

Abstract

On the order of hundreds of absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) models have been described in the literature in the past decade which are more often than not inaccessible to anyone but their authors. Public accessibility is also an issue with computational models for bioactivity, and the ability to share such models still remains a major challenge limiting drug discovery. We describe the creation of a reference implementation of a Bayesian model-building software module, which we have released as an open source component that is now included in the Chemistry Development Kit (CDK) project, as well as implemented in the CDD Vault and in several mobile apps. We use this implementation to build an array of Bayesian models for ADME/Tox, in vitro and in vivo bioactivity, and other physicochemical properties. We show that these models possess cross-validation receiver operator curve values comparable to those generated previously in prior publications using alternative tools. We have now described how the implementation of Bayesian models with FCFP6 descriptors generated in the CDD Vault enables the rapid production of robust machine learning models from public data or the user’s own datasets. The current study sets the stage for generating models in proprietary software (such as CDD) and exporting these models in a format that could be run in open source software using CDK components. This work also demonstrates that we can enable biocomputation across distributed private or public datasets to enhance drug discovery.
Introduction
For well over a decade, the cost of in vitro and in vivo screening of absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) properties of molecules has motivated efforts to develop various in silico methods to efficiently pre-filter candidates for actual physical testing.1−29 By relying on very large, internally consistent datasets, large pharmaceutical companies have succeeded in developing highly predictive but ultimately proprietary models.29−33 At one pharmaceutical company, for example, many of these models (e.g., volume of distribution, aqueous kinetic solubility, acid dissociation constant, distribution coefficient, microsomal clearance, CYP3A4 time-dependent inhibition)30−36 as well as other endpoints15,22 have achieved such high accuracy that they have essentially put the experimental assays out of business. It is likely that most large pharmaceutical companies can now perform experimental assays for a small fraction of compounds pre-filtered through the proprietary ADME/Tox and physicochemical property computational models, thus improving cost efficiency while minimizing in vitro and animal experimentation. Extra-pharma computational efforts have not been so successful, largely because they have, by necessity, drawn upon considerably smaller datasets, in many cases trying to combine information from the literature.37−43 This situation, however, has improved with larger datasets publicly available in PubChem,44,45 ChEMBL,46−48 CDD,49 and others, and some drug companies depositing their data (e.g., the recently deposited AstraZeneca data in ChEMBL), which can be useful for model building.50−53
ADME/Tox properties have been modeled by us1,54−81 and many other groups29,82 using an array of machine learning algorithms such as support vector machines,59 Bayesian modeling,19 Gaussian processes,83 and many others.84 A more exhaustive review of the different machine learning approaches is outside the scope of this work. These combined efforts at ADME/Tox model building have likely resulted in hundreds of published models which are, unfortunately, inaccessible to anyone but their authors in most cases. This limited access problem for published models is also likely the case with computational models for bioactivity or other physicochemical properties of interest. The ability to share such models freely still remains a major challenge when dealing with issues of proprietary samples or data, as repercussions for such for-profit pharmaceutical companies could be severe. The current development of technologies for open models and descriptors builds on established methodologies.85−88 Datasets for quantitative structure–activity relationships (QSAR) have previously been represented in a reproducible way via QSAR-ML.85 These methods also come with a reference implementation for the Bioclipse workbench,86,87 which provides a graphical interface. There have been several early efforts at cheminformatics Web services; e.g., Indiana University provides access to cheminformatics methods (fingerprints, 2D depiction, and various molecular descriptors) and statistical techniques. These have been used to develop models for the NCI60 cancer cell lines.89,90 In addition, there are Web tools for the prediction of bioactivities and physicochemical properties, like the Chemistry Activity Predictor (GUSAR).91 Also, the Open Notebook Science (ONS) project92 has developed models for solubility and melting point using web services based on open descriptors and algorithms. These tools all enable parties to collaborate publicly but do not facilitate private or selective collaboration.
We have previously demonstrated a proof of concept using open descriptors and modeling tools to model very large ADME datasets at Pfizer.22 Models were constructed with open descriptors and keys (CDK + SMARTS) using open software (C5.0) and performed essentially identically to expensive proprietary descriptors and models (MOE2D + SMARTS + Rulequest’s Cubist) across all metrics of performance when evaluated on human liver microsomal stability (HLM), RRCK passive permeability, P-gp efflux, and aqueous solubility datasets.22 Pfizer’s HLM dataset, used in this study, contained more than 230,000 compounds and covered a diverse range of chemistry space as well as addressing many therapeutic areas. The HLM dataset was split into a training set (80%) and a test set (20%) using the venetian blind splitting method. In addition, a newly screened set of 2310 compounds was evaluated as a blind dataset. All the key metrics of model performance, e.g., R2, RMSE, kappa, sensitivity, specificity, and positive predictive value (PPV), were nearly identical for the open source approach vs proprietary software (e.g., PPV = 0.80 vs 0.82).
Our goal is to enable extra-pharma drug discovery projects to exploit in silico machine learning methods that have, until now, been confined in practice to pharma and to a few knowledgeable academics. These methods better exploit limited screening resources and will enable such projects to cover more unexplored chemical space and to address ADME/Tox earlier in the discovery process. Extra-pharma projects represent a growing trend for commercial drug discovery93−95 to be the principal efforts to find cures for many neglected diseases (e.g., tuberculosis, malaria, Chagas disease, visceral leishmaniasis, etc.), and thousands of orphan indications96,97 will require more collaborations and data, and therefore model sharing. This approach has the potential to accelerate the discovery of promising drug-like lead compounds with acceptable properties in vivo and ultimately yield a significant impact on global health.
We now describe the creation of a reference implementation of a Bayesian model-building software module, which we have released as an open source component that is now included in the Chemistry Development Kit (CDK) project98,99 and incorporated using the FCFP6 descriptors in the CDD Vault, which was also recently made open source.100 We make use of the CDD Public database,49 which has over 100 public datasets that can be used to generate community-based models, including extensive neglected infectious disease SAR datasets (malaria, tuberculosis, Chagas disease, etc.), and ADMEdata.com datasets that are broadly applicable to many projects. An accompanying paper uses this software to develop models on a much larger scale.101
Experimental Section
Data and Materials Availability
All computational models are available from the authors upon request. All molecules for malaria, tuberculosis, and cholera datasets from Table 1 are available in CDD Public (https://app.collaborativedrug.com/register), and the models from Table 2 are available from http://molsync.com/bayesian1.
Table 1. Datasets Used for Bayesian Models Created with CDD Models Using FCFP6 Fingerprints.
| model | datasets used and refs | cutoff for active | no. of molecules | three-fold ROCa |
|---|---|---|---|---|
| malaria (Plasmodium falciparum) | CDD Public datasets (MMV, St. Jude, Novartis, and TCAMS)127−129 | 3D7 EC50 <10 nM | 184 actives, 19,824 inactives | 0.97 |
| TB (Mycobacterium tuberculosis) | CDD Public datasets from NIAID/SRI (MLSMR, CB2, kinase)138−140 | Mtb inhibition >90% | 6891 actives, 210,190 inactives | 0.88 |
| TB (Mycobacterium tuberculosis) | CDD Public datasets from NIAID/SRI (MLSMR, CB2, kinase, and ARRA)138−141 | Mtb IC50 or IC90 <10 μM | 3712 actives, 1145 inactives | 0.89 |
| TB (Mycobacterium tuberculosis) | CDD Public MLSMR single-point data | Mtb inhibition >90% | 3986 actives, 210,447 inactives | 0.87 |
| TB (Mycobacterium tuberculosis) | CDD Public MLSMR dose–response | Mtb IC50 <10 μM and classed as active | 624 actives, 1649 inactives | 0.75 |
| TB (Mycobacterium tuberculosis) efficacy in vivo mouse | CDD Public112 | described in ref (112) | 371 actives, 407 inactives | 0.73 |
| cholera | CDD Public in the TB ARRA dataset141 | IC50 <5 μM | 50 actives, 1874 inactives | 0.93 |
| Ames mutagenicity | ref (142) | Ames positive, active = 1 | 3501 actives, 3007 actives | 0.83 |
| mouse intrinsic clearance | data from ChEMBL | <10 μL/(min·g) | 52 actives, 312 inactives | 0.82 |
| human intrinsic clearance | data from ChEMBL | ≤10 μL/(min·g) | 105 actives, 638 inactives | 0.92 |
| human intrinsic clearance | AZ data from ChEMBL143 | ≤10 μL/(min·mg) | 496 actives, 604 inactives | 0.80 |
| Caco-2 | proprietary data from ADMEdata.com | pH 6.5, cutoff >1×10–5 | 181 actives, 325 inactives | 0.79 |
| Caco-2 | data from ChEMBL | cutoff >1×10–5 | 60 actives, 399 inactives | 0.89 |
| 5-HT2B | ref (144) | active = 1, described in ref (144) | 146 actives, 607 inactives | 0.89 |
| solubility | ref (145) | Log solubility = −5 | 1136 actives, 154 inactives | 0.87 |
| PXR activation | ref (146) | described in ref (146) | 174 actives, 143 inactives | 0.80 |
| maximum recommended therapeutic dose | ref (147) | >10 mg/(kg·day) | 350 actives, 813 inactives | 0.85 |
| blood brain barrier permeability | ref (28) | BBB positive, described in ref (28) | 1472 actives, 432 inactives | 0.92 |
ROC = receiver operator characteristic integral.
