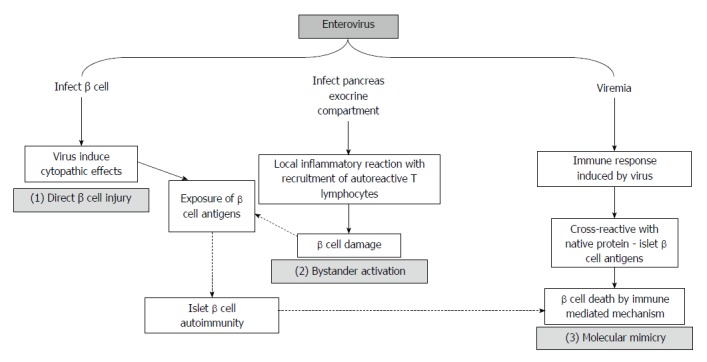
Schematic representation of possible injury mechanisms from enterovirus infection in type 1 diabetes mellitus development (adapted from Roivainen[40]).
Alternatively, β-cell damage may result from a virus-induced inflammatory reaction in the exocrine pancreas compartment. Viral infections often lead to the production of proinflammatory cytokines and the activation of antigen-presenting cells (APCs). In addition, these infections may cause tissue damage and may expose endogenous antigens that are presented by APCs. In individuals who are genetically predisposed to T1DM, viral infections may result in the impaired activation of self-reactive T cells through a mechanism that is independent of specific T-cell receptor (TCR) stimulation[98,99]. This process, called “bystander activation”, does not require specific TCR stimulation and was supported by a study of CVB4 infection in transgenic mice that resulted in the activation of circulating naive islet-specific T cells and clinical diabetes development[100].
Furthermore, the mechanism of cell destruction may be based on molecular mimicry[101]. The activation of a T-cell population against an environmental antigen results in the development of autoimmune disease if the epitope recognized shows sequence or structural similarity with a self-protein. Although virus-specific T lymphocytes are activated during an infection, antibody responses are critical in the defense against enteroviruses and are responsible for the clearance of the infection. Neutralizing antibodies are directed against the capsid surface of CVB and nonstructural proteins. These proteins are produced exclusively during the replication of the virus and are released as a consequence of the lysis of the infected cells. Directly linked to T1DM triggering was an observation of the amino acid sequence similarity between CVB4 nonstructural protein 2C and GAD65 (PEVEKEK), which suggests that the cellular anti-viral response may cross-react with the native protein, inducing an autoimmune response[102].
However, in several studies, the molecular mimicry hypothesis has been controversial because healthy control groups presented reactivity to the molecular section of GAD65[103]. In addition, molecular mimicry is extremely common in nature, which suggests that an impaired immune response may be crucial for β-cell damage in contrast with molecular mimicry[103].
All these mechanisms described may occur simultaneously. In fact, inflammatory conditions induced by virus infection will trigger autoimmunity resulting in T1DM only in susceptible individuals[38]. This hypothesis, Fertile Field, postulates that following the inflammation caused by virus infection, autoreactive T cells may be generated by bystander activation or molecular mimicry or both. The damage of beta cells and its presentation to immune system lead to antigenic epitope spreading, which explains the broad autoreactive T-cell repertoire in T1DM patients. This hypothesis may be of interest because enterovirus infection activates a strong innate immune response[104,105].
Innate and acquired immunity
Immune responses against infection by microbes are highly complex, and recent advances in the understanding of innate immunity components have elucidated the integration of innate immunity and acquired immunity. Innate immunity has limited specificity for microbes and works in a similar manner against most infectious agents. The main components of the innate immune system are physical and chemical barriers, blood proteins, including the complement system and other inflammatory mediators, such as phagocytes (neutrophils and macrophages) and natural killer cells. In contrast with the innate immune system, the acquired immune system consists of defense mechanisms with remarkable specificity for distinguishing molecules. The acquired immune response increases in magnitude with successive exposure to a specific microbe. Acquired immunity develops as a response to infection. The components of the acquired immune system are lymphocytes and their products, which are known as antibodies.
The innate immune response provides the initial defense against infectious agents. Pathogen recognition by the innate immune system is usually mediated by receptors that recognize the molecular patterns of different organisms. Recent studies have demonstrated that this recognition is mainly mediated by Toll-like receptors (TLR)[106,107]. TLR2 detects lipoproteins, lipoteichoic acid and zymosan. TLR3 recognizes dsRNA, and TLR4 recognizes lipopolysaccharide (LPS). Additionally, flagellin is detected by TLR5, ssRNA by TLR7/8 and CpG DNA by TLR9. TLRs are expressed in multiple tissues, predominantly in the cells of the immune system, particularly APCs[108].
TLR signaling induces the expression of costimulatory molecules and the production of inflammatory cytokines, such as interferons (IFNs) and interleukins (ILs), which activate both the innate and acquired immune responses. An intact innate immune response is critical for host survival during enterovirus infection. The rapid induction of IFNs is important for protection against infection. A recent study found a high rate of mortality in mice that were unresponsive to type I IFNs or lacked IFN-β after CVB infection. In addition, mortality occurred early in these mice[109].
Viral infection, innate immunity and T1DM
Viruses activate the innate immune response and induce the production of proinflammatory cytokines[98,110,111]. Enteroviruses demonstrate a tropism for the pancreas. However, the mechanism of β-cell damage is unclear[112].
