

Abstract
The JC polyomavirus (JCPyV) infects approximately 50% of the human population. In healthy individuals the infection remains dormant and asymptomatic, but in immuno-suppressed patients it can cause progressive multifocal leukoencephalopathy (PML), a potentially fatal demyelinating disease. Currently, there are no drugs against JCPyV infection, nor for the treatment of PML. Here, we report the development of small molecule inhibitors of JCPyV that target the initial interaction between the virus and host cell and thereby block viral entry. Utilizing a combination of computational and NMR-based screening techniques, we target the LSTc tetrasaccharide binding site within the VP1 pentameric coat protein of JCPyV. Four of the compounds from the screen effectively block viral infection in our in vitro assays using SVG-A cells. For the most potent compound, we used saturation transfer difference NMR to determine the mode of binding to purified pentamers of JCPyV VP1. Collectively these results demonstrate the viability of this class of compounds for eventual development of JCPyV-antiviral therapeutics.
JCPyV is a polyomavirus, a family of double-stranded DNA-based viruses enclosed in small (40 nm) capsids, and the etiological agent for progressive multifocal leukoencephalopathy (PML). Seroepidemiological studies have detected anti-JCPyV antibodies in a high percentage of humans, varying from 35–70% depending on socio-economic and ethnic factors[1–3]. In most cases, JCPyV is benign and asymptomatic as a latent infection in the kidneys [4], from which it only flares up in immune-compromised patients. In these rare cases, the virus also changes its tropism, infecting the astrocytes and oligodendrocytes in the central nervous system. The death of oligodendrocytes consequently leads to the demyelination of the axons, resulting in PML. PML presents a very similar pathology to multiple sclerosis (MS) except it progresses much faster, resulting in death in only 6–12 months.
While naturally occurring PML is very rare, the use of immune-suppressors, such as tacrolimus [5] and belatacept (Nulojix) [6], used for reducing graft rejection in transplant patients, has been documented to cause the disease [7]. Cancer medications, such as infliximab (Remicade), have also been documented to lead to PML [8,9]. Likewise, immunosuppressive treatments for MS have been associated with PML. Although encouraging results have been achieved with mefloquine [10], and with a combination of cidofovir and recombinant IL-7 [11], there is no effective treatment against JCPyV.
Here, we describe our efforts to block the initial step of JCPyV infection, the association of the viral capsid with the target cell. The JCPyV capsid is composed of 72 icosahedral capsomers, whose main component is the VP1 pentameric coat protein (360 copies). The binding of VP1 to the host cell surface is mediated by the cellular lactoseries tetrasaccharide c (LSTc) as shown by Neu et al. [12]. The co-crystal structure of the VP1 pentamer with LSTc reveals the features of this association in high resolution and provides important insight into the site of this initial interaction [12]. The binding site is nestled between the loops of two VP1 monomers; making contacts with residues from loops DE, HI, and BC1 of one monomer, and BC2 of the adjacent monomer. Many of the mutations previously shown to correlate with PML including L54F, N264D/T, S266F/L, and S268/F/Y/C [13], were shown to be involved in LSTc binding. Using the structural insight afforded by the structure and a combination of computational screening and NMR structural characterization, we have identified a small molecular weight compound that potently blocks JCPyV infectivity.
Materials & Methods
Virtual Screening
Computational screening was carried out using AutoDock 4.0.1 [14]. The coordinates for the JCPyV pentamer were obtained from the Protein Data Bank (3NXG). A virtual library of 3486 small molecules from Life Chemicals Inc. was selected based on viral targeting properties and a high diversity (Tanimoto factor greater than .90). Hydrogen atoms and atomic charges were added to all the ligands and pentamer using AutoDock Tools 1.5.4. All ligands were considered as flexible while the pentamer was kept rigid. The grid box was centered on the LSTc binding site identified by the x-ray structure of the pentamer complexed with two of the binding pockets occupied (the other three pockets are occluded by proten packing) [12]. The Lamarkian genetic algorithm was used for 250,000 evaluations of each of initial 50 poses generated fro each of the small molecules. The lowest energy conformations (denoted as AY1-AY11) from the screening were selected for further experimental characterization and purchased from Life Chemicals Inc. (Burlington, ON, Canada).