Table 2. Datasets Used for Bayesian Models Created for Use by MMDS, with ECFP6 Fingerprintsa.
| model | datasets used and refs | cutoff for active | no. of molecules | five-fold ROC |
|---|---|---|---|---|
| solubility | ref (145) | Log solubility = −5 | 1144 actives, 155 inactives | 0.86 |
| probe-like | ref (148) | described in ref (148) | 253 actives, 69 inactives | 0.76 |
| hERG | ref (149) | described in ref (149) | 373 actives, 433 inactives | 0.85 |
| KCNQ1 | PubChem BioAssay: AID 2642150 | using actives assigned in PubChem | 301,737 actives, 3878 inactives | 0.84 |
| Bubonic plague (Yersina pestis) | PubChem single-point screen BioAssay: AID 898 | active when inhibition ≥50% | 223 actives, 139,710 inactives | 0.81 |
| Chagas disease (Typanosoma cruzi) | Pubchem BioAssay: AID 2044 | with EC50 <1 μM, >10-fold difference in cytotoxicity as active | 1692 actives, 2363 inactives | 0.8 |
| TB (Mycobacterium tuberculosis) | in vitro bioactivity and cytotoxicity data from MLSMR, CB2, kinase, and ARRA datasets110 | Mtb activity and acceptable Vero cell cytotoxicity selectivity index = (MIC or IC90)/CC50 ≥10 | 1434 actives, 5789 inactives | 0.73 |
| malaria (Plasmodium falciparum) | CDD Public datasets (MMV, St. Jude, Novartis, and TCAMS)127−129 | 3D7 EC50 <10 nM | 175 actives, 19,604 inactives | 0.98 |
All eight models are ECFP6, with folding into 32,768 slots.
Laplacian-Modified Naïve Bayesian Definition and Pseudocode
Bayesian models have been a useful part of computer-aided drug discovery for many years and were popularized in Pipeline Pilot.19,102,103 The statistical method is particularly useful for correlated structure-derived fingerprint bit strings with an activity measurement that has been classified as active or inactive on the basis of a selected threshold. Variants on the original Bayes theorem can be used to produce an estimate of the likelihood of activity for proposed compounds. For reproducing binary classifications, Bayesian methods have several appealing features. The model creation process is typically very fast and can be implemented in O(N) time, which means that an ordinary desktop computer can build and evaluate models with hundreds of thousands of compounds with minimal delay. When general purpose structure-derived fingerprints are used, the methods tend to be quite robust, which is in contrast to methods such as QSAR, which require some expertise to select appropriate descriptors, avoid overtraining, and ensure domain applicability.
Unlike many other applications of Bayesian methods, the use of chemical structure fingerprints as inputs means priors often numbers in the thousands, which produces scale distortions. Even if the prior probabilities are approximately 0.5 in each case, multiplying thousands of such values together tends to warp the distribution of the posterior probabilities to being asymptotically close to 0 or 1, which introduces numerical precision issues. By summing the logarithms of the ratios rather than multiplying the fractions, and incrementing the numerator and denominator, the precision issues are eliminated, and the resulting predictions tend to follow a linear distribution.
The main drawback with this particular Bayesian variant is that the resulting prediction is not a probability, but rather an arbitrary number that has no particular upper or lower bound. The results can be converted into a two-state classification by selecting a threshold, or into a probability-like value by picking a linear transformation function, but these are post-Bayesian calibrations that must be made by applying judgment criteria that are not an intrinsic part of the method.
We have used our work100 on the reference extended connectivity ECFP and FCFP fingerprints to create a software class that allows Bayesian models to be created from a collection of molecules and activity data, used to predict probabilities for new molecules, and to serialize/deserialize the model as a structure text string that can be saved to a file or shared with any other package that implements the same functionality.
The Laplacian-modified naïve Bayesian (which we call Bayesian models for simplicity) formula uses a simple definition, which pre-supposes that each molecule has been described by enumerating a list of fingerprints that applies to it, and has a determination of whether it is active or inactive. For each fingerprint code in the entire dataset:
where Ci is the contribution associated with the presence of a fingerprint hash code i, which is in turn derived from Ai and Ti, which are respectively the number of active molecules with the fingerprint and the total number of molecules with the fingerprint, while R is the overall fraction of actives.
Building the Bayesian model is a simple matter of determining the total set of fingerprints in the dataset and, for each of them, calculating the value of Ci. Any fingerprint that is theoretically possible but not encountered in the training set has an implied value of 0. Any fingerprint hash code that is observed equally often in active and inactive molecules (or not at all in either) has a ratio of 1, for which the log value is zero.
When making a prediction for an incoming molecule, the value is determined by adding up the contributions for each fingerprint hash code for the molecule:
The resulting prediction, Pm, is an uncalibrated value: unlike for the conventional Bayes theorem, the result is not a probability and is generally not directly interpretable, meaning that there is no significance to either the scale or offset. Methods for interpreting these values will be discussed subsequently.
Creating a Bayesian model using this method is very fast and has favorable scalability properties, because it requires just two passes through the input collection: the total number of actives and inactives needs to be summed, and after that, each compound needs to be considered only individually. The total memory required to build the Bayesian model is bounded by the theoretical number of fingerprints. For each possible unique fingerprint hash code, it is necessary to store two integers (Ai, Ti) and derive one floating point value per fingerprint (Ci). For small, relatively dense fingerprint schemes, these can be stored in a flat array (e.g., when folding fingerprints into 1024 possible values), but for larger schemes with sparse occupancy it is better to use a dictionary object (e.g., when the full 32-bit range of ECFP6 or FCFP6 fingerprints is allowed).
The pseudocode for the model building is as follows:
let T = empty dictionary (key: hash code i, value: total t)
let A = empty dictionary (key: hash code i, value: actives a)
- for m in all molecules in training set:
- determine list of fingerprints F for molecule m
- for fingerprint hash code i in F:
- increment Ti
- if molecule m is active: increment Ai
let R = total actives/total molecules
let C = empty dictionary (key: hash code i, value: contribution v)
let L = unique list of keys for T
- for i in L:
- put Ci = log([Ai + 1]/[Ti·R + 1])
For making a prediction for an incoming molecule:
let m = molecular structure
determine list of fingerprints F for molecule m
let Pm = 0
-
for i in L:
-
if i is one of the fingerprints in F:
let Pm = Pm + Ci
-
Implementing these algorithms using a flat array rather than a dictionary object is analogous and differs only in the way indices are looked up.
Chemistry Development Kit
The method described in this article is implemented in the Chemistry Development Kit (CDK) project and made available under the terms of the Lesser Gnu Public License (LGPL). The latest version of the project can be obtained from its SourceForge host and underlying Git repository (http://sourceforge.net/p/cdk/code/ci/master/tree). The Bayesian modeling capabilities are available within the tools section, the main class for which is org.openscience.cdk.fingerprint.model.Bayesian.
Using the CDK library to create a new Bayesian model from
a collection of molecule objects and boolean activity values is straightforward.
For example, given the filename for an MDL SDfile with a field called
“pIC50”, for which any molecule with a value of 6 or
greater is considered active, the following Java code snippet can
be used to create a serialized model:
The resulting serialized form can be stored for future use.
If
it is stored in a file, it can be easily retrieved and used to apply
to a different SDfile, e.g.:
The first step is to read the model from the pre-existing file (“activity.bayesian”). The second step iterates over the input SDfile, while writing to another SDfile with two extra fields appended: “RawPrediction”, which contains the uncalibrated outcome from the modified Bayesian method, and “ScaledPrediction”, which contains the prediction that has been scaled using metrics originally derived from the internal cross-validation.
These two examples demonstrate the ‘create and consume’ use cases and can be easily adapted to scenarios besides reading and writing from files. Serialized Bayesian models can be embedded in any kind of text-friendly data structure, e.g., XML documents, JSON messages, SQL tables, etc. Use of models to provide predictions can be applied to a variety of invocations, such as command line tools, incorporation into modeling packages with a graphical interface, Web services accessible via API, etc.
File Format
For saving models for subsequent reuse, the information necessary to apply the model to make predictions for new molecules can be stored in a text-based file format. The molecules that were used to build the model are not included in the serialized form, nor are the fingerprints that were generated from them. This means that sharing a serialized model allows the recipient to make inferences on the basis of the original data without explicitly having access to it. For confidentiality purposes, sharing models without the underlying data is useful in a number of situations, but it should be noted that this cannot be considered as entirely foolproof: a determined hacker with some context would likely be able to make a well educated guess as to the actives contained in the training set.
Figure 1 shows an example of a serialized file. The default file extension is .bayesian, and the MIME type is chemical/x-bayesian. The text should be encoded as UTF-8 unicode, for which all of the content is limited to the ASCII subset, except for the freeform text notes. End of line should be encoded Unix-style, and floating point numbers can be encoded with a decimal point (e.g., 1.23, with a period symbol for the separator, invariant of localization) or scientific notation (e.g., 1.23 × 10–9). The format is case- and whitespace-sensitive. The body of the format consists of individual lines, each of which encodes a discrete property, and is of arbitrary length.
Figure 1.

Example of a serialized file containing a very small Bayesian model. The default file extension is .bayesian, and the MIME type is chemical/x-bayesian.
The first line contains the header, which consists of the recognition sequence and essential information about the model. The first nine characters are always set to the ASCII characters for the string “Bayesian!” (hex: 42 61 79 65 73 69 61 6E 21), which can be used as a recognition sequence. This is useful for situations such as embedding in streams, within whitespace-padded subfields such as XML elements, or when the file extension or MIME type is unavailable or unreliable.
The recognition sequence is followed by four comma-separated fields: fingerprint type, folding length, calibration minimum, and maximum. Only the first two fields are mandatory, and parsers should ignore additional fields, in case the format is subsequently extended.
The fingerprint type must be one of ECFPn or FCFPn, where n is 0, 2, 4, or 6. These correspond to the eight different permutations of circular fingerprints that are implemented in the CDK library. The most commonly used values are ECFP6 and FCFP6. The variety of fingerprints may be extended at a later date. When a parser encounters a fingerprint type that it does not recognize, it should invoke an error pathway if there is any intention of applying the model to new molecules, since the ability to produce the exact same fingerprints is a pre-requisite. The folding length should either be 0 (no folding, i.e., full range of 32-bit integers) or a power of 2 (e.g., 512, 1024, 2048, etc.). The parser should fail for invalid folding lengths. The calibration minimum and maximum values are used to transform raw predictions into a probability-like range. Since this is calculated by analyzing the cross-validation metrics, which is an optional step, the information may not be available. If not included, then the model can only be used to generate raw prediction values. Note that sometimes the minimum and maximum values are equal, which can occur for datasets that are small or trivial. In this case, the degenerate value should be treated like a simple threshold, giving results of 0 or 1, rather than a probability-like transform.