CVB4 infection induces the production of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor α (TNF-α)[113]. These cytokines are involved in the host defense response against infection; however, recent studies have suggested that pro-inflammatory cytokines that are activated in response to viral infections may play a role in the pathogenesis of T1DM[114,115]. TNF-α may be involved in β-cell damage[116].
Using RT-PCR, Wen et al[117] recently isolated pancreatic islet β-cells from different species of mice and detected TLR2, 3, 4 and 9 in the islet cells of normal mice but a higher expression of TLR2, 3 and 4. The same methodology was applied in a study of pancreatic β-cells from three healthy human donors, and a higher expression of TLR3 was found in these cells. However, after treatment with microbial stimuli, LPS from gram-negative bacteria and CpG oligonucleotide DNA increased the expression of TLR2, 4 and 9. Moreover, viral stimulation with poly (I:C), a synthetic dsRNA that is capable of triggering immune responses, TLR2, 3, 4 and 9 expression was observed. The same microbial stimuli were assessed in vivo, and only the viral stimulus poly (I:C) triggered diabetes mellitus.
In NOD mice, CVB can induce diabetes because of the framework that is established by insulitis, which is caused by the increased expression of TLR4 and 8 by dendritic cells[118]. A study in human pancreatic cells demonstrated that CVB4 leads to the production of inflammatory cytokines, particularly IL-6 and TNFα. TLR4 is necessary for triggering this immune response; however, this response is independent of CVB4 internalization and replication. Therefore, the interaction between TLR4 and CBV4 in pancreatic cells may induce an innate immune response[119].
Studies in experimental models have demonstrated that the innate immune response is crucial for host survival during enterovirus infection. A weak innate immune response may allow unrestricted replication and the systemic spread of the virus. Tissue damage may ensue as a direct effect of the infecting virus, and an inefficient immune response may enable viral persistence. A robust innate immune response will limit early viral replication and diminish virally instigated damage, thereby allowing the host to mount an adaptive immune response. However, autoimmune pathologies may arise in the wake of a very potent immune response. In this scenario, the innate immune response to infection triggers the activation of self-reactive T cells[120]. Many autoimmune diseases are associated with the excessive production of IFNs[121]. Therefore, Lien et al[122] postulated that the relationship between viruses and TLRs in the development of diabetes in BioBreeding diabetes-resistant (BBDR) rats is a complex process that involves the modulation of auto-reactive T cells. In response to pathogenic microorganisms, the innate immune response that is mediated by TLR may lead to the bystander activation of auto-reactive T cells, the release of pro-inflammatory cytokines and the impairment of pancreatic islet cells[123,124].
In contrast, a strong immune response is more likely to prevent the virus from productively infecting host cells and is more likely to hinder virus access to the pancreas. However, an inefficient response enhances the risk for systemic viral spread and the induction of an innate immune response in tissues that are targeted by the virus. Therefore, viral infection with a weak initial immune response may result in higher systemic levels of pro-inflammatory cytokines and the activation of auto-reactive T cells[120] (Figure 3).
Figure 3.
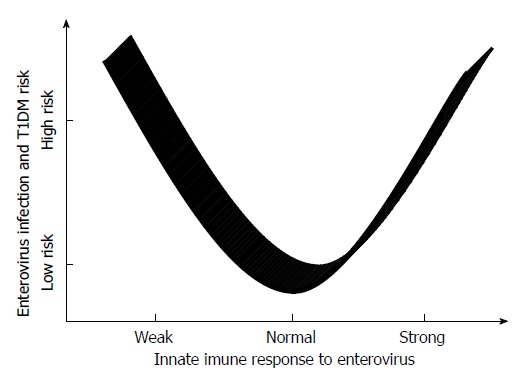
Hypothetic relationship between innate immune response to enterovirus and type 1 diabetes mellitus risk. T1DM: Type 1 diabetes mellitus.
Interferon transcriptional signature
Two studies were recently conducted with children from the BABYDIET cohort[125] and the DIPP study[126] who were genetically predisposed to T1DM. Using targeted and genome-wide transcriptomics, the expression of IFN signatures during the onset of autoimmunity and the progression to clinical T1DM was investigated. An IFN signature was first characterized in systemic lupus erythematosus disease in which IFN was found to be correlated with disease severity.
In these two studies, both research groups identified an IFN signature in the children before the development of islet autoimmunity. Ferreira et al[125] found that the IFN signature was increased before seroconversion in predisposed patients and corresponded with two or more episodes of respiratory infection, suggesting that the IFN signature is related to viral infections. The expression was intermediate in samples that were collected postseroconversion and in children with clinical T1DM.
The increased expression of type 1 IFN is a normal response to viral and bacterial infections in healthy individuals, but the pattern of expression and the presence of an IFN signature in peripheral blood may be a marker of a recent antiviral immune response. In individuals with a genetic risk for T1DM, an altered response to viral infection with unbalanced effector and regulatory cells may result in an overreaction. Importantly, the expression of an IFN signature is consistent with the activation of innate immune pathways during the initiation of islet autoimmunity and may be the first sign of this process.