Protein Expression and Purification
A non-capsid forming, truncated version of the JC virus VP1 coat protein (residues G23–N290) was cloned into pET15 vector (Novagen), with an N-terminal hexa-histidine affinity tag (gift of Prof. Dr. T. Steele, University of Tubingen). The protein was expressed and purified as previously described [12]. Briefly, protein grown in E.coli BL21(DE3) (Invitrogen), was purified by nickel affinity chromatography followed by size exclusion chromatography on a Superdex-200 column (GE Healthcare), and dialyzed into phosphate-buffered saline pH 7.4 (PBS: 10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl). Aliquots of 100 μM concentration were stored at −80°C, and diluted and used as needed.
STD NMR
The AY4 compound was solubilized in DMSO-d6 to a stock concentration of 200 mM. The pure VP1 protein was used at a final concentration of 20 μM in PBS, with 200 μM AY4, in the presence of 0.1% D38-dodecylphosphocholine (DPC), and 5% D2O. Before acquisition, the samples were incubated for 3 hours, to establish equilibrium and for AY4 to adopt the most stable configurational isomer. The control sample was prepared identically, but in absence of VP1. All NMR experiments were acquired at 25 °C on a 700 MHz Bruker Avance III NMR spectrometer, using either a TCI cryogenic probe or a TXI probe. In all experiments water suppression was achieved with excitation sculpting [15]. Resonance assignment of the AY4 compound was carried out on the control sample utilizing a 2D TOCSY spectrum (DIPSI-2 mixing sequence of 65 ms) [16] with 8–32 scans aquired with 2048 and 740 points in F2 and F1, respectively (Supplemental Information, Figure S1) The saturation transfer difference experiment acquired on- and off-resonance data in an interleaved manner (alternating at each scan). For the on-resonance experiment, the protein was saturated at 540 Hz or at 200 Hz, for 3 seconds utilizing a train of 50 ms Gaussian pulses. For the off-resonance spectrum, the sample was irradiated at −2000 Hz. The total relaxation delay was 8 seconds. A spin lock sequence of 50 ms was utilized to filter residual VP1 signals. Data acquisition consisted of 512 scans and 32k points. Controls in the absence of VP1 were performed in the same conditions. Quantitation of the STD-effect was carried out by measuring the intensity of each assigned peak in the on- and off-resonance spectra:
JCPyV and Pseudovirus Preparations
JCPyV was produced and purified as previously described [12]. Briefly, the cells were subjected to 3 freeze-thaw cycles, followed by treatment with type-V neuraminidase and deoxycholate to release the virus, and sonication to disrupt the cells. The debris was removed by centrifugation, and then the virus was pelleted by a second centrifugation through 20% sucrose at 150,000g, for 3 hours at 4°C. The viral pellet was resuspended in buffer A (10mM Tris–HCl, 50mM NaCl, 0.1mM CaCl2), and then further purified by centrifuging through a CsCl step-gradient (1.29–1.35 g/mL) at 115,000g, for 18 hours at 4°C. The band containing the virus was collected, and the CsCl was removed by dialysis. The virus was further purified through a S500HR size-exclusion column. Virus concentration was measured by the Bradford assay.
Alexa fluor-488 (Invitrogen cat#A10235) labeling of the JC virus was achieved in bicarbonate buffer pH 8.0, according to the manufacturer’s instructions. For quality check, the proteins were UV-visualized after SDS-PAGE.