The model specification ends with a line beginning with the string “!End”. This should be considered as the terminator sequence regardless of trailing characters or whitespace. All lines in between the header and footer can be examined out of order.
Lines that match the template {bit index}={contribution}, where bit index is an integer and contribution is a floating point number, make up the payload of the model. The contributions-per-bit are typically stored in a dictionary object, since the bit coverage is sparse, i.e. usually not all of the possible bits are represented. If the fingerprints are folded, then the bit indices range from 0 to folding-1. If not folded, the indices are represented as signed integers: approximately half of the values will be negative.
For generating raw Bayesian predictions, all that is needed is fingerprint type, folding, and contribution list. All remaining lines are optional and must follow the general format of {category}:{key}={value}, whereby category and key must be plain ASCII without whitespace, category must begin with an alphanumeric character, and value may contain any unicode characters, except for end-of-line. Duplicates are allowed. When software needs to parse, modify then write a model file, any optional lines that are not understood should be preserved as-is.
The optional data that are currently used by the CDK implementation include the following:
-
•
training:size and training:actives: the total number of compounds in the training set, and the number of actives, respectively
-
•
roc:auc: integral of the receiver operator characteristic, which is a number between 0 and 1
-
•
roc:type: the method used to partition the data for internal cross-validation, which is one of leave-one-out, three-fold, or five-fold.
-
•
roc:x and roc:y: two comma-separated lists of numbers from 0 to 1 which can be used to recreate the ROC curve visually. Note that for large datasets, the total number of points may be reduced in order to limit the impact on file size. This means that while the resolution is indistinguishable for graph plotting purposes, recalculating the integral from these points is less precise than using the stored roc:auc value.
-
•
note:title: ideally a short free-text description of the model that communicates to a scientist what data were being modeled. It should be expected to be displayed in a single line.
-
•
note:origin: a short free-text description that provides information about the provider of the model, be it the software algorithm or the source of the data, or both.
-
•
note:comment: a completely freeform field, which may be of any length. Since newlines are disallowed, multiple paragraphs of comments should be encoded by having multiple comment lines.
The file size for a serialized model depends on the size and diversity of the molecules. One of the main reasons for opting to fold the fingerprints is that it places a reasonable maximum limit on the file size. For example, a collection of 7000 molecules with experimentally determined activity against Mycobacterium tuberculosis using ECFP6 with no folding produced a file size of 1.4 Mb. Folding the fingerprints into 32,768 bits reduced the file size to 646 kb, and into 2048 down to 67 kb.101
CDD Models
The CDD Vault104 product makes use of the fingerprinting functionality in the CDK to provide Bayesian model-building capabilities, which we have termed CDD Models. While the Bayesian implementation is proprietary, the underlying algorithm for model generation is equivalent to the method described in this article. Models can be created and used within the CDD Vault environment, and at any time they can be exported using the format described above, which means that they can be utilized by any software that either implements the requisite algorithms described in this article or makes use of the CDK library.
The CDD Models extension is part of CDD Vision in CDD Vault. A model is created by separating a set of molecules into two collections: those that could be considered ‘actives’ and those that can be considered ‘inactives’. These classes are then used to train the model, after a series of steps are taken to ensure logical consistency. These include ensuring that duplicates do not appear in either collection, that there is no overlap between the collections, and that each collection contains at least two molecules. The Standard InChIKey105 of each molecule is used as the criteria for detecting and removing duplicates. These precautions are addressed in a series of pre-processing steps in the CDD Ruby on Rails application, where the modeling process and molecule management system is hosted, wherein the training sets are algorithmically curated via optimized raw SQL code.
Once the training set has passed these checks and pre-processing, the model is generated. CDD Vault uses the FCFP6100,102,106 structural fingerprints to build the Bayesian statistical model.107 This machine learning model is stored as a special type of protocol (category = Machine-Learning model), which provides an ROC plot generated by stratified three-fold cross-validation. This ROC plot is interactive, allowing the user to explore the sensitivity, specificity, and corresponding score cutoff at each point along the curve (Figure 2A). After the model has been created, each molecule in the user’s selected ‘project’ receives a relative score, applicability number (fraction of structural features shared with the training set), and maximum similarity number (maximum Tanimoto/Jaccard similarity to any of the “good” molecules). The model can be subsequently shared with both other users and the user’s other ‘projects’ to score any molecule of interest.
Figure 2.
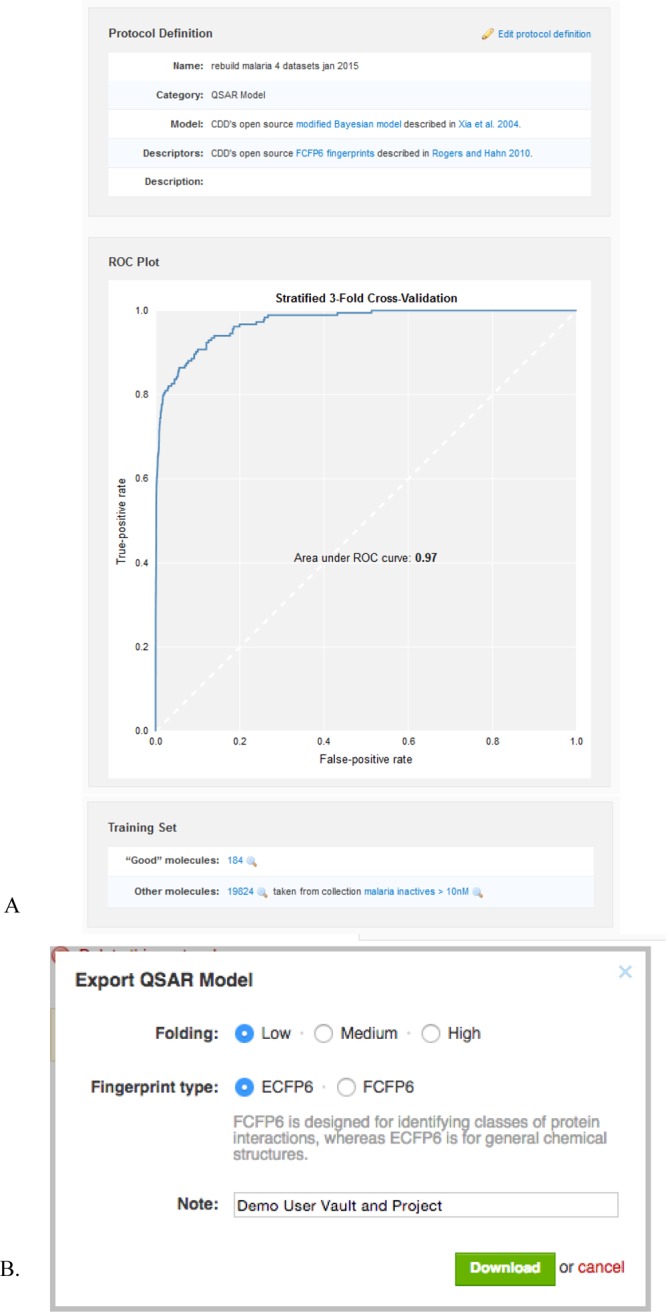
Example of the model output in CDD Models. (A) Model derived from whole-cell datasets from antimalarial screening across four CDD Public datasets (MMV, St. Jude, Novartis, and TCAMS), ∼20,000 EC50 values, cutoff < 10 nM. (B) Options for exporting a model from CDD.
The model can also be exported from CDD Vault by making use of the aforementioned .bayesian file format (Figure 2B). To render a serialized version of a model, CDD Vault feeds the training set structures into the serialization implementation described in the previous section. The connection between the Ruby code in CDD Vault and the Java-based serialization code is accomplished using RJB (Ruby-Java Bridge). Further details on using CDD Models are described in the Supporting Information.
Mobile Apps
Once the Bayesian model building was formalized as part of the CDK project, with a rigorously defined file format, it became a straightforward matter to implement the algorithm on other platforms. We have previously described the implementation of ECFP and FCFP fingerprints in a way that is agnostic to the specifics of any particular cheminformatics toolkit and so can be easily ported to other platforms, such that it is literally compatible with the original reference. We have taken the same approach with the Bayesian modeling and ported enough of the functionality to the iOS mobile platform such that models created with the CDK or CDD Vault using ECFP6 fingerprints can be parsed from within mobile apps and used to make predictions. Currently Bayesian model prediction capabilities have been incorporated into the Mobile Molecular DataSheet (MMDS) app (Figure 3), Approved Drugs, and MolPrime+ (all apps produced by Molecular Materials Informatics). Several useful Bayesian models have been packaged with the apps as default functionality, and it is also possible to import user-created models in order to make structure-based predictions within the mobile app.
Figure 3.

Example of the Bayesian model implemented in the MMDS mobile app. (a) hERG model, based on literature data. (b) A molecule from a hERG paper.151 (c) Results scored with this model (hERG measured IC50 = 24 nM) showing a visually intuitive atom coloring for this and other Bayesian models. This compound would appear to be an inhibitor of hERG and possibly KCNQ1 potassium channels.
Application to Datasets
To illustrate the utility of CDD Models we evaluated several datasets available in CDD public as well as in our own CDD Vaults (Table 1). These include screening datasets for malaria, tuberculosis, and cholera from whole-cell screens, in vivo data from mice treated with potential antituberculars, as well as several ADME/Tox properties such as Ames mutagenicity, mouse and human intrinsic clearance, Caco-2, 5-HT2B, solubility, PXR activation, maximum recommended therapeutic dose, and blood brain barrier permeability data. In all cases, three-fold ROC data were collated.