CONCLUSION
Extended and cumulative discussions have been conducted on the role of enterovirus infection in the etiopathogenesis of T1DM, and substantial supporting evidence has been found. Improvements in enterovirus detection methods and randomized studies with patient follow-up have confirmed the importance of HEV in T1DM development and progression. However, the frequency, mechanisms, and pathways of virally induced autoimmunity and β-cell destruction in T1DM remain to be determined. In this way, the causal link between EV and T1DM involves a complex interplay between viruses, β-cells, innate and acquired immune systems in the particular genetic context of an individual. The influence of these several concomitant mechanisms by which the virus damages pancreatic β-cells, which, consequently, may lead to T1DM establishment makes investigating the role of enterovirus infection quite difficult. It will be extremely important to design prospective studies with large study populations, more frequent sampling of various specimens and a standardized methodology to detect enterovirus infection, linking it to islet autoimmunity or T1DM and controlling for confounding factors. Advances in molecular and genomic studies may facilitate the identification of pathways at the earlier stages of autoimmunity when preventive and therapeutic approaches may be more effective.
REVIEW CRITERIA
The databases that were searched in this study included PubMed and Embase. Papers that were published from 2004-2014 were included in the study, with a particular interest in papers that were published after 2009. The search terms were “virus”, “enterovirus”, “coxsackievirus”, “type 1 diabetes mellitus”, “auto-immune diabetes”, and “insulin-dependent diabetes”. The selected publications were full-text papers that were published in English. Additional references were selected from the reference lists of the selected article.
Footnotes
Conflict-of-interest: None.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 4, 2014
First decision: December 26, 2014
Article in press: April 9, 2015
P- Reviewer: Chi-Ho C, Cosmi E, Nishio K S- Editor: Tian YL L- Editor: A E- Editor: Wang CH
References
- 1.Çekin Y, Özkaya E, Gülkesen H, Akçurin S, Çolak D. Investigation of enterovirus infections, autoimmune factors and HLA genotypes in patients with T1DM. Minerva Endocrinol. 2014;39:67–74. [PubMed] [Google Scholar]
- 2.Kantárová D, Buc M. Genetic susceptibility to type 1 diabetes mellitus in humans. Physiol Res. 2007;56:255–266. doi: 10.33549/physiolres.930956. [DOI] [PubMed] [Google Scholar]
- 3.Ghazarian L, Diana J, Simoni Y, Beaudoin L, Lehuen A. Prevention or acceleration of type 1 diabetes by viruses. Cell Mol Life Sci. 2013;70:239–255. doi: 10.1007/s00018-012-1042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boettler T, von Herrath M. Protection against or triggering of Type 1 diabetes? Different roles for viral infections. Expert Rev Clin Immunol. 2011;7:45–53. doi: 10.1586/eci.10.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gamble DR, Taylor KW. Seasonal incidence of diabetes mellitus. Br Med J. 1969;3:631–633. doi: 10.1136/bmj.3.5671.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karvonen M, Viik-Kajander M, Moltchanova E, Libman I, LaPorte R, Tuomilehto J. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care. 2000;23:1516–1526. doi: 10.2337/diacare.23.10.1516. [DOI] [PubMed] [Google Scholar]
- 7.Jaïdane H, Hober D. Role of coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008;34:537–548. doi: 10.1016/j.diabet.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Hyöty H, Hiltunen M, Lönnrot M. Enterovirus infections and insulin dependent diabetes mellitus--evidence for causality. Clin Diagn Virol. 1998;9:77–84. doi: 10.1016/s0928-0197(98)00007-5. [DOI] [PubMed] [Google Scholar]
- 9.Jarosz-Chobot P, Polanska J, Szadkowska A, Kretowski A, Bandurska-Stankiewicz E, Ciechanowska M, Deja G, Mysliwiec M, Peczynska J, Rutkowska J, et al. Rapid increase in the incidence of type 1 diabetes in Polish children from 1989 to 2004, and predictions for 2010 to 2025. Diabetologia. 2011;54:508–515. doi: 10.1007/s00125-010-1993-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rewers M, Zimmet P. The rising tide of childhood type 1 diabetes--what is the elusive environmental trigger? Lancet. 2004;364:1645–1647. doi: 10.1016/S0140-6736(04)17368-6. [DOI] [PubMed] [Google Scholar]
- 11.Patterson CC, Dahlquist GG, Gyürüs E, Green A, Soltész G. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet. 2009;373:2027–2033. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- 12.Szopa TM, Titchener PA, Portwood ND, Taylor KW. Diabetes mellitus due to viruses--some recent developments. Diabetologia. 1993;36:687–695. doi: 10.1007/BF00401138. [DOI] [PubMed] [Google Scholar]
- 13.Forlenza GP, Rewers M. The epidemic of type 1 diabetes: what is it telling us? Curr Opin Endocrinol Diabetes Obes. 2011;18:248–251. doi: 10.1097/MED.0b013e32834872ce. [DOI] [PubMed] [Google Scholar]
