

Abstract
Background
Basic understanding of linkage disequilibrium (LD) and population structure, as well as the consistency of gametic phase across breeds is crucial for genome-wide association studies and successful implementation of genomic selection. However, it is still limited in goats. Therefore, the objectives of this research were: (i) to estimate genome-wide levels of LD in goat breeds using data generated with the Illumina Goat SNP50 BeadChip; (ii) to study the consistency of gametic phase across breeds in order to evaluate the possible use of a multi-breed training population for genomic selection and (iii) develop insights concerning the population history of goat breeds.
Results
Average r2 between adjacent SNP pairs ranged from 0.28 to 0.11 for Boer and Rangeland populations. At the average distance between adjacent SNPs in the current 50 k SNP panel (~0.06 Mb), the breeds LaMancha, Nubian, Toggenburg and Boer exceeded or approached the level of linkage disequilibrium that is useful (r2 > 0.2) for genomic predictions. In all breeds LD decayed rapidly with increasing inter-marker distance. The estimated correlations for all the breed pairs, except Canadian and Australian Boer populations, were lower than 0.70 for all marker distances greater than 0.02 Mb. These results are not high enough to encourage the pooling of breeds in a single training population for genomic selection. The admixture analysis shows that some breeds have distinct genotypes based on SNP50 genotypes, such as the Boer, Cashmere and Nubian populations. The other groups share higher genome proportions with each other, indicating higher admixture and a more diverse genetic composition.
Conclusions
This work presents results of a diverse collection of breeds, which are of great interest for the implementation of genomic selection in goats. The LD results indicate that, with a large enough training population, genomic selection could potentially be implemented within breed with the current 50 k panel, but some breeds might benefit from a denser panel. For multi-breed genomic evaluation, a denser SNP panel also seems to be required.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-015-0220-1) contains supplementary material, which is available to authorized users.
Keywords: Effective population size, Genomic selection, Goat breeds, Goat 50 k panel, LD
Background
Goats are highly adaptable to different environmental conditions being raised all over the world for milk, meat and fibre production. Although they present reasonable reproductive and productive performance, it is necessary to improve their production efficiency to become more competitive with other livestock industries. In this regard, genetic selection plays a very important role and substantial genetic gain has been achieved using traditional breeding methods. However, there are some important traits that are difficult or expensive to measure (e.g. resistance to diseases, carcass traits, etc.), measured late in life or sex limited (e.g. milk production and composition). The development of genomic technologies means that new methods have become available such as genomic selection (GS) proposed by Meuwissen et al. [1].
GS has been successfully implemented in dairy cattle breeding programs and it is either under development or in the process of being implemented in other animal species. In dairy cattle the main advantage of GS is that it reduces the generation interval increasing the genetic gain per year. In goats, the generation interval is relatively lower than cattle, but could still be reduced. GS could also help to increase the selection intensity, which would increase productivity and reduce costs in breeding programs. As a first step for goat breeders, a 50 K SNP panel [2] has been developed by the International Goat Genome Consortium (IGGC), facilitating both genome wide association studies (GWAS) and the opportunity to implement GS.
One relevant parameter to the implementation of genomic selection in a breeding program is the extent to which linkage disequilibrium (LD) persists across the genome and how it varies between populations. LD is defined as a non-random association of alleles at two or more loci and is influenced by population history breeding system and the pattern of geographic subdivision [3]. The marker density required for successful GWAS and subsequently genomic selection, depends on the extent of LD across the genome [4]. A low LD level would require a higher marker density to enable markers to capture most of the genetic variation in the population. The persistence of LD has been evaluated in a number of domesticated animal species including pigs [5–7], horses [8], cattle [9–11] and sheep [12, 13]. A preliminary evaluation has also been conducted in goats using French dairy breeds [14]. Given the persistence of LD varies considerably between breeds in other species [13], it is important to characterise LD in a diverse collection of goat populations.
In addition to linkage disequilibrium accuracy of genomic selection also depends on the number of records available to estimate marker effects (training population). This may be a limitation factor for implementation of GS in goats because the genotyping costs are still relatively high compared to the economic value of the animals. An alternative to increase the number of animals in the training population is combining data from multi-breed populations. To obtain good accuracies of predictions using multi-breed populations it is required not only high LD between the markers and the quantitative trait loci (QTL) in each breed, but also high consistency of gametic phase between the markers and the QTL across breeds. Consistency of gametic phase is a measure of the degree of agreement of gametic phase for pairs of markers between two populations [6] that is also dependent of the difference on allele frequencies and relatedness between the two populations.
A variety of evolutionary phenomena impact observed allele frequencies distributions and the persistence of linkage disequilibrium. These include forces such as genetic drift migration, natural selection, and mutation rate. Therefore, population history strongly influences the extent of LD, particularly in domestic animal populations which have undergone bottlenecks during both domestication and the subsequent formation of breeds. The strength of these forces is likely to be different across the farm yard animal species, and indeed between breeds within each species. This prompted the investigation, in this study, of aspects of population history including ancestral effective population size, which can be inferred from the observed extent of LD [15–17].
There are many goat breeds been raised commercially all over the world and during the years they were characterized by high levels of admixture followed by animal movement. For instance goats were carried by the early explorers to America and Oceania [18] and some African breeds were also introduced more recently, such as South African Boer [19]. In order to better understand how modern goat breeds developed historically and to what degree they may have been mixed in the past, one alternative is to look at their breed composition through an analysis of admixture and/or principal components analysis (PCA).