Several datasets were also selected for integration into mobile apps (including MMDS and Approved Drugs). These datasets included solubility, probe-like,108 hERG, KCNQ1, screening data for whole-cell phenotypic screens against Bubonic plague as well as Chagas disease. In all cases, five-fold ROC data were collated, summarized, and compared to published results.
Results
Chemistry Development Kit
As mentioned previously, the reference implementation for the Bayesian algorithm and the underlying ECFP/FCFP fingerprints is available in the CDK library, which can be freely downloaded from Github. The open source implementation is also accompanied by a testing library, which runs a battery of tests to ensure that the basic functionality is operating as-described.
CDD Bayesian Models
CDD Models using FCFP6 fingerprints have been demonstrated using diverse datasets, such as from public phenotypic screening, and published ADME/Tox datasets (Table 1). In most cases, the three-fold cross-validation ROC values are >0.75. We have also illustrated with the tuberculosis (M. tuberculosis) and malaria (P. falciparum) screening datasets that very large dose–response or single-point datasets can be constructed by combining datasets in CDD Public. Other datasets were collected for this study by manual mining of ChEMBL (mouse intrinsic clearance, human intrinsic clearance, Caco-2). Although these datasets are generated from many different published datasets, the ROC values are a good starting point (>0.80) and comparable to those obtained from proprietary datasets. Several of these datasets (blood brain barrier permeability and PXR) were used recently for a comparison across SVM and Bayesian methods,109 and the three-fold cross-validation ROC values were similar to those obtained with five-fold cross-validation in this study. The use of the ROC value in this way is a reasonable method to evaluate the utility of the computational models. However, ideally the use of an additional external test set would provide further confidence. The ROC values for the M. tuberculosis models are comparable to those published recently using a commercial tool. For example, in this study MLSMR single-point model three-fold ROC = 0.87 (Figure S1, five-fold ROC 0.87,110 leave out 50% × 100 cross-validation ROC = 0.86111) and MLSMR dose–response model three-fold cross-validation ROC = 0.75 (leave out 50% × 100 cross-validation ROC = 0.73111), M. tuberculosis efficacy in mouse three-fold ROC = 0.73 (five-fold ROC = 0.73112), and Ames mutagenicity three-fold ROC = 0.83 (five-fold ROC = 0.84109).
Mobile App Bayesian Models
The models developed using the same underlying code in mobile apps used the ECFP6 descriptors, and all eight models described had five-fold ROC values >0.75 (Table 2). Several of these datasets have previously been used to generate SVM and Bayesian methods with FCFP6 descriptors109 using other software. For example, the three-fold ROC for the probe-like dataset in this study was 0.76 (five-fold ROC = 0.73108), the three-fold ROC for the hERG dataset was 0.85 (five-fold ROC = 0.84109), and the three-fold ROC for the KCNQ1 dataset was 0.84 (five-fold ROC = 0.86109). The models derived with FCFP6 (Table 1) and ECFP6 descriptors (Table 2) can be compared; e.g., the three-fold ROC for the malaria dataset using FCFP6 was 0.97 (Table 1) (using ECFP6 for the same dataset five-fold ROC = 0.98, Table 2).
These examples of models generated previously and now with open source descriptors and algorithms suggest they are likely comparable (based on ROC values) and will be evaluated prospectively in future studies. We have also made the models in the mobile app freely accessible via the link http://molsync.com/bayesian1, which is summarized in Figure 4.
Figure 4.

Screenshots summarizing the ROC plots and active and inactive compounds for eight models implemented in MMDS.
Discussion
We have recently suggested how providing computational models tightly integrated in software used for storing and sharing chemistry and biology data will be useful for decision making.113 Some resources exist such as qsardb.org and ochem.eu for public model sharing and development,114,115 while another, Chembench, provides a resource for creating and using models and other cheminformatics tools privately.116 Our work, proposing that open source descriptors and algorithms are comparable to commercial software in performance,22 will ideally lead to more sharing of computational models. At approximately the same time, QSAR-ML85 was developed to enable standards for interoperability of QSAR models.85,117 We now build on this prior work as the current study sets the stage for being able to generate a model in proprietary software such as CDD Vault and export a model in a format that could be run in open source software using CDK components. This is a significant advance, because it means that a shared Bayesian model can in principle be used by anyone, regardless of which commercial software packages they have licenses to, since the model capabilities are implemented by an open source toolkit that runs on essentially every desktop platform (CDK is written in cross-platform Java). The creation of additional products that implement the same identical reference algorithm, e.g., mobile apps,100,118−123 makes use of shared models increasingly convenient. None of the existing Web sites for creating or storing QSAR models appear to offer this capability.
In terms of willingness to share models, sharing with collaborators is one thing, while sharing models openly with the community at large is another, but we have at least removed the main technology hurdle for fingerprint-based Bayesian models in this study. As previously noted, the shared models do not contain chemical structures or the fingerprints corresponding directly to them. However, the direct correlation between structural features and fingerprint does provide clues as to what active molecules an organization may have been using to build their models, and so this caveat must be taken into account when trustworthiness cannot be assumed. While additional security measures are appropriate for the world of proprietary high-value disease targets, this is much less of an issue for rare or neglected diseases, which is where we believe that open model sharing will have the greatest impact.113 There has been considerable research and discussion on efforts to securely share chemistry data,124−126 and some of these approaches could be implemented to encrypt models in future.
We have now described how implementation of Bayesian models with FCFP6 descriptors generated in the CDD vault enables the rapid creation of machine learning models from public datasets or the user’s own proprietary data. We also enable the resultant models to be selectively shared (or not) within CDD without having to disclose the underlying data—this represents a practical middle ground, where a trusted broker (CDD) allows a research group to share some of the benefits of their results, but not necessarily full access to the raw data, nor sufficient detail to reverse engineer it. Since sharing is not mandatory, and the option exists to export a model in an open format that can be used by anyone, this means that the full spectrum of model sharing options is available. Providing researchers with greater flexibility to designate and share models with specified collaborators, over particular time intervals, and with clear rights encourages data exchange by allowing researchers to share on terms they control. More fine-grained access control will expand the boundary of what models can be shared to fit the comfort levels of scientists (and their management and lawyers). From having been involved in a number of collaborations large and small, we have observed considerable variation in the need for security and desired degree of openness.
The possibility of using such models to further drug discovery for neglected diseases is of considerable interest, since the available software has traditionally catered to the proprietary market that provides most of the funding. There is now a significant amount of SAR data for rare and neglected diseases that is publicly available, and following up on open data with open source modeling algorithms is an important step. For example, pharmaceutical companies and other research groups have performed high-throughput screens on likely millions of compounds in the search for antimalarials, but they have generally only offered up the active compounds,127−130 some of which are available in CDD Public. By selecting a cut-off for activity for the antimalarial data that is very stringent (e.g., <10 nM) in CDD Models one can construct a Bayesian model with a three-fold ROC = 0.97 upon combining four public datasets (Table 1). This model may be useful for virtual screening of future compound libraries and complements our other efforts at machine learning models for antimalarial research.131 Ideally having access to the millions of other inactive compounds would also be useful, although one could imagine a company could just make a model available by selecting a cut-off for inactives as we have demonstrated herein.
Any efficiencies that can be gained in drug discovery would be highly desirable as it is widely known it is both time-consuming and very costly.93,94 Therefore, the use of tools like computational models that can point out drug candidate liabilities earlier will have considerable value.1,132,133 With a considerable percentage of drug failures attributed to ADME/Tox issues,1,43 it is still important to assess these qualities early in the drug development process. Running experimental ADME/Tox assays on each compound for initial screening of chemical libraries is cost- and labor-intensive,1,43 while computational approaches that rapidly and reliably predict these qualities are gaining more acceptance in the drug discovery community. It is therefore possible to exclude compounds that are most likely to exhibit undesirable ADME or toxicity problems sooner. We present an approach to drug discovery using computational methods for predicting whole-cell activity as well as ADME/Tox and physicochemical properties that can be broadly applied and do not have to be restricted to large companies with sophisticated software and big budgets. For example, modeling of microsomal metabolism has been used with large datasets,22,29,35 and such models are now more accessible through availability of public data. The results summarized in Tables 1 and 2 suggest that reliable Bayesian models for various bioactivity and ADME/Tox models can be generated with simple fingerprint descriptors (FCFP6 and ECFP6) and the same Bayesian algorithm. This is enabled in such a way that experience in building computational models, while valuable, may not be essential to facilitate model generation, compound scoring, and interpretation.
The models described in Table 2 which are available in MMDS are now also freely accessible (http://molsync.com/bayesian1). Our main motivation for creating and disseminating this work is to enable the sharing of Bayesian models between a diverse set of toolkits and computing platforms. We have previously described our open source implementation of ECFP6 and FCFP6 fingerprints,100 inspired by the original commercial implementation that was partially reported in the literature without the disclosure of key details, which remain a trade secret.102,106 While there are several other examples of the general approach, our intent was to create a reference implementation and document it so that identical results could be reproduced. The algorithm herein is explicitly documented in a stepwise fashion, and the reference method is available publicly in source code, and hence can be used to compare against when re-implementing in another environment. We believe that taking such care to ensure that the algorithms can be implemented in a way that is 100% compatible with the formal reference removes a major barrier to scientific progress, since building and using models is no longer an isolated activity. We have deliberately taken a two-prong approach: by releasing a fully functional implementation as part of a popular open source toolkit, and also taking the effort to document the algorithm in fine grained detail, to encourage creators of commercial software to consider the advantages of interoperability within their own proprietary products.