- 14.Gerstein HC. Cow’s milk exposure and type I diabetes mellitus. A critical overview of the clinical literature. Diabetes Care. 1994;17:13–19. doi: 10.2337/diacare.17.1.13. [DOI] [PubMed] [Google Scholar]
- 15.Couper JJ, Steele C, Beresford S, Powell T, McCaul K, Pollard A, Gellert S, Tait B, Harrison LC, Colman PG. Lack of association between duration of breast-feeding or introduction of cow’s milk and development of islet autoimmunity. Diabetes. 1999;48:2145–2149. doi: 10.2337/diabetes.48.11.2145. [DOI] [PubMed] [Google Scholar]
- 16.Jankosky C, Deussing E, Gibson RL, Haverkos HW. Viruses and vitamin D in the etiology of type 1 diabetes mellitus and multiple sclerosis. Virus Res. 2012;163:424–430. doi: 10.1016/j.virusres.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Hyppönen E, Läärä E, Reunanen A, Järvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;358:1500–1503. doi: 10.1016/S0140-6736(01)06580-1. [DOI] [PubMed] [Google Scholar]
- 18.Sarmiento L, Galvan JA, Cabrera-Rode E, Aira L, Correa C, Sariego S, Fonseca M, Cubas-Dueñas I, Hung LH, Resik S, et al. Type 1 diabetes associated and tissue transglutaminase autoantibodies in patients without type 1 diabetes and coeliac disease with confirmed viral infections. J Med Virol. 2012;84:1049–1053. doi: 10.1002/jmv.23305. [DOI] [PubMed] [Google Scholar]
- 19.Ganda OP, Rossini AA, Like AA. Studies on streptozotocin diabetes. Diabetes. 1976;25:595–603. doi: 10.2337/diab.25.7.595. [DOI] [PubMed] [Google Scholar]
- 20.Myers MA, Mackay IR, Rowley MJ, Zimmet PZ. Dietary microbial toxins and type 1 diabetes--a new meaning for seed and soil. Diabetologia. 2001;44:1199–1200. doi: 10.1007/s001250100617. [DOI] [PubMed] [Google Scholar]
- 21.Hägglöf B, Blom L, Dahlquist G, Lönnberg G, Sahlin B. The Swedish childhood diabetes study: indications of severe psychological stress as a risk factor for type 1 (insulin-dependent) diabetes mellitus in childhood. Diabetologia. 1991;34:579–583. doi: 10.1007/BF00400277. [DOI] [PubMed] [Google Scholar]
- 22.Filippi CM, von Herrath MG. Viral trigger for type 1 diabetes: pros and cons. Diabetes. 2008;57:2863–2871. doi: 10.2337/db07-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gale EA. Congenital rubella: citation virus or viral cause of type 1 diabetes? Diabetologia. 2008;51:1559–1566. doi: 10.1007/s00125-008-1099-4. [DOI] [PubMed] [Google Scholar]
- 24.Blomqvist M, Juhela S, Erkkila S, Korhonen S, Simell T, Kupila A, Vaarala O, Simell O, Knip M, Ilonen J. Rotavirus infections and development of diabetes-associated autoantibodies during the first 2 years of life. Clin Exp Immunol. 2002;128:511–515. doi: 10.1046/j.1365-2249.2002.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coppieters KT, Boettler T, von Herrath M. Virus infections in type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2:a007682. doi: 10.1101/cshperspect.a007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roivainen M, Klingel K. Virus infections and type 1 diabetes risk. Curr Diab Rep. 2010;10:350–356. doi: 10.1007/s11892-010-0139-x. [DOI] [PubMed] [Google Scholar]
- 27.Hober D, Alidjinou EK. Enteroviral pathogenesis of type 1 diabetes: queries and answers. Curr Opin Infect Dis. 2013;26:263–269. doi: 10.1097/QCO.0b013e3283608300. [DOI] [PubMed] [Google Scholar]
- 28.Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol. 2010;6:279–289. doi: 10.1038/nrendo.2010.27. [DOI] [PubMed] [Google Scholar]
- 29.Tracy S, Drescher KM, Chapman NM. Enteroviruses and type 1 diabetes. Diabetes Metab Res Rev. 2011;27:820–823. doi: 10.1002/dmrr.1255. [DOI] [PubMed] [Google Scholar]
- 30.Diaz-Horta O, Baj A, Maccari G, Salvatoni A, Toniolo A. Enteroviruses and causality of type 1 diabetes: how close are we? Pediatr Diabetes. 2012;13:92–99. doi: 10.1111/j.1399-5448.2011.00790.x. [DOI] [PubMed] [Google Scholar]
- 31.Jaïdane H, Sauter P, Sane F, Goffard A, Gharbi J, Hober D. Enteroviruses and type 1 diabetes: towards a better understanding of the relationship. Rev Med Virol. 2010;20:265–280. doi: 10.1002/rmv.647. [DOI] [PubMed] [Google Scholar]
- 32.Filippi C, von Herrath M. How viral infections affect the autoimmune process leading to type 1 diabetes. Cell Immunol. 2005;233:125–132. doi: 10.1016/j.cellimm.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Sané F, Moumna I, Hober D. Group B coxsackieviruses and autoimmunity: focus on Type 1 diabetes. Expert Rev Clin Immunol. 2011;7:357–366. doi: 10.1586/eci.11.11. [DOI] [PubMed] [Google Scholar]
- 34.Christen U, Hintermann E, Holdener M, von Herrath MG. Viral triggers for autoimmunity: is the ‚glass of molecular mimicry‘ half full or half empty? J Autoimmun. 2010;34:38–44. doi: 10.1016/j.jaut.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol. 2012;42:5–15. doi: 10.1007/s12016-011-8285-8. [DOI] [PubMed] [Google Scholar]
- 36.In’t Veld P. Insulitis in the human endocrine pancreas: does a viral infection lead to inflammation and beta cell replication? Diabetologia. 2011;54:2220–2222. doi: 10.1007/s00125-011-2224-3. [DOI] [PubMed] [Google Scholar]