Pseudovirus was produced by transfecting 293FT cells with HsOpJCVP1pwP, HsOpJCVP2ph2p, HsOpJCVP3ph3p and a reporter plasmid, phGluc (Addgene) (JC pseudovirus) or HsOpBK(Dunlop)VP1, HsOpBKVP2/ph2P, HsOpBKVP3/ph2P and phGluc (VP1, lab generated; Addgene) (BK pseudovirus) using Fugene (Promega) in T-75 flasks, according to the manufacturer’s protocol. 24h post transfection, cells were scaled up to 2.5x the plating surface area in T-150 flasks. Following 24h of growth cells were harvested by scraping. Cells were subjected to 3 freeze-thaw cycles and sonication to disrupt the cells membrane, followed by treatment with 0.25% deoxycholate (30min at 37°C) and type-V neuraminidase (1h at 37°C) to release the pseudovirus. Pseudoviruses were then purified over an iodixanol gradient, and spun for 3.5 h at 15 °C at 50,000 rpm (234,000 × g) in an SW55ti rotor. The band containing pseudovirus was extracted by syringe.
Cell Lines
293FT cells are derived from human embryonic kidney cells transformed with the SV40 large TAg (Invitrogen Life Technologies). 293FT were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented to contain 10% fetal bovine serum (FBS) (Mediatech, Inc.), 0.1 mM non-essential amino acids (NEAA), 6 mM l-glutamine, 1 mM sodium pyruvate, and 500 mg/ml geneticin. SVGA cells are subclones of the SV40 origin-defective transformed human glial cell line SVG. SVGA cells were cultured in Modified Eagle Medium (MEM) supplimented with 10% FBS. Cos-7 cells are an African green monkey kidney cell line and were cultured in DMEM supplimented with 10% FBS. All lines were grown in a humidified chamber at 37°C and 5% CO2.
Pseudovirus Infection Assays
24h prior to infection, SVGA cells (JC-PSV) or Cos-7 cells (BK-PSV) were seeded to 96w dishes at a density of 10,000 cells/well in phenol red-free MEM + 10% FBS. Phenol red-free media was used through the duration of the experiment. Compounds incubated at the indicated concentrations for 1h on ice with purified PSV. Compound + PSV mixture was used to infect pre-chilled cells for an additional 1h on ice. Following infection, cells were fed complete media (phenol red free MEM + 10% FBS) to a total volume of 100ul. 24h post infection, media was removed, cells washed, and fed to a total volume of 125ul complete media. 72h post infection, sample supernatant was measured for secreted luciferase (Glomax plate reader, Promega; Gaussia Luciferase Kit, New England Biolabs) according to manufacturer instructions. Compounds AY4 through AY7 inhibited infection of SVG cells by JC-PSV in a dose dependent manner and did not inhibit BK-PSV infection in Cos-7 cells. Cell viability was measured from the same samples for pseudovirus infection cells using the Cell Titer 96® Aqueous Non-Radioactive Cell Proliferation Assay (MTS) kit (Promega cat # G5421) according to the manufacturer’s instructions. Metabolically active cells release a product into the supernatant which is measured by absorbance at 490 nm and is directly proportional to the number of cells. Secreted luciferase (RLU) was normalized to the number of viable cells in each sample. The reduction in infection was not due to toxicity.
Viral Infection Assays
24h prior to infection, SVGA cells were seeded to 96w dishes at a density of 10,000 cells/well in MEM + 10% FBS. Compounds were incubated with JCPyV in 100ul 1x PBS for 1h on ice. Compound + virus mixture was used to infect pre-chilled SVGA cells for an additional 1h on ice. Following infection, complexes were aspirated, cells washed twice in complete media, and fed complete media appropriate to each cell type. 72h post infection, samples were fixed in ice cold methanol and stained for V-antigen (V-ag) using Pab597 and Alexa fluor 488. Infection was scored by V-ag positive nuclei per 10x field of view. Compounds were able to inhibit JCPyV infection in a dose dependent manner.