Because the source code is a part of the CDK, the modeling functionality that we describe can be used in a variety of scenarios as-is. Any software environment that is capable of linking to a Java Virtual Machine (either directly or through a pipe) can make use of this functionality. Since the CDK is made available under the LGPL license, it can be incorporated into proprietary products as long as it is linked as a separate library, but for internal projects, back-end services for which the software is not distributed, or open source projects with a compatible license, it can be used essentially without restrictions. For wholly closed-source products, and platforms that are not compatible with the Java Virtual Machine, the methodology can be re-implemented without difficulty. The exact implementation of Bayesian model building and subsequent calibration is straightforward, and we have represented it in pseudocode form (see the accompanying paper for details of algorithms for additional analysis101). The CDK version is readily available to verify literal compatibility and can be used as a limitless source of validation data for direct comparison. Thus far, the method has been ported to Objective-C, in order to enable the use of Bayesian models within several different mobile apps (Figure 3) and CDD Vault as CDD Models. The use of CDD Models online in the CDD Vault data sharing platform to create Bayesian models, the use of mobile apps to apply them to small collections of proposed compounds, and integration into other products and scripts via the CDK library present a number of opportunities for making computational modeling potentially more useful and widespread. Currently structure–activity models are generally only able to be created and used by one specific platform, or if they have some portability, they often suffer from serious compatibility issues due to differences in the underlying technology (e.g., aromaticity models, ylide representations, SMARTS implementations, partial charge models, etc.) By releasing a well-documented reference implementation as open source and building powerful and useful functionality on top of it, we hope to encourage computational chemists and software creators to make use of this increased inter-operability.
Future work related to this project will include the implementation of further measures to assess model quality and the applicability115,134−137 of a model to a test compound. In the accompanying paper,101 we describe several additional algorithms, including calibration of raw Bayesian results to a probability-like scale, the effects of folding fingerprints into a smaller range, methods for extracting suitable validation test sets from large public datasets, automated determination of thresholds for active/inactive, and the impact of training set selection on internal cross-validation metrics. As others begin to use the new CDK functionality, CDD Models, and Bayesian functionality implemented in various mobile apps, we will expect to see further prospective and retrospective testing of the underlying technology and descriptions of the utility and limitations.
Acknowledgments
We gratefully acknowledge Dr. Barry Bunin and all our colleagues at CDD for their support and development of CDD Models. S.E. and A.M.C. kindly acknowledge many valuable discussions with Antony J. Williams on sharing models and mobile apps for chemistry. This project was supported by Award No. 9R44TR000942-02, “Biocomputation across distributed private datasets to enhance drug discovery”, from the NIH National Center for Advancing Translational Sciences. The Chagas disease datasets were collected with funding from the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID), Grant R41-AI108003-01, “Identification and validation of targets of phenotypic high throughput screening”. The CDD TB database was made possible with funding from the Bill and Melinda Gates Foundation (Grant No. 49852, “Collaborative drug discovery for TB through a novel database of SAR data optimized to promote data archiving and sharing”). R.C.R. acknowledges the American Reinvestment and Recovery Act Grant 1RC1AI086677-01 that provided support for the presented study (NIH, NIAID, “Targeting MDR-Tuberculosis”). J.S.F. also thanks Rutgers University for financial support.
Glossary
Abbreviations
- ADME/Tox
absorption, metabolism, distribution, excretion, and toxicity
- CDD
Collaborative Drug Discovery
- CDK
Chemistry Development Kit
- ECFP6
extended connectivity
- FCFP6
molecular function class fingerprints of maximum diameter 6
- hERG
human ether-a-go-go related gene
- HLM
human liver microsomal stability
- HTS
high-throughput screening
- LGPL
Lesser Gnu Public License
- MMDS
Mobile Molecular DataSheet
- Mtb
Mycobacterium tuberculosis
- ONS
Open Notebook Science
- PPV
positive predictive value
- PXR
pregnane X-receptor
- QSAR
quantitative structure–activity relationship
- ROC
receiver operator curve
- SAR
structure–activity relationship
- SVM
support vector machine
Supporting Information Available
Description of how to use CDD Models, and one supporting figure. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.5b00143.
Author Contributions
A.M.C. developed the software; S.E. developed the CDD Models; all authors wrote the manuscript.
The authors declare the following competing financial interest(s): S.E. is a consultant for Collaborative Drug Discovery Inc. A.M.C. is the founder of Molecular Materials Informatics, Inc.
Supplementary Material
References
- Ekins S.; Waller C. L.; Swaan P. W.; Cruciani G.; Wrighton S. A.; Wikel J. H. Progress in predicting human ADME parameters in silico. J. Pharmacol. Toxicol. Methods 2000, 44, 251–272. [DOI] [PubMed] [Google Scholar]
- Wessel M. D.; Mente S. ADME by computer. Annu. Rep. Med. Chem. 2001, 36, 257–266. [Google Scholar]
- Boobis A.; Gundert-Remy U.; Kremers P.; Macheras P.; Pelkonen O. In silico prediction of ADME and pharmacokinetics. Report of an expert meeting organised by COST B15. Eur. J. Pharm. Sci. 2002, 17, 183–193. [DOI] [PubMed] [Google Scholar]
- Butina D.; Segall M. D.; Frankcombe K. Predicting ADME properties in silico: methods and models. Drug Discov. Today 2002, 7, S83–S88. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Boulanger B.; Swaan P. W.; Hupcey M. A. Towards a new age of virtual ADME/TOX and multidimensional drug discovery. Mol. Divers. 2002, 5, 255–275. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Rose J. P. In Silico ADME/TOX: The state of the art. J. Mol. Graphics 2002, 20, 305–309. [DOI] [PubMed] [Google Scholar]
- Klein C.; Kaiser D.; Kopp S.; Chiba P.; Ecker G. F. Similarity based SAR (SIBAR) as tool for early ADME profiling. J. Comput. Aided Mol. Des. 2002, 16, 785–793. [DOI] [PubMed] [Google Scholar]
- Krejsa C. M.; Horvath D.; Rogalski S. L.; Penzotti J. E.; Mao B.; Barbosa F.; Migeon J. C. Predicting ADME properties and side effects: the BioPrint approach. Curr. Opin. Drug Discov. Devel. 2003, 6, 470–480. [PubMed] [Google Scholar]
- van de Waterbeemd H.; Gifford E. ADMET in silico modelling: towards prediction paradise?. Nat. Rev. Drug Discov. 2003, 2, 192–204. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Swaan P. W. Computational models for enzymes, transporters, channels and receptors relevant to ADME/TOX. Rev. Comput. Chem. 2004, 20, 333–415. [Google Scholar]
- Smith P. A.; Sorich M. J.; Low L. S.; McKinnon R. A.; Miners J. O. Towards integrated ADME prediction: past, present and future directions for modelling metabolism by UDP-glucuronosyltransferases. J. Mol. Graph. Model. 2004, 22, 507–517. [DOI] [PubMed] [Google Scholar]
- Stoner C. L.; Gifford E.; Stankovic C.; Lepsy C. S.; Brodfuehrer J.; Prasad J. V.; Surendran N. Implementation of an ADME enabling selection and visualization tool for drug discovery. J. Pharm. Sci. 2004, 93, 1131–1141. [DOI] [PubMed] [Google Scholar]
- Yamashita F.; Hashida M. In silico approaches for predicting ADME properties of drugs. Drug Metab. Pharmacokinet. 2004, 19, 327–338. [DOI] [PubMed] [Google Scholar]
- Balakin K. V.; Ivanenkov Y. A.; Savchuk N. P.; Ivaschenko A. A.; Ekins S. Comprehensive computational assessment of ADME properties using mapping techniques. Curr. Drug Discov. Technol. 2005, 2, 99–113. [DOI] [PubMed] [Google Scholar]
- O’Brien S. E.; de Groot M. J. Greater than the sum of its parts: combining models for useful ADMET prediction. J. Med. Chem. 2005, 48, 1287–1291. [DOI] [PubMed] [Google Scholar]
- Chang C.; Ekins S.. Pharmacophores for human ADME/Tox-related proteins. In Pharmacophores and pharmacophore searches; Langer T., Hoffman R. D., Eds.; Wiley-VCH: Weinheim, 2006; Chapter 14, pp 299–324. [Google Scholar]
- Ekins S. Systems-ADME/Tox: Resources and network approaches. J. Pharmacol. Toxicol. Methods 2006, 53, 38–66. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Bugrim A.; Brovold L.; Kirillov E.; Nikolsky Y.; Rakhmatulin E.; Sorokina S.; Ryabov A.; Serebryiskaya T.; Melnikov A.; Metz J.; Nikolskaya T. Algorithms for network analysis in systems-ADME/Tox using the MetaCore and MetaDrug platforms. Xenobiotica 2006, 36, 877–901. [DOI] [PubMed] [Google Scholar]
- Klon A. E.; Lowrie J. F.; Diller D. J. Improved naive Bayesian modeling of numerical data for absorption, distribution, metabolism and excretion (ADME) property prediction. J. Chem. Inf. Model. 2006, 46, 1945–1956. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Honeycutt J. D.; Metz J. T. Evolving molecules using multi-objective optimization: applying to ADME. Drug Discov. Today 2010, 15, 451–460. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Williams A. J. Precompetitive Preclinical ADME/Tox Data: Set It Free on The Web to Facilitate Computational Model Building to Assist Drug Development. Lab Chip 2010, 10, 13–22. [DOI] [PubMed] [Google Scholar]