- 37.Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19:80–94. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile? Nat Rev Microbiol. 2003;1:151–157. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- 39.King R, Mills D. Coxsackie B virus. The great pretender. Aust Fam Physician. 2000;29:51–52. [PubMed] [Google Scholar]
- 40.Roivainen M. Enteroviruses: new findings on the role of enteroviruses in type 1 diabetes. Int J Biochem Cell Biol. 2006;38:721–725. doi: 10.1016/j.biocel.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 41.Glimåker M. Enteroviral meningitis. Diagnostic methods and aspects on the distinction from bacterial meningitis. Scand J Infect Dis Suppl. 1992;85:1–64. doi: 10.3109/inf.1992.24.suppl-85.01. [DOI] [PubMed] [Google Scholar]
- 42.Nix WA, Berger MM, Oberste MS, Brooks BR, McKenna-Yasek DM, Brown RH, Roos RP, Pallansch MA. Failure to detect enterovirus in the spinal cord of ALS patients using a sensitive RT-PCR method. Neurology. 2004;62:1372–1377. doi: 10.1212/01.wnl.0000123258.86752.51. [DOI] [PubMed] [Google Scholar]
- 43.Glimåker M, Abebe A, Johansson B, Ehrnst A, Olcén P, Strannegård O. Detection of enteroviral RNA by polymerase chain reaction in faecal samples from patients with aseptic meningitis. J Med Virol. 1992;38:54–61. doi: 10.1002/jmv.1890380112. [DOI] [PubMed] [Google Scholar]
- 44.Craighead JE, McLane MF. Diabetes mellitus: induction in mice by encephalomyocarditis virus. Science. 1968;162:913–914. doi: 10.1126/science.162.3856.913. [DOI] [PubMed] [Google Scholar]
- 45.Yoon JW, Onodera T, Notkins AL. Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J Exp Med. 1978;148:1068–1080. doi: 10.1084/jem.148.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoon JW, Jun HS. Viruses cause type 1 diabetes in animals. Ann N Y Acad Sci. 2006;1079:138–146. doi: 10.1196/annals.1375.021. [DOI] [PubMed] [Google Scholar]
- 47.Tracy S, Drescher KM, Jackson JD, Kim K, Kono K. Enteroviruses, type 1 diabetes and hygiene: a complex relationship. Rev Med Virol. 2010;20:106–116. doi: 10.1002/rmv.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Craig ME, Nair S, Stein H, Rawlinson WD. Viruses and type 1 diabetes: a new look at an old story. Pediatr Diabetes. 2013;14:149–158. doi: 10.1111/pedi.12033. [DOI] [PubMed] [Google Scholar]
- 49.Gamble DR, Taylor KW, Cumming H. Coxsackie viruses and diabetes mellitus. Br Med J. 1973;4:260–262. doi: 10.1136/bmj.4.5887.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- 51.Hober D, Sane F, Jaïdane H, Riedweg K, Goffard A, Desailloud R. Immunology in the clinic review series; focus on type 1 diabetes and viruses: role of antibodies enhancing the infection with Coxsackievirus-B in the pathogenesis of type 1 diabetes. Clin Exp Immunol. 2012;168:47–51. doi: 10.1111/j.1365-2249.2011.04559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Green J, Casabonne D, Newton R. Coxsackie B virus serology and Type 1 diabetes mellitus: a systematic review of published case-control studies. Diabet Med. 2004;21:507–514. doi: 10.1111/j.1464-5491.2004.01182.x. [DOI] [PubMed] [Google Scholar]
- 53.Oikarinen S, Tauriainen S, Hober D, Lucas B, Vazeou A, Sioofy-Khojine A, Bozas E, Muir P, Honkanen H, Ilonen J, et al. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes. 2014;63:655–662. doi: 10.2337/db13-0620. [DOI] [PubMed] [Google Scholar]
- 54.Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, Ruokoranta T, Lecouturier V, André P, Harju R, et al. Coxsackievirus B1 is associated with induction of β-cell autoimmunity that portends type 1 diabetes. Diabetes. 2014;63:446–455. doi: 10.2337/db13-0619. [DOI] [PubMed] [Google Scholar]
- 55.Atkinson MA, Bowman MA, Campbell L, Darrow BL, Kaufman DL, Maclaren NK. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J Clin Invest. 1994;94:2125–2129. doi: 10.1172/JCI117567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tian J, Lehmann PV, Kaufman DL. T cell cross-reactivity between coxsackievirus and glutamate decarboxylase is associated with a murine diabetes susceptibility allele. J Exp Med. 1994;180:1979–1984. doi: 10.1084/jem.180.5.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature. 1993;366:72–75. doi: 10.1038/366072a0. [DOI] [PubMed] [Google Scholar]
- 58.Tian J, Clare-Salzler M, Herschenfeld A, Middleton B, Newman D, Mueller R, Arita S, Evans C, Atkinson MA, Mullen Y, et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat Med. 1996;2:1348–1353. doi: 10.1038/nm1296-1348. [DOI] [PubMed] [Google Scholar]
- 59.Kaufman DL, Erlander MG, Clare-Salzler M, Atkinson MA, Maclaren NK, Tobin AJ. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:283–292. doi: 10.1172/JCI115573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, De Camilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–156. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 61.Hou J, Said C, Franchi D, Dockstader P, Chatterjee NK. Antibodies to glutamic acid decarboxylase and P2-C peptides in sera from coxsackie virus B4-infected mice and IDDM patients. Diabetes. 1994;43:1260–1266. doi: 10.2337/diab.43.10.1260. [DOI] [PubMed] [Google Scholar]