Quantification of Virus Binding by Flow Cytometry
Directly labeled JCPyV-488 was incubated with each compound in 100ul 1x PBS for 1h on ice. Following binding, complexes were incubated with 100,000 SVGA cells for an additional 1h on ice. Controls were left untreated (cells alone), bound with JC-488, or with secondary antibody (AF488, Invitrogen). Samples were washed twice in 1x PBS to remove unbound virus, fixed in 1% paraformaldehyde, an analyzed by flow cytometry (BD FacsCanto II). All compounds were able to inhibit binding of JCPyV-488 to SVG cells in a dose dependent manner.
Results
Identification of inhibitors of LSTc binding
Employing computational screening we examined the ability of a 3000 member targeted library to bind the LSTc binding region of the VP1 pentamer. The best eleven compounds targeting the LSTc binding region, share important structural similarities, including a benzenesulfonamide core in which two other aromatic or non-aromatic heterocyclic groups are connected via sulfonamide group and para- position of the benzene (Figure 1). One half of the hits share a dimethylamine-propyl arm via para-benzamide.
Figure 1.

Structure of compounds from virtual screening against JCPyV VP1 crystal structure, targeting the LSTc binding site.
Compounds AY4 - AY7 block pseudovirus infectivity in SVG-A cells
Anti-viral activities of the 11 compounds identified from the screening were tested in the JC-pseudovirus (PSV) model. JC pseudoviruses were produced in 293FT cells, and then used to infect SVG-A cells. Each compound was titrated in a dose dependent manner from 100 μM up to 1 mM (Figure 2). The level of pseudovirus infection was quantified by measuring the amount of secreted luciferase in each sample. AY4 – AY7, inhibited infection in a dose dependent manner. AY4 and AY5 significantly blocked infection at a concentration of 500 μM, and completely blocked infection at 1 mM concentration, while preserving cell viability. Importantly the four compounds demonstrated no activity in the inhibition of the BK pseudovirus in a similar assay (Supplementary Information, Figure S2). Compounds AY1 and AY10 demonstrated a slight increase in infectivity at the highest dose but was not concentration dependent and therefore not further examined.
Figure 2.
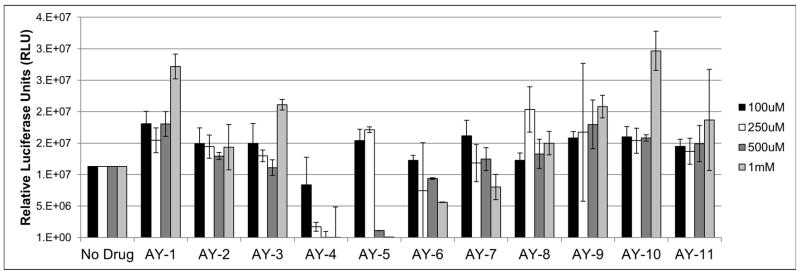
Compounds AY4 to AY7 block infection by the JC pseudovirus. The 11 compounds, AY1-AY11, were tested in the JC-PSV assay. Purified pseudoviruses were added to SVG-A cell cultures, in the presence of the compounds, and 72 h post infection, supernatants from each sample were measured for secreted luciferase.
To validate the results from the pseudoviral model, the hit compounds AY4 through −7 were then tested in a JCPyV assay. JC virions were produced as previously described [12,17], and the level of infection was quantified 72 hour post infection by scoring the number of V-antigen positive cells (Figure 3). This experiment recapitulated the results observed with the pseudovirus model: a clear dose response is observed, with compound AY4 showing the best inhibitory activity.
Figure 3.