Basic understanding of LD and population structure as well as the consistency of gametic phase across breeds is crucial for the implementation of genomic selection and is still limited in goats. Therefore, the objectives of this research were to estimate genome-wide levels of LD in Australian and Canadian goat breeds using data generated with the Illumina Goat SNP50 BeadChip to study the consistency of gametic phase between different breeds in order to evaluate the possible use of a multi-breed training population for genomic selection and develop insights concerning the population history of goat breeds.
Methods
The Canadian animals included in this study were managed in accordance with the Recommended Code of Practice for the Care and Handling of Farm Animals - GOATS (Canadian Agri - Food Research Council) [20]. All the samples were collected from commercial farms and the animal owners agreed to be involved in the project through their respective associations i.e. Ontario Goat and Société des éleveurs de chèvres laitières de race du Quebec. Samples were collected by well trained staff following industry best practices. Animal handling and sample collection from Australian animals were performed in accordance with Animal Ethics, CSIRO Brisbane Animal Ethics Committee.
Animals
The data analyzed in this study included genotypes of goats raised for milk meat and fibre production from two sources: i) a set of 976 Canadian goats from six breeds (Alpine, Boer, LaMancha, Nubian, Saanen and Toggenburg) and ii) 175 Australian goats from three breeds (Boer, Cashmere and Rangeland). The total number of genotyped animals for each breed by country is described in Table 1. The Canadian animals were from 25 commercial herds located in the provinces of Ontario and Quebec, two artificial insemination (AI) centres, and the Agriculture and Agri-Food Canada (AAFC) Centre for Animal Genetic Resources (Saskatoon, Saskatchewan). Most of the samples were ear notches (76 %), but also included extracted DNA samples from older animals (13 %), blood samples (9 %) and semen straws (2 %).
Table 1.
Number of animals and amount of SNPs excluded during the quality control procedure of the genotype data
| Breed | Excluded SNPs* | Remaining SNPs | |||||
|---|---|---|---|---|---|---|---|
| N | MAF < 0.05 | SNP CR < 0.90 | HWE | Het. | Total3 | ||
| AL1 | 403 | 3,828 | 2,358 | 39 | 1 | 6,820 | 45,268 |
| SA1 | 318 | 4,155 | 2,358 | 45 | 2 | 7,140 | 44,946 |
| MAF < 0.15 | SNP CR < 0.90 | HWE | Het. | Total3 | |||
| LN1 | 81 | 10,321 | 2,358 | 54 | 156 | 13,436 | 38,650 |
| NU1 | 54 | 16,013 | 2,379 | 21 | 242 | 19,233 | 32,853 |
| TO1 | 53 | 15,388 | 2,362 | 63 | 870 | 19,233 | 32,863 |
| BO1 | 67 | 13,282 | 2,374 | 17 | 185 | 16,438 | 35,648 |
| BO2 | 61 | 11,562 | 55 | 81 | 471 | 15,029 | 37,057 |
| CA2 | 48 | 9,508 | 147 | 33 | 689 | 13,272 | 38,814 |
| RL2 | 66 | 4,695 | 57 | 52 | 274 | 7,980 | 44,106 |
1Canada, 2Australia, 3It was excluded 2,958 SNPs without chromosome number and/or position information or SNPs located in the sexual chromosomes. AL: Alpine, SA: Saanen, LN: LaMancha, NU: Nubian, TO: Toggenburg, BO: Boer, CA: Cashmere, RL: Rangeland; *some SNPs were excluded due to more than one criterion,, MAF = minor allele frequency; CR = call rate; HWE = χ 2 test for Hardy-Weinberg equilibrium (p-value < 10−6). Het: excess of heterozigosity (>0.15)
The Australian populations and the genotypes derived from them have been described previously [21]. In brief animals were sampled from three different regions: 61 Boer goats from the Yarrabee goat herd in Queensland, 66 Rangeland goats from outback New South Wales and 48 Cashmere goats from Queensland. DNA was extracted from whole blood using the Qiagen Blood and Tissue extraction kit following the manufacturer’s instructions.
SNP genotyping and data filters
All the animals were genotyped using the Illumina goat SNP50 BeadChip (Illumina Inc. San Diego, CA) containing 53,347 single nucleotide polymorphisms (SNPs). SNP filtering and quality control conducted on the Australian populations resulted in analysis of a final marker set containing 52,088 loci [21]. The Canadian and Australian datasets were merged and only the 52,088 SNPs present in both datasets were kept for further analysis.
The genotyping quality control was performed within breed to remove SNPs and/or samples that could bias the LD estimates. SNPs with MAF lower than 5 % (for Alpine and Saanen breeds) or 15 % (for other breeds which have a much smaller number of genotyped animals) were removed prior to estimation of LD to prevent monomorphic loci inflating LD. SNPs were also excluded if the call rate was lower than 90 %, if they deviated significantly from Hardy–Weinberg equilibrium (HWE, p < 10−6) or if they presented a heterozygosity excess (>0.15, [22]). Only mapped autosomal SNPs were included for further analyses. Missing SNP genotypes were not imputed due to the limited number of genotyped animals in each breed. Besides the SNPs quality control, we also performed a quality control to animals, where individuals that had SNP call rate < 0.90 were removed. The number of SNPs excluded during the quality control procedure by each criterion is presented in Table 1. The number of SNPs per breed remaining after exclusions ranged from 32,853 to 45,268 out of 52,088 SNPs.
Extent of linkage disequilibrium
The extent of LD between markers was measured using r2 as proposed by Hill and Robertson [23], which is the squared correlation between alleles at two loci. It can be expressed as:
where D = f (AB) – f (A) f (B) and f (AB), f (A), f (a), f (B), and f (b), are observed frequencies of haplotype AB and alleles A, a, B, and b, respectively. However, the number of animals genotyped for this study was not enough to reconstruct haplotypes accurately. Thus, a D estimate suggested by Lynch and