- Gupta R. R.; Gifford E. M.; Liston T.; Waller C. L.; Bunin B.; Ekins S. Using open source computational tools for predicting human metabolic stability and additional ADME/TOX properties. Drug Metab. Dispos. 2010, 38, 2083–2090. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Li W.; Zhou Y.; Shen J.; Wu Z.; Liu G.; Lee P. W.; Tang Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Wrighton S. A. Application of in silico approaches to predicting drug–drug interactions. J. Pharmacol. Toxicol. Methods 2001, 45, 65–69. [DOI] [PubMed] [Google Scholar]
- Ekins S. In silico approaches to predicting metabolism, toxicology and beyond. Biochem. Soc. Trans. 2003, 31, 611–614. [DOI] [PubMed] [Google Scholar]
- Kemp C. A.; Flanagan J. U.; van Eldik A. J.; Marechal J. D.; Wolf C. R.; Roberts G. C.; Paine M. J.; Sutcliffe M. J. Validation of model of cytochrome P450 2D6: an in silico tool for predicting metabolism and inhibition. J. Med. Chem. 2004, 47, 5340–5346. [DOI] [PubMed] [Google Scholar]
- de Graaf C.; Vermeulen N. P.; Feenstra K. A. Cytochrome P450 in silico: an integrative modeling approach. J. Med. Chem. 2005, 48, 2725–2755. [DOI] [PubMed] [Google Scholar]
- Martins I. F.; Teixeira A. L.; Pinheiro L.; Falcao A. O. A Bayesian approach to in silico blood-brain barrier penetration modeling. J. Chem. Inf. Model. 2012, 52, 1686–1697. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Unwalla R.; Denny R. A.; Bikker J.; Di L.; Humblet C. Development of QSAR models for microsomal stability: identification of good and bad structural features for rat, human and mouse microsomal stability. J. Comput. Aided Mol. Des. 2010, 24, 23–35. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Obach R. S.; Dicapua F. M.; Bakken G. A.; Lu J.; Potter D. M.; Gao F.; Miller M. D.; Zhang Y. A hybrid mixture discriminant analysis-random forest computational model for the prediction of volume of distribution of drugs in human. J. Med. Chem. 2006, 49, 2262–2267. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Obach R. S.; Shalaeva M. Y.; Gao F. Prediction of human volume of distribution values for neutral and basic drugs. 2. Extended data set and leave-class-out statistics. J. Med. Chem. 2004, 47, 1242–1250. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Obach R. S.; Shalaeva M. Y.; Gao F. Prediction of volume of distribution values in humans for neutral and basic drugs using physicochemical measurements and plasma protein binding. J. Med. Chem. 2002, 45, 2867–2876. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Shalaeva M. Y.; Tupper K. A.; Gao F. ElogDoct: A tool for lipophilicity determination in drug discovery. 2. Basic and neutral compounds. J. Med. Chem. 2001, 44, 2490–2497. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Shalaeva M. Y.; Tupper K. A.; Gao F.; Abraham M. H. ElogPoct A tool for lipophilicity determination in drug discovery. J. Med. Chem. 2000, 43, 2922–2928. [DOI] [PubMed] [Google Scholar]
- Chang C.; Duignan D. B.; Johnson K. D.; Lee P. H.; Cowan G. S.; Gifford E. M.; Stankovic C. J.; Lepsy C. S.; Stoner C. L. The development and validation of a computational model to predict rat liver microsomal clearance. J. Pharm. Sci. 2009, 98, 2857–2867. [DOI] [PubMed] [Google Scholar]
- Zientek M.; Stoner C.; Ayscue R.; Klug-McLeod J.; Jiang Y.; West M.; Collins C.; Ekins S. Integrated in silico-in vitro strategy for addressing cytochrome P450 3A4 time-dependent inhibition. Chem. Res. Toxicol. 2010, 23, 664–676. [DOI] [PubMed] [Google Scholar]
- Lagorce D.; Sperandio O.; Galons H.; Miteva M. A.; Villoutreix B. O. FAF-Drugs2: free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinformatics 2008, 9, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villoutreix B. O.; Renault N.; Lagorce D.; Sperandio O.; Montes M.; Miteva M. A. Free resources to assist structure-based virtual ligand screening experiments. Curr. Protein Pept. Sci. 2007, 8, 381–411. [DOI] [PubMed] [Google Scholar]
- Ekins S.Computational Toxicology: risk assessment for pharmaceutical and environmental chemicals; John Wiley and Sons: Hoboken, NJ, 2007. [Google Scholar]
- Balani S. K.; Miwa G. T.; Gan L. S.; Wu J. T.; Lee F. W. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top. Med. Chem. 2005, 5, 1033–1038. [DOI] [PubMed] [Google Scholar]
- van De Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [DOI] [PubMed] [Google Scholar]
- Walters W. P.; Murcko M. A. Prediction of ‘drug-likeness’. Adv. Drug Deliv. Rev. 2002, 54, 255–271. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Ring B. J.; Grace J.; McRobie-Belle D. J.; Wrighton S. A. Present and future in vitro approaches for drug metabolism. J. Pharm. Toxicol. Methods 2000, 44, 313–324. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Bryant S. H. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Bolton E.; Dracheva S.; Karapetyan K.; Shoemaker B. A.; Suzek T. O.; Wang J.; Xiao J.; Zhang J.; Bryant S. H. An overview of the PubChem BioAssay resource. Nucleic Acids Res. 2010, 38, D255–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento A. P.; Gaulton A.; Hersey A.; Bellis L. J.; Chambers J.; Davies M.; Kruger F. A.; Light Y.; Mak L.; McGlinchey S.; Nowotka M.; Papadatos G.; Santos R.; Overington J. P. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014, 42, D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadatos G.; Overington J. P. The ChEMBL database: a taster for medicinal chemists. Future Med. Chem. 2014, 6, 361–364. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Bunin B. A. The Collaborative Drug Discovery (CDD) database. Methods Mol. Biol. 2013, 993, 139–154. [DOI] [PubMed] [Google Scholar]
- Sun H.; Veith H.; Xia M.; Austin C. P.; Tice R. R.; Huang R. Prediction of Cytochrome P450 Profiles of Environmental Chemicals with QSAR Models Built from Drug-like Molecules. Mol. Inform. 2012, 31, 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Veith H.; Xia M.; Austin C. P.; Huang R. Predictive models for cytochrome p450 isozymes based on quantitative high throughput screening data. J. Chem. Inf. Model. 2011, 51, 2474–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veith H.; Southall N.; Huang R.; James T.; Fayne D.; Artemenko N.; Shen M.; Inglese J.; Austin C. P.; Lloyd D. G.; Auld D. S. Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat. Biotechnol. 2009, 27, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur R.; Leister W.; Veith H.; Shinn P.; Southall N.; Austin C. P.; Inglese J.; Auld D. S. Monitoring compound integrity with cytochrome P450 assays and qHTS. J. Biomol. Screen. 2009, 14, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Diao L.; Polli J. E. A Substrate Pharmacophore for the Human Organic Cation/Carnitine Transporter Identifies Compounds Associated with Rhabdomyolysis. Mol. Pharmaceutics 2012, 9, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y.; Li L.; Kim G.; Ekins S.; Wang H.; Swaan P. W. Identification and Validation of Novel hPXR Activators Amongst Prescribed Drugs via Ligand-Based Virtual Screening. Drug Metab. Dispos. 2011, 39, 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Williams A. J.; Xu J. J. A Predictive Ligand-Based Bayesian Model for Human Drug Induced Liver Injury. Drug Metab. Dispos. 2010, 38, 2302–2308. [DOI] [PubMed] [Google Scholar]
- Ivanenkov Y. A.; Savchuk N. P.; Ekins S.; Balakin K. V. Computational mapping tools for drug discovery. Drug Discov. Today 2009, 14, 767–775. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Kortagere S.; Iyer M.; Reschly E. J.; Lill M. A.; Redinbo M.; Krasowski M. D. Challenges Predicting Ligand-Receptor Interactions of Promiscuous Proteins: The Nuclear Receptor PXR. PLoS Comput. Biol. 2009, 5, e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortagere S.; Chekmarev D. S.; Welsh W. J.; Ekins S. New predictive models for blood brain barrier permeability of drug-like molecules. Pharm. Res. 2008, 25, 1836–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal A.; Krasowski M. D.; Reschly E. J.; Sinz M. W.; Swaan P. W.; Ekins S. Machine learning methods and docking for predicting human pregnane X receptor activation. Chem. Res. Toxicol. 2008, 21, 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Kholodovych V.; Ai N.; Sinz M.; Gal J.; Gera L.; Welsh W. J.; Bachmann K.; Mani S. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Mol. Pharmacol. 2008, 74, 662–672. [DOI] [PubMed] [Google Scholar]
- Chekmarev D. S.; Kholodovych V.; Balakin K. V.; Ivanenkov Y.; Ekins S.; Welsh W. J. Shape signatures: new descriptors for predicting cardiotoxicity in silico. Chem. Res. Toxicol. 2008, 21, 1304–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal A.; Bahadduri P.; Chang C.; Polli J. E.; Swaan P.; Ekins S. Computational Models to Assign Biopharmaceutics Drug Disposition Classification from Molecular Structure. Pharm. Res. 2007, 24, 2249–2262. [DOI] [PubMed] [Google Scholar]
- Jones D. R.; Ekins S.; Li L.; Hall S. D. Computational approaches that predict metabolic intermediate complex formation with CYP3A4 (+b5). Drug Metab. Dispos. 2007, 35, 1466–1475. [DOI] [PubMed] [Google Scholar]
- Embrechts M. J.; Ekins S. Classification of metabolites with kernel-partial least squares (K-PLS). Drug Metab. Dispos. 2007, 35, 325–327. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Embrechts M. J.; Breneman C. M.; Jim K.; Wery J.-P.. Novel applications of Kernel-partial least squares to modeling a comprehensive array of properties for drug discovery. In Computational Toxicology: Risk assessment for pharmaceutical and environmental chemicals; Ekins S., Ed.; Wiley-Interscience: Hoboken, NJ, 2007; pp 403–432. [Google Scholar]