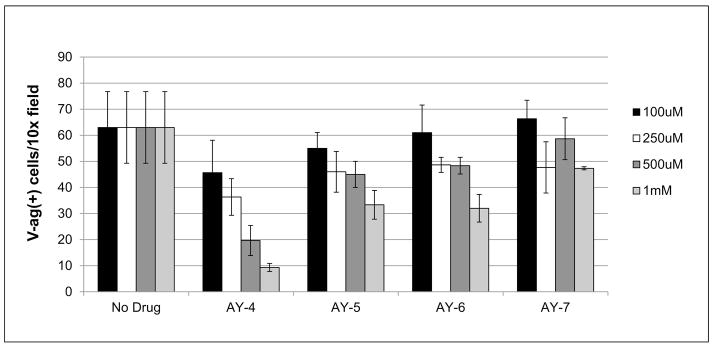
JCPyV antiviral activity inhibited by AY4 to AY7 compounds. Purified JC virus preparation was used to infect SVG-A cells, in the presence of increasing amounts of the compounds. Post-infection cells were fixed and stained for the presence of nuclear VP1 protein. Compound AY4 shows an outstanding inhibitory activity. (error bars – SD, P<0.05)
Compounds AY4 – AY7 bind directly to the viral surface, and block viral attachment to cells
Our next goal was to verify the mechanism of the AY4 to AY7 antiviral activity. Our original hypothesis was that the antiviral activity was mediated by blockage of viral attachment to host cells, as inferred by targeting the key regions of the viral capsid. Alexa Fluor 488 labeled JCPyV (JC-488) was pre-incubated with AY4 to AY7, as previously described [17]. The drug-preincubated JC-488 preparations were then added to SVG-A cells and the percentage of cells bound to JC-488 in the presence of AY4 to −7 after incubation was analyzed by flow cytometry. The data clearly shows that the compound-bound JC-488 has significantly impaired host cell binding abilities even at the lower doses tested, when compared to JC-488 alone (Figure 4).
Figure 4. Compounds AY4 to AY7 block the interaction of labeled JC virus with SVG-A cells.

JC virions were labeled with Alexa Fluor 488 and the SVG-A cells were infected in the presence of the compounds. The cell populations were fixed and quantitated by flow cytometry, scoring for surface bound JCPyV-488. The compounds significantly diminish the cell binding abilities of the virus.
Of the compounds tested, AY4 was most efficient at blocking binding of JC-488 to host cells. While the virus binding is already blocked at a compound concentration of 100 μM, the infection assays (Figures 2 and 3) show that viral infection is decreased in a dose dependent manner.
Compound AY4 directly binds to the purified VP1 pentamer
To characterize the mode of binding of AY4 to the VP1 pentamer, we turned to high resolution NMR and saturation transfer difference (STD) experiments [18,19]. First, two technical challenges had to be overcome. As the compounds of this series are only poorly soluble in water, we utilized zwitterionic dodecyl-phosphocoline (DPC) as a solubilizing agent and obtained high quality 1D 1H NMR spectra for AY4 in aqueous buffer. In addition, the trisubstituted aryl-amide functionality of AY4 generated cis/trans isomers leading to two sets of signals exchanging on the NMR-timescale. However, the system slowly equilibrates in favor of the trans isomer after a 3 hour equilibration.
The STD-NMR experiments were carried out in the presence of perdeuterated DPC and the VP1 pentamer. Control experiments were acquired under identical conditions in the presence of DPC but with no VP1, to exclude any non-specific effect due to the environment. The STD-NMR experiment shows protein-ligand interaction (in the form of a residual difference spectrum, Figure 5) and allows group epitope mapping [20] to characterize the binding interactions at an atomic level. The strongest STD signal is exhibited by the fluorinated benzothiazole, and by the tetrahydro-quinoline groups, indicating a strong interaction with VP1, possibly binding to internal hydrophobic pockets. The weakest STD signal is obtained from the dimethyl-amino-propyl arm, indicating that this is projecting away from the protein. Next, we re-examined the results from computational docking to find binding poses consistent with the experimental data. The lowest energy structure, in accord with the STD data, is shown in Figure 6.
Figure 5. Compound AY4 directly binds to the JCPyV VP1 pentamer.