- 62.Richter W, Mertens T, Schoel B, Muir P, Ritzkowsky A, Scherbaum WA, Boehm BO. Sequence homology of the diabetes-associated autoantigen glutamate decarboxylase with coxsackie B4-2C protein and heat shock protein 60 mediates no molecular mimicry of autoantibodies. J Exp Med. 1994;180:721–726. doi: 10.1084/jem.180.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oikarinen S, Martiskainen M, Tauriainen S, Huhtala H, Ilonen J, Veijola R, Simell O, Knip M, Hyöty H. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes. 2011;60:276–279. doi: 10.2337/db10-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35. doi: 10.1136/bmj.d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hyöty H, Hiltunen M, Knip M, Laakkonen M, Vähäsalo P, Karjalainen J, Koskela P, Roivainen M, Leinikki P, Hovi T. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes. 1995;44:652–657. doi: 10.2337/diab.44.6.652. [DOI] [PubMed] [Google Scholar]
- 66.Lönnrot M, Salminen K, Knip M, Savola K, Kulmala P, Leinikki P, Hyypiä T, Akerblom HK, Hyöty H. Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: a prospective study. Childhood Diabetes in Finland (DiMe) Study Group. J Med Virol. 2000;61:214–220. [PubMed] [Google Scholar]
- 67.Lönnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, Savola K, Muona P, Simell T, Koskela P, et al. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes. 2000;49:1314–1318. doi: 10.2337/diabetes.49.8.1314. [DOI] [PubMed] [Google Scholar]
- 68.Salminen K, Sadeharju K, Lönnrot M, Vähäsalo P, Kupila A, Korhonen S, Ilonen J, Simell O, Knip M, Hyöty H. Enterovirus infections are associated with the induction of beta-cell autoimmunity in a prospective birth cohort study. J Med Virol. 2003;69:91–98. doi: 10.1002/jmv.10260. [DOI] [PubMed] [Google Scholar]
- 69.Sadeharju K, Hämäläinen AM, Knip M, Lönnrot M, Koskela P, Virtanen SM, Ilonen J, Akerblom HK, Hyöty H. Enterovirus infections as a risk factor for type I diabetes: virus analyses in a dietary intervention trial. Clin Exp Immunol. 2003;132:271–277. doi: 10.1046/j.1365-2249.2003.02147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Füchtenbusch M, Irnstetter A, Jäger G, Ziegler AG. No evidence for an association of coxsackie virus infections during pregnancy and early childhood with development of islet autoantibodies in offspring of mothers or fathers with type 1 diabetes. J Autoimmun. 2001;17:333–340. doi: 10.1006/jaut.2001.0550. [DOI] [PubMed] [Google Scholar]
- 71.Simonen-Tikka ML, Pflueger M, Klemola P, Savolainen-Kopra C, Smura T, Hummel S, Kaijalainen S, Nuutila K, Natri O, Roivainen M, et al. Human enterovirus infections in children at increased risk for type 1 diabetes: the Babydiet study. Diabetologia. 2011;54:2995–3002. doi: 10.1007/s00125-011-2305-3. [DOI] [PubMed] [Google Scholar]
- 72.Graves PM, Rotbart HA, Nix WA, Pallansch MA, Erlich HA, Norris JM, Hoffman M, Eisenbarth GS, Rewers M. Prospective study of enteroviral infections and development of beta-cell autoimmunity. Diabetes autoimmunity study in the young (DAISY) Diabetes Res Clin Pract. 2003;59:51–61. doi: 10.1016/s0168-8227(02)00198-5. [DOI] [PubMed] [Google Scholar]
- 73.Stene LC, Oikarinen S, Hyöty H, Barriga KJ, Norris JM, Klingensmith G, Hutton JC, Erlich HA, Eisenbarth GS, Rewers M. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: the Diabetes and Autoimmunity Study in the Young (DAISY) Diabetes. 2010;59:3174–3180. doi: 10.2337/db10-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tapia G, Cinek O, Rasmussen T, Witsø E, Grinde B, Stene LC, Rønningen KS. Human enterovirus RNA in monthly fecal samples and islet autoimmunity in Norwegian children with high genetic risk for type 1 diabetes: the MIDIA study. Diabetes Care. 2011;34:151–155. doi: 10.2337/dc10-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salminen KK, Vuorinen T, Oikarinen S, Helminen M, Simell S, Knip M, Ilonen J, Simell O, Hyöty H. Isolation of enterovirus strains from children with preclinical Type 1 diabetes. Diabet Med. 2004;21:156–164. doi: 10.1111/j.1464-5491.2004.01097.x. [DOI] [PubMed] [Google Scholar]
- 76.Sarmiento L, Cubas-Dueñas I, Cabrera-Rode E. Evidence of association between type 1 diabetes and exposure to enterovirus in Cuban children and adolescents. MEDICC Rev. 2013;15:29–32. doi: 10.37757/MR2013V15.N1.7. [DOI] [PubMed] [Google Scholar]
- 77.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 79.Ramsey CD, Celedón JC. The hygiene hypothesis and asthma. Curr Opin Pulm Med. 2005;11:14–20. doi: 10.1097/01.mcp.0000145791.13714.ae. [DOI] [PubMed] [Google Scholar]
- 80.Romagnani S. The increased prevalence of allergy and the hygiene hypothesis: missing immune deviation, reduced immune suppression, or both? Immunology. 2004;112:352–363. doi: 10.1111/j.1365-2567.2004.01925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Viskari HR, Koskela P, Lönnrot M, Luonuansuu S, Reunanen A, Baer M, Hyöty H. Can enterovirus infections explain the increasing incidence of type 1 diabetes? Diabetes Care. 2000;23:414–416. doi: 10.2337/diacare.23.3.414. [DOI] [PubMed] [Google Scholar]