- Ekins S.; Chang C.; Mani S.; Krasowski M. D.; Reschly E. J.; Iyer M.; Kholodovych V.; Ai N.; Welsh W. J.; Sinz M.; Swaan P. W.; Patel R.; Bachmann K. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol. Pharmacol. 2007, 72, 592–603. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Balakin K. V.; Savchuk N.; Ivanenkov Y. Insights for human Ether-a-Go-Go-Related Gene Potassium Channel inhibition using recursive partitioning, Kohonen and Sammon mapping Techniques. J. Med. Chem. 2006, 49, 5059–5071. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Nikolsky Y.; Nikolskaya T. Techniques: Application of Systems Biology to Absorption, Distribution, Metabolism, Excretion, and Toxicity. Trends Pharmacol. Sci. 2005, 26, 202–209. [DOI] [PubMed] [Google Scholar]
- Ekins S. Predicting undesirable drug interactions with promiscuous proteins in silico. Drug Discov. Today 2004, 9, 276–285. [DOI] [PubMed] [Google Scholar]
- Balakin K. V.; Ekins S.; Bugrim A.; Ivanenkov Y. A.; Korolev D.; Nikolsky Y.; Skorenko S. A.; Ivashchenko A. A.; Savchuk N. P.; Nikolskaya T. Kohonen maps for prediction of binding to human cytochrome P450 3A4. Drug Metab. Dispos. 2004, 32, 1183–1189. [DOI] [PubMed] [Google Scholar]
- Balakin K. V.; Ekins S.; Bugrim A.; Ivanenkov Y. A.; Korolev D.; Nikolsky Y.; Ivashchenko A. A.; Savchuk N. P.; Nikolskaya T. Quantitative structure-metabolism relationship modeling of the metabolic N-dealkylation rates. Drug Metab. Dispos. 2004, 32, 1111–1120. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Berbaum J.; Harrison R. K. Generation and validation of rapid computational filters for CYP2D6 and CYP3A4. Drug Metab. Dispos. 2003, 31, 1077–1080. [DOI] [PubMed] [Google Scholar]
- Ethell B. T.; Ekins S.; Wang J.; Burchell B. Quantitative structure activity relationships for the glucuronidation of simple phenols by expressed human UGT1A6 and UGT1A9. Drug Metab. Dispos. 2002, 30, 734–738. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Mirny L.; Schuetz E. G. A ligand-based approach to understanding selectivity of nuclear hormone receptors PXR, CAR, FXR, LXRα and LXRβ. Pharm. Res. 2002, 19, 1788–1800. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Kim R. B.; Leake B. F.; Dantzig A. H.; Schuetz E.; Lan L. B.; Yasuda K.; Shepard R. L.; Winter M. A.; Schuetz J. D.; Wikel J. H.; Wrighton S. A. Three dimensional quantitative structure-activity relationships of inhibitors of P-glycoprotein. Mol. Pharmacol. 2002, 61, 964–973. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Kim R. B.; Leake B. F.; Dantzig A. H.; Schuetz E.; Lan L. B.; Yasuda K.; Shepard R. L.; Winter M. A.; Schuetz J. D.; Wikel J. H.; Wrighton S. A. Application of three dimensional quantitative structure-activity relationships of P-glycoprotein inhibitors and substrates. Mol. Pharmacol. 2002, 61, 974–981. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Crumb W. J.; Sarazan R. D.; Wikel J. H.; Wrighton S. A. Three dimensional quantitative structure activity relationship for the inhibition of the hERG (human ether-a-gogo related gene) potassium channel. J. Pharmacol. Exp. Ther. 2002, 301, 427–434. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Boulanger B.; Swaan P. W.; Hupcey M. A. Z. Towards a new age of virtual ADME/TOX and multidimensional drug discovery. J. Comput. Aided Mol. Des 2002, 16, 381–401. [DOI] [PubMed] [Google Scholar]
- Ekins S.; de Groot M.; Jones J. P. Pharmacophore and three dimensional quantitative structure activity relationship methods for modeling cytochrome P450 active sites. Drug Metab. Dispos. 2001, 29, 936–944. [PubMed] [Google Scholar]
- Ekins S.; Ring B. J.; Bravi G.; Wikel J. H.; Wrighton S. A.. Predicting drug-drug interactions in silico using pharmacophores: a paradigm for the next millennium. In Pharmacophore perception, development, and use in drug design; Guner O. F., Ed.; IUL: San Diego, 2000; pp 269–299. [Google Scholar]
- Paranjpe P. V.; Grass G. M.; Sinko P. J. In Silico Tools for Drug Absorption Prediction: Experience to Date. Am. J. Drug Deliv. 2003, 1, 133–148. [Google Scholar]
- Obrezanova O.; Csanyi G.; Gola J. M.; Segall M. D. Gaussian processes: a method for automatic QSAR modeling of ADME properties. J. Chem. Inf. Model. 2007, 47, 1847–1857. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhu H.; Oprea T. I.; Golbraikh A.; Tropsha A. QSAR modeling of the blood-brain barrier permeability for diverse organic compounds. Pharm. Res. 2008, 25, 1902–1914. [DOI] [PubMed] [Google Scholar]
- Spjuth O.; Willighagen E. L.; Guha R.; Eklund M.; Wikberg J. E. Towards interoperable and reproducible QSAR analyses: Exchange of datasets. J. Cheminform. 2010, 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spjuth O.; Alvarsson J.; Berg A.; Eklund M.; Kuhn S.; Masak C.; Torrance G.; Wagener J.; Willighagen E. L.; Steinbeck C.; Wikberg J. E. Bioclipse 2: a scriptable integration platform for the life sciences. BMC Bioinformatics 2009, 10, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spjuth O.; Helmus T.; Willighagen E. L.; Kuhn S.; Eklund M.; Wagener J.; Murray-Rust P.; Steinbeck C.; Wikberg J. E. Bioclipse: an open source workbench for chemo- and bioinformatics. BMC Bioinformatics 2007, 8, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck C.; Hoppe C.; Kuhn S.; Floris M.; Guha R.; Willighagen E. L. Recent developments of the chemistry development kit (CDK)—an open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006, 12, 2111–2120. [DOI] [PubMed] [Google Scholar]
- Dong X.; Gilbert K. E.; Guha R.; Heiland R.; Kim J.; Pierce M. E.; Fox G. C.; Wild D. J. Web service infrastructure for chemoinformatics. J. Chem. Inf. Model. 2007, 47, 1303–1307. [DOI] [PubMed] [Google Scholar]
- Guha R.; Schurer S. C. Utilizing high throughput screening data for predictive toxicology models: protocols and application to MLSCN assays. J. Comput. Aided Mol. Des. 2008, 22, 367–384. [DOI] [PubMed] [Google Scholar]
- Zakharov A. V.; Peach M. L.; Sitzmann M.; Nicklaus M. C. QSAR modeling of imbalanced high-throughput screening data in PubChem. J. Chem. Inf. Model. 2014, 54, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J.-C.http://usefulchem.blogspot.com/2011/06/open-melting-points-on-iphone-via-mmds.html, June 10, 2011.
- Paul S. M.; Mytelka D. S.; Dunwiddie C. T.; Persinger C. C.; Munos B. H.; Lindborg S. R.; Schacht A. L. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [DOI] [PubMed] [Google Scholar]
- Munos B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959–968. [DOI] [PubMed] [Google Scholar]
- Munos B. Can open-source R&D reinvigorate drug research?. Nat. Rev. Drug Discov. 2006, 5, 723–729. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Williams A. J.; Krasowski M. D.; Freundlich J. S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Williams A. J. Finding promiscuous old drugs for new uses. Pharm. Res. 2011, 28, 1786–1791. [DOI] [PubMed] [Google Scholar]
- May J. W.; Steinbeck C. Efficient ring perception for the Chemistry Development Kit. J. Cheminform. 2014, 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisken S.; Meinl T.; Wiswedel B.; de Figueiredo L. F.; Berthold M.; Steinbeck C. KNIME-CDK: Workflow-driven cheminformatics. BMC Bioinformatics 2013, 14, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. M.; Sarker M.; Ekins S. New target predictions and visualization tools incorporating open source molecular fingerprints for TB Mobile 2.0. J. Cheminform. 2014, 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. M.; Ekins S. Open Source Bayesian Models. 2. Mining a “Big Dataset” To Create and Validate Models with ChEMBL. J. Chem. Inf. Model. 2015, (following paper in this issue) 10.1021/acs.jcim.5b00144. [DOI] [PubMed] [Google Scholar]
- Rogers D.; Brown R. D.; Hahn M. Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up. J. Biomol. Screen. 2005, 10, 682–686. [DOI] [PubMed] [Google Scholar]
- Chen B.; Sheridan R. P.; Hornak V.; Voigt J. H. Comparison of random forest and Pipeline Pilot Naive Bayes in prospective QSAR predictions. J. Chem. Inf. Model. 2012, 52, 792–803. [DOI] [PubMed] [Google Scholar]
- Hohman M.; Gregory K.; Chibale K.; Smith P. J.; Ekins S.; Bunin B. Novel web-based tools combining chemistry informatics, biology and social networks for drug discovery. Drug Discov. Today 2009, 14, 261–270. [DOI] [PubMed] [Google Scholar]
- Klinger R.; Kolarik C.; Fluck J.; Hofmann-Apitius M.; Friedrich C. M. Detection of IUPAC and IUPAC-like chemical names. Bioinformatics 2008, 24, i268–i276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D.; Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [DOI] [PubMed] [Google Scholar]
- Xia X.; Maliski E. G.; Gallant P.; Rogers D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [DOI] [PubMed] [Google Scholar]