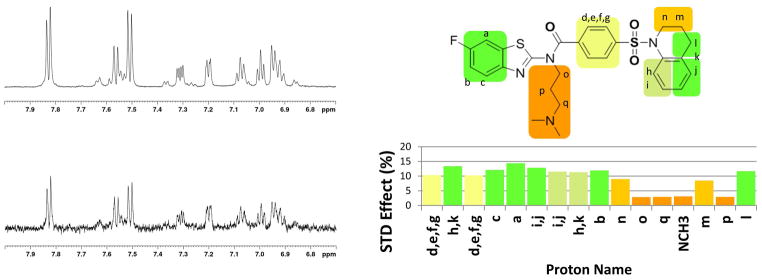
The direct interaction between the VP1 pentamer and AY4 was ascertained via saturation transfer difference NMR. (left) top: reference spectrum with off-resonance saturation showing the AY4 1H aromatic region resonances; bottom: difference spectrum resulting from saturation transfer to the aromatic protons of AY4. (right) the STD effect was quantified and mapped onto the structure of AY4.
Figure 6. Model of the binding of AY4 to the JCPyV VP1 pentamer.

The vdW surface of the VP1 pentamer is color coded: BC loop (light green), HI loop (blue), DE loop (yellow) and BC2 from the other pentamer (dark green). In white sticks the lowest energy structure of AY4 in agreement with the experimental STD-NMR data. In orange sticks, for reference, is depicted LSTc as in the crystal structure.
Discussion
Antiviral drugs can be generally categorized into replication inhibitors, which suppress viral multiplication, and entry inhibitors, which block the spread of infection to new cells. Most replication inhibitors are older generation drugs, which came into existence as the fruition of classical cell culture screens. However, the availability of detailed molecular structures and advanced computational screening methods have allowed the shift from non-mechanistic, empirical screens to more focused, mechanism-based drug design strategies [21]. As a result of this trend, many of the new generation antiviral drugs, and drug candidates, are entry inhibitors, which either target the capsid assembly or association to the cells [22–27].
The LSTc binding pocket was chosen as a target since binding to this site of glycosylation mediates the infection of host cells with the JC virus.12 Of the initial 11 hit compounds, 4 have shown antiviral activity in the in vitro tests, the best of which was AY4. All four of the compounds block viral attachment to cells in a dose dependent manner, as evidenced by pseudovirus (PSV) and virus assays. Interestingly, the compounds inhibit most of the virion capsid attachment to cells by 100 μM concentration (Figure 4). However, additional titration of the compounds continues to decrease the viral load in cells (Figures 2). This may indicate that the compounds have an additional inhibitory activity besides inhibition of entry. Whether they can act as replication inhibitors, for example, is currently under investigation.
A comparison of the structural features of the compounds targeting the LSTc site provides important insight into the requirements for activity. It is interesting to note that the AY2, 8, 10 and 11 have significant structural similarity to the active compounds. AY2, 8 and 10 contain a 4-(3,4-dihydroquinolin-1(2H)-ylsulfonyl)-benzamide group, but are unable to block JCPyV infectivity. This partially implies the importance of the benzothiazole ring for anti-viral activity. All four active compounds are halogenated, while AY8, which is structurally very similar to the most active compound AY4, but lacks the 6-fluoro group (6-fluorobenzothiazol) (replaced with 4-methyl (4-methylbenzothiazol group)) lacks activity. The location of the halogenation seems to be critical as well: AY11 is a structural isomer of AY7, only differing by the position of the halogen atom in the benzothiazole ring, and yet is not active. These structure-activity relationship data highlight the specificity of VP1 binding. The compounds displayed no inhibitory activity in a BK pseudovirus assay, further confirming the compounds specificity for the LSTc binding site of JCPyV.