- 82.Borchers AT, Uibo R, Gershwin ME. The geoepidemiology of type 1 diabetes. Autoimmun Rev. 2010;9:A355–A365. doi: 10.1016/j.autrev.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 83.Viskari H, Ludvigsson J, Uibo R, Salur L, Marciulionyte D, Hermann R, Soltesz G, Füchtenbusch M, Ziegler AG, Kondrashova A, et al. Relationship between the incidence of type 1 diabetes and maternal enterovirus antibodies: time trends and geographical variation. Diabetologia. 2005;48:1280–1287. doi: 10.1007/s00125-005-1780-9. [DOI] [PubMed] [Google Scholar]
- 84.Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest. 2009;119:1515–1523. doi: 10.1172/JCI38503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Juhela S, Hyöty H, Roivainen M, Härkönen T, Putto-Laurila A, Simell O, Ilonen J. T-cell responses to enterovirus antigens in children with type 1 diabetes. Diabetes. 2000;49:1308–1313. doi: 10.2337/diabetes.49.8.1308. [DOI] [PubMed] [Google Scholar]
- 86.Juhela S, Hyöty H, Uibo R, Meriste SH, Uibo O, Lönnrot M, Halminen M, Simell O, Ilonen J. Comparison of enterovirus-specific cellular immunity in two populations of young children vaccinated with inactivated or live poliovirus vaccines. Clin Exp Immunol. 1999;117:100–105. doi: 10.1046/j.1365-2249.1999.00954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Christen U, Wolfe T, Möhrle U, Hughes AC, Rodrigo E, Green EA, Flavell RA, von Herrath MG. A dual role for TNF-alpha in type 1 diabetes: islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J Immunol. 2001;166:7023–7032. doi: 10.4049/jimmunol.166.12.7023. [DOI] [PubMed] [Google Scholar]
- 88.Bach JF. Infections and autoimmune diseases. J Autoimmun. 2005;25 Suppl:74–80. doi: 10.1016/j.jaut.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 89.Christen U, von Herrath MG. Do viral infections protect from or enhance type 1 diabetes and how can we tell the difference? Cell Mol Immunol. 2011;8:193–198. doi: 10.1038/cmi.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tracy S, Drescher KM. Coxsackievirus infections and NOD mice: relevant models of protection from, and induction of, type 1 diabetes. Ann N Y Acad Sci. 2007;1103:143–151. doi: 10.1196/annals.1394.009. [DOI] [PubMed] [Google Scholar]
- 91.Jenson AB, Rosenberg HS, Notkins AL. Pancreatic islet-cell damage in children with fatal viral infections. Lancet. 1980;2:354–358. [PubMed] [Google Scholar]
- 92.Oikarinen M, Tauriainen S, Honkanen T, Vuori K, Karhunen P, Vasama-Nolvi C, Oikarinen S, Verbeke C, Blair GE, Rantala I, et al. Analysis of pancreas tissue in a child positive for islet cell antibodies. Diabetologia. 2008;51:1796–1802. doi: 10.1007/s00125-008-1107-8. [DOI] [PubMed] [Google Scholar]
- 93.Skog O, Korsgren O, Frisk G. Modulation of innate immunity in human pancreatic islets infected with enterovirus in vitro. J Med Virol. 2011;83:658–664. doi: 10.1002/jmv.21924. [DOI] [PubMed] [Google Scholar]
- 94.Dotta F, Galleri L, Sebastiani G, Vendrame F. Virus infections: lessons from pancreas histology. Curr Diab Rep. 2010;10:357–361. doi: 10.1007/s11892-010-0137-z. [DOI] [PubMed] [Google Scholar]
- 95.Coppieters KT, von Herrath M. Antibody cross-reactivity and the viral aetiology of type 1 diabetes. J Pathol. 2013;230:1–3. doi: 10.1002/path.4174. [DOI] [PubMed] [Google Scholar]
- 96.Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Del Prato S, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elshebani A, Olsson A, Westman J, Tuvemo T, Korsgren O, Frisk G. Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res. 2007;124:193–203. doi: 10.1016/j.virusres.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 98.Yeung WC, Al-Shabeeb A, Pang CN, Wilkins MR, Catteau J, Howard NJ, Rawlinson WD, Craig ME. Children with islet autoimmunity and enterovirus infection demonstrate a distinct cytokine profile. Diabetes. 2012;61:1500–1508. doi: 10.2337/db11-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grieco FA, Sebastiani G, Spagnuolo I, Patti A, Dotta F. Immunology in the clinic review series; focus on type 1 diabetes and viruses: how viral infections modulate beta cell function. Clin Exp Immunol. 2012;168:24–29. doi: 10.1111/j.1365-2249.2011.04556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781–785. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- 101.Kaufman DL, Clare-Salzler M, Tian J, Forsthuber T, Ting GS, Robinson P, Atkinson MA, Sercarz EE, Tobin AJ, Lehmann PV. Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes. Nature. 1993;366:69–72. doi: 10.1038/366069a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Varela-Calvino R, Sgarbi G, Arif S, Peakman M. T-Cell reactivity to the P2C nonstructural protein of a diabetogenic strain of coxsackievirus B4. Virology. 2000;274:56–64. doi: 10.1006/viro.2000.0446. [DOI] [PubMed] [Google Scholar]