- Litterman N.; Lipinski C. A.; Bunin B. A.; Ekins S. Computational Prediction and Validation of an Expert’s Evaluation of Chemical Probes. J. Chem. Inf. Model. 2014, 54, 2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S. Progress in computational toxicology. J. Pharmacol. Toxicol. Methods 2014, 69, 115–140. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Freundlich J. S.; Reynolds R. C. Are Bigger Data Sets Better for Machine Learning? Fusing Single-Point and Dual-Event Dose Response Data for Mycobacterium tuberculosis. J. Chem. Inf. Model. 2014, 54, 2157–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Bradford J.; Dole K.; Spektor A.; Gregory K.; Blondeau D.; Hohman M.; Bunin B. A Collaborative Database And Computational Models For Tuberculosis Drug Discovery. Mol. BioSyst. 2010, 6, 840–851. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Pottorf R.; Reynolds R. C.; Williams A. J.; Clark A. M.; Freundlich J. S. Looking back to the future: predicting in vivo efficacy of small molecules versus Mycobacterium tuberculosis. J. Chem. Inf. Model. 2014, 54, 1070–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Clark A. M.; Swamidass S. J.; Litterman N.; Williams A. J. Bigger data, collaborative tools and the future of predictive drug discovery. J. Comput. Aided Mol. Des. 2014, 28, 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruoja V.; Moosus M.; Kahru A.; Sihtmae M.; Maran U. Measurement of baseline toxicity and QSAR analysis of 50 non-polar and 58 polar narcotic chemicals for the alga Pseudokirchneriella subcapitata. Chemosphere 2014, 96, 23–32. [DOI] [PubMed] [Google Scholar]
- Sushko I.; Novotarskyi S.; Korner R.; Pandey A. K.; Rupp M.; Teetz W.; Brandmaier S.; Abdelaziz A.; Prokopenko V. V.; Tanchuk V. Y.; Todeschini R.; Varnek A.; Marcou G.; Ertl P.; Potemkin V.; Grishina M.; Gasteiger J.; Schwab C.; Baskin I. I.; Palyulin V. A.; Radchenko E. V.; Welsh W. J.; Kholodovych V.; Chekmarev D.; Cherkasov A.; Aires-de-Sousa J.; Zhang Q. Y.; Bender A.; Nigsch F.; Patiny L.; Williams A.; Tkachenko V.; Tetko I. V. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker T.; Grulke C. M.; Pozefsky D.; Tropsha A. Chembench: a cheminformatics workbench. Bioinformatics 2010, 26, 3000–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A. J.; Ekins S.; Spjuth O.; Willighagen E. L. Accessing, using, and creating chemical property databases for computational toxicology modeling. Methods Mol. Biol. 2012, 929, 221–241. [DOI] [PubMed] [Google Scholar]
- Williams A. J.; Ekins S.; Clark A. M.; Jack J. J.; Apodaca R. L. Mobile apps for chemistry in the world of drug discovery. Drug Discov. Today 2011, 16, 928–939. [DOI] [PubMed] [Google Scholar]
- Clark A. M.; Ekins S.; Williams A. J. Redefining cheminformatics with intuitive collaborative mobile apps. Mol. Informatics 2012, 31, 569–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Clark A. M.; Williams A. J. Open Drug Discovery Teams: A Chemistry Mobile App for Collaboration. Mol. Informatics 2012, 31, 585–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. M.; Williams A. J.; Ekins S. Cheminformatics workflows using mobile apps. Chem-Bio Informatics J. 2013, 13, 1–18. [Google Scholar]
- Ekins S.; Clark A. M.; Sarker M. TB Mobile: A Mobile App for Anti-tuberculosis Molecules with Known Targets. J. Cheminform. 2013, 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Clark A. M.; Williams A. J. Incorporating Green Chemistry Concepts into Mobile Chemistry Applications and Their Potential Uses. ACS Sustain Chem. Eng. 2013, 1, 8–13. [Google Scholar]
- Swamidass S. J.; Matlock M.; Rozenblit L. Securely Measuring the Overlap between Private Datasets with Cryptosets. PLoS One 2015, 10, e0117898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamidass S. J.; Schillebeeckx C. N.; Matlock M.; Hurle M. R.; Agarwal P. Combined Analysis of Phenotypic and Target-Based Screening in Assay Networks. J. Biomol. Screen. 2014, 19, 782–790. [DOI] [PubMed] [Google Scholar]
- Matlock M.; Swamidass S. J. Sharing chemical relationships does not reveal structures. J. Chem. Inf. Model. 2014, 54, 37–48. [DOI] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jimenez-Diaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; El Mazouni F.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; Derisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F.-J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J.-L.; Vanderwall D. E.; Green D. V. S.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [DOI] [PubMed] [Google Scholar]
- Gagaring K.; Borboa R.; Francek C.; Chen Z.; Buenviaje J.; Plouffe D.; Winzeler E.; Brinker A.; Diagena T.; Taylor J.; Glynne R.; Chatterjee A.; Kuhen K.. Novartis-GNF Malaria Box. ChEMBL-NTD (www.ebi.ac.uk/chemblntd).
- Ekins S.; Williams A. J. Meta-analysis of molecular property patterns and filtering of public datasets of antimalarial “hits” and drugs. MedChemComm 2010, 1, 325–330. [Google Scholar]
- Zhang L.; Fourches D.; Sedykh A.; Zhu H.; Golbraikh A.; Ekins S.; Clark J.; Connelly M. C.; Sigal M.; Hodges D.; Guiguemde A.; Guy R. K.; Tropsha A. Discovery of Novel Antimalarial Compounds Enabled by QSAR-Based Virtual Screening. J. Chem. Inf. Model. 2013, 53, 475–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Mestres J.; Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Mestres J.; Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov S.; Dimitrova G.; Pavlov T.; Dimitrova N.; Patlewicz G.; Niemela J.; Mekenyan O. A stepwise approach for defining the applicability domain of SAR and QSAR models. J. Chem. Inf. Model. 2005, 45, 839–849. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Bruneau P.; Mewes H. W.; Rohrer D. C.; Poda G. I. Can we estimate the accuracy of ADME-Tox predictions?. Drug Discov. Today 2006, 11, 700–707. [DOI] [PubMed] [Google Scholar]
- Tropsha A.; Golbraikh A. Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [DOI] [PubMed] [Google Scholar]
- Tetko I. V.; Sushko I.; Pandey A. K.; Zhu H.; Tropsha A.; Papa E.; Oberg T.; Todeschini R.; Fourches D.; Varnek A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [DOI] [PubMed] [Google Scholar]
- Ananthan S.; Faaleolea E. R.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Laughon B. E.; Maddry J. A.; Mehta A.; Rasmussen L.; Reynolds R. C.; Secrist J. A. III; Shindo N.; Showe D. N.; Sosa M. I.; Suling W. J.; White E. L. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb.) 2009, 89, 334–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddry J. A.; Ananthan S.; Goldman R. C.; Hobrath J. V.; Kwong C. D.; Maddox C.; Rasmussen L.; Reynolds R. C.; Secrist J. A. III; Sosa M. I.; White E. L.; Zhang W. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis (Edinb.) 2009, 89, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds R. C.; Ananthan S.; Faaleolea E.; Hobrath J. V.; Kwong C. D.; Maddox C.; Rasmussen L.; Sosa M. I.; Thammasuvimol E.; White E. L.; Zhang W.; Secrist J. A. III. High throughput screening of a library based on kinase inhibitor scaffolds against Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb.) 2012, 92, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S.; Freundlich J. S.; Hobrath J. V.; Lucile White E.; Reynolds R. C. Combining computational methods for hit to lead optimization in Mycobacterium tuberculosis drug discovery. Pharm. Res. 2014, 31, 414–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.; Mika S.; Schroeter T.; Sutter A.; ter Laak A.; Steger-Hartmann T.; Heinrich N.; Muller K. R. Benchmark data set for in silico prediction of Ames mutagenicity. J. Chem. Inf. Model. 2009, 49, 2077–2081. [DOI] [PubMed] [Google Scholar]
- Temesi D. G.; Martin S.; Smith R.; Jones C.; Middleton B. High-throughput metabolic stability studies in drug discovery by orthogonal acceleration time-of-flight (OATOF) with analogue-to-digital signal capture (ADC). Rapid Commun. Mass Spectrom. 2010, 24, 1730–1736. [DOI] [PubMed] [Google Scholar]
- Hajjo R.; Grulke C. M.; Golbraikh A.; Setola V.; Huang X. P.; Roth B. L.; Tropsha A. Development, validation, and use of quantitative structure-activity relationship models of 5-hydroxytryptamine (2B) receptor ligands to identify novel receptor binders and putative valvulopathic compounds among common drugs. J. Med. Chem. 2010, 53, 7573–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huuskonen J. Estimation of aqueous solubility for a diverse set of organic compounds based on molecular topology. J. Chem. Inf. Comput. Sci. 2000, 40, 773–777. [DOI] [PubMed] [Google Scholar]
- Kortagere S.; Chekmarev D.; Welsh W. J.; Ekins S. Hybrid scoring and classification approaches to predict human pregnane X receptor activators. Pharm. Res. 2009, 26, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E. J.; Kruhlak N. L.; Benz R. D.; Contrera J. F. Assessment of the health effects of chemicals in humans: I. QSAR estimation of the maximum recommended therapeutic dose (MRTD) and no effect level (NOEL) of organic chemicals based on clinical trial data. Curr. Drug Discov. Technol. 2004, 1, 61–76. [DOI] [PubMed] [Google Scholar]
- Litterman N. K.; Lipinski C. A.; Bunin B. A.; Ekins S. Computational Prediction and Validation of an Expert’s Evaluation of Chemical Probes. J. Chem. Inf. Model. 2014, 54, 2996–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Li Y.; Wang J.; Chen L.; Zhang L.; Yu H.; Hou T. ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharmaceutics 2012, 9, 996–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F.; Yu H.; Zou B.; Babcock J.; Long S.; Li M. hERGCentral: a large database to store, retrieve, and analyze compound-human Ether-a-go-go related gene channel interactions to facilitate cardiotoxicity assessment in drug development. Assay Drug Dev Technol. 2011, 9, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Kameda M.; Ando M.; Miyazoe H.; Sekino E.; Ito S.; Masutani K.; Kamijo K.; Takezawa A.; Moriya M.; Ito M.; Ito J.; Nakase K.; Matsushita H.; Ishihara A.; Takenaga N.; Tokita S.; Kanatani A.; Sato N.; Fukami T. Discovery of novel diarylketoxime derivatives as selective and orally active melanin-concentrating hormone 1 receptor antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 5339–5345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.