Our proposed model for the binding of AY4 to the VP1-pentamer is shown in Figure 6. The topological orientation of the ligand is fully consistent with the NMR-derived STD data and the structure-activity relationship derived from the analysis of the 12 compounds. For example, the benzothiazole ring is within a small hydrophobic cleft which would not tolerate modifications beyond the 6-fluoro modification of AY4. The ligand makes a number of interactions with residues previously shown to be important for LSTc binding and JCPyV infectivity, including L54, F67, N123, and S268 [12,13]. Unfortunately, all attempts to demonstrate the ability of AY4 to displace LSTc binding are inconclusive, largely due to the low affinity of LSTc for the VP1 pentamer, (5 mM) [12]. The poor affinity of LSTc is consistent with its inability to inhibit infection at a concentration of 1 mM, where our compound demonstrates almost complete protection.
Here, we present a novel class of compounds that represent a first step in development of inhibitors for the initial binding event of virus to host cell. Although optimization is required to enhance affinity and physicochemical properties (solubility), activities which are on-going in our laboratories, the inhibitory activity is significantly better than the natural substrate for cellular association.
Supplementary Material
Acknowledgments
We wish to thank Dr. Christian Nelson (Brown University), and members of Dr. Thilo Stehle’s laboratory (University of Tübingen, Germany) for the fruitful discussions. Gretchen Gee developed the protocol for production of JC and BK pseudoviruses, and generated BK-PSV VP1 (Dunlop). This work was support in part by the National Institutes of Health through the award P01 NS 065719 and the core facilities supported by P30 GM 103410.
ABBREVIATIONS
- JCPyV
JC-polyomavirus
- PML
progressive multifocal leukoencephalopathy
- STD
saturation transfer difference spectroscopy
References
- 1.Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DW, Miller E. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71:115–23. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- 2.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis. 2009;199:837–46. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 4.Dorries K. Molecular biology and pathogenesis of human polyomavirus infections. Dev Biol Stand. 1998;94:71–9. [PubMed] [Google Scholar]
- 5.Singh N, Bonham A, Fukui M. Immunosuppressive-associated leukoencephalopathy in organ transplant recipients. Transplantation. 2000;69:467–72. doi: 10.1097/00007890-200002270-00001. [DOI] [PubMed] [Google Scholar]
- 6.Grinyo J, Charpentier B, Pestana JM, Vanrenterghem Y, Vincenti F, Reyes-Acevedo R, Apanovitch AM, Gujrathi S, Agarwal M, Thomas D, Larsen CP. An integrated safety profile analysis of belatacept in kidney transplant recipients. Transplantation. 2010;90:1521–7. doi: 10.1097/TP.0b013e3182007b95. [DOI] [PubMed] [Google Scholar]
- 7.Berger JR. Progressive multifocal leukoencephalopathy and newer biological agents. Drug Saf. 2010;33:969–83. doi: 10.2165/11537510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Giannecchini S, Clausi V, Vultaggio A, Macera L, Maggi F, Martelli F, Azzi A, Maggi E, Matucci A. Assessment of the risk of polyomavirus JC reactivation in patients with immune-mediated diseases during long-term treatment with infliximab. J Neurovirol. 2012;18:55–61. doi: 10.1007/s13365-012-0078-1. [DOI] [PubMed] [Google Scholar]
- 9.Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leukoencephalopathy in a patient treated with infliximab. Arthritis Rheum. 2010;62:3191–5. doi: 10.1002/art.27687. [DOI] [PubMed] [Google Scholar]
- 10.Nevin RL. Pharmacokinetic considerations in the repositioning of mefloquine for treatment of progressive multifocal leukoencephalopathy. Clin Neurol Neurosurg. 2012;114:1204–5. doi: 10.1016/j.clineuro.2012.02.046. [DOI] [PubMed] [Google Scholar]