- 103.Hansson SF, Korsgren S, Pontén F, Korsgren O. Enteroviruses and the pathogenesis of type 1 diabetes revisited: cross-reactivity of enterovirus capsid protein (VP1) antibodies with human mitochondrial proteins. J Pathol. 2013;229:719–728. doi: 10.1002/path.4166. [DOI] [PubMed] [Google Scholar]
- 104.Zipris D. Toll-like receptors and type 1 diabetes. Adv Exp Med Biol. 2010;654:585–610. doi: 10.1007/978-90-481-3271-3_25. [DOI] [PubMed] [Google Scholar]
- 105.Schulte BM, Lanke KH, Piganelli JD, Kers-Rebel ED, Bottino R, Trucco M, Huijbens RJ, Radstake TR, Engelse MA, de Koning EJ, et al. Cytokine and chemokine production by human pancreatic islets upon enterovirus infection. Diabetes. 2012;61:2030–2036. doi: 10.2337/db11-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 107.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 108.Bortell R, Pino SC, Greiner DL, Zipris D, Rossini AA. Closing the circle between the bedside and the bench: Toll-like receptors in models of virally induced diabetes. Ann N Y Acad Sci. 2008;1150:112–122. doi: 10.1196/annals.1447.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Deonarain R, Cerullo D, Fuse K, Liu PP, Fish EN. Protective role for interferon-beta in coxsackievirus B3 infection. Circulation. 2004;110:3540–3543. doi: 10.1161/01.CIR.0000136824.73458.20. [DOI] [PubMed] [Google Scholar]
- 110.Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- 111.Zipris D. Innate immunity in type 1 diabetes. Diabetes Metab Res Rev. 2011;27:824–829. doi: 10.1002/dmrr.1256. [DOI] [PubMed] [Google Scholar]
- 112.Smura T, Ylipaasto P, Klemola P, Kaijalainen S, Kyllönen L, Sordi V, Piemonti L, Roivainen M. Cellular tropism of human enterovirus D species serotypes EV-94, EV-70, and EV-68 in vitro: implications for pathogenesis. J Med Virol. 2010;82:1940–1949. doi: 10.1002/jmv.21894. [DOI] [PubMed] [Google Scholar]
- 113.Vreugdenhil GR, Schloot NC, Hoorens A, Rongen C, Pipeleers DG, Melchers WJ, Roep BO, Galama JM. Acute onset of type I diabetes mellitus after severe echovirus 9 infection: putative pathogenic pathways. Clin Infect Dis. 2000;31:1025–1031. doi: 10.1086/318159. [DOI] [PubMed] [Google Scholar]
- 114.Pino SC, Kruger AJ, Bortell R. The role of innate immune pathways in type 1 diabetes pathogenesis. Curr Opin Endocrinol Diabetes Obes. 2010;17:126–130. doi: 10.1097/MED.0b013e3283372819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nair S, Leung KC, Rawlinson WD, Naing Z, Craig ME. Enterovirus infection induces cytokine and chemokine expression in insulin-producing cells. J Med Virol. 2010;82:1950–1957. doi: 10.1002/jmv.21900. [DOI] [PubMed] [Google Scholar]
- 116.Bopegamage SA, Petrovicová A. Tumour necrosis factor alfa and glucose levels in sera of mice infected with coxsackie B4 and A7 viruses. Acta Virol. 1998;42:409–411. [PubMed] [Google Scholar]
- 117.Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol. 2004;172:3173–3180. doi: 10.4049/jimmunol.172.5.3173. [DOI] [PubMed] [Google Scholar]
- 118.Triantafilou K, Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J Virol. 2004;78:11313–11320. doi: 10.1128/JVI.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Trucco M. Regeneration of the pancreatic beta cell. J Clin Invest. 2005;115:5–12. doi: 10.1172/JCI23935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lind K, Hühn MH, Flodström-Tullberg M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: the innate immune response to enteroviruses and its possible role in regulating type 1 diabetes. Clin Exp Immunol. 2012;168:30–38. doi: 10.1111/j.1365-2249.2011.04557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Crow MK. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther. 2010;12 Suppl 1:S5. doi: 10.1186/ar2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lien E, Zipris D. The role of Toll-like receptor pathways in the mechanism of type 1 diabetes. Curr Mol Med. 2009;9:52–68. doi: 10.2174/156652409787314453. [DOI] [PubMed] [Google Scholar]
- 123.Filippi CM, Ehrhardt K, Estes EA, Larsson P, Oldham JE, von Herrath MG. TLR2 signaling improves immunoregulation to prevent type 1 diabetes. Eur J Immunol. 2011;41:1399–1409. doi: 10.1002/eji.200939841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shibasaki S, Imagawa A, Tauriainen S, Iino M, Oikarinen M, Abiru H, Tamaki K, Seino H, Nishi K, Takase I, et al. Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr J. 2010;57:211–219. doi: 10.1507/endocrj.k09e-291. [DOI] [PubMed] [Google Scholar]
- 125.Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, Cutler AJ, Doecke JD, Flint S, McKinney EF, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes. 2014;63:2538–2550. doi: 10.2337/db13-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kallionpää H, Elo LL, Laajala E, Mykkänen J, Ricaño-Ponce I, Vaarma M, Laajala TD, Hyöty H, Ilonen J, Veijola R, et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes. 2014;63:2402–2414. doi: 10.2337/db13-1775. [DOI] [PubMed] [Google Scholar]