- 11.Patel A, Patel J, Ikwuagwu J. Treatment of progressive multifocal leukoencephalopathy and idiopathic CD4+ lymphocytopenia. J Antimicrob Chemother. 2010;65:2489–92. doi: 10.1093/jac/dkq389. [DOI] [PubMed] [Google Scholar]
- 12.Neu U, Maginnis MS, Palma AS, Ströh1 LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe. 2010;8:309–19. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunyaev SR, Lugovskoy A, Simon K, Gorelik L. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML) PLoS Genet. 2009;5:e1000368. doi: 10.1371/journal.pgen.1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stott K, Stonehouse J, Keeler J, Hwang TL, Shaka AJ. Excitation Sculpting in High-Resolution Nuclear Magnetic Resonance Spectroscopy: Application to Selective NOE Experiments. J Am Chem Soc. 1995;117:4199–4200. [Google Scholar]
- 16.Shaka AJ, Lee CJ, Pines A. Iterative schemes for bilinear operators; application to spin decoupling. J Magn Reson. 1988;77:274–293. [Google Scholar]
- 17.Nelson CD, Derdowski A, Maginnis MS, O’Hara BA, Atwood WJ. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology. 2012;428:30–40. doi: 10.1016/j.virol.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer M, Meyer B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew Chem Int Ed Engl. 1999;38:784–788. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 19.Meyer B, Klein J, Mayer M, Meinecke R, Moller H, Neffe A, Schuster O, Wulfken J, Ding Y, Knaie O, Labbe J, Palcic MM, Hindsgaul O, Wagner B, Ernst B. Saturation Transfer Difference NMR Spectroscopy for Identifying Ligand Epitopes and Binding Specificities. Ernst Schering Res Found Workshop. 2004:149–67. doi: 10.1007/978-3-662-05397-3_9. [DOI] [PubMed] [Google Scholar]
- 20.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123:6108–17. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 21.Jain AN. Virtual screening in lead discovery and optimization. Curr Opin Drug Discov Devel. 2004;7:396–403. [PubMed] [Google Scholar]
- 22.Altmeyer R. Virus attachment and entry offer numerous targets for antiviral therapy. Curr Pharm Des. 2004;10:3701–12. doi: 10.2174/1381612043382729. [DOI] [PubMed] [Google Scholar]
- 23.Basu A, Li B, Mills DM, Panchal RG, Cardinale SC, Butler MM, Peet NP, Majgier-Baranowska H, Williams JD, Patel I, Moir DT, Bavari S, Ray R, Farzan MR, Rong L, Bowlin TL. Identification of a small-molecule entry inhibitor for filoviruses. J Virol. 2011;85:3106–19. doi: 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berkhout B, Eggink D, Sanders RW. Is there a future for antiviral fusion inhibitors? Curr Opin Virol. 2012;2:50–9. doi: 10.1016/j.coviro.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Krepstakies M, Lucifora J, Nagel CH, Zeisel MB, Holstermann B, Hohenberg H, Kowalski I, Gutsmann T, Baumert TF, Brandenburg K, Hauber J, Protzer U. A new class of synthetic peptide inhibitors blocks attachment and entry of human pathogenic viruses. J Infect Dis. 2012;205:1654–64. doi: 10.1093/infdis/jis273. [DOI] [PubMed] [Google Scholar]
- 26.Thomas CJ, Casquilho-Gray HE, York J, DeCamp DL, Dai D, Petrilli EB, Boger DL, Slayden RA, Amberg SM, Sprang SR, Nunberg JH. A specific interaction of small molecule entry inhibitors with the envelope glycoprotein complex of the Junín hemorrhagic fever arenavirus. J Biol Chem. 2011;286:6192–200. doi: 10.1074/jbc.M110.196428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang QY, Patel SJ, Vangrevelinghe E, Xu HY, Rao R, Jaber D, Schul W, Gu F, Heudi O, Ma NL, Poh MK, Phong WY, Keller TH, Jacoby E, Vasudevan SG. A small-molecule dengue virus entry inhibitor. Antimicrob Agents Chemother. 2009;53:1823–31. doi: 10.1128/AAC.01148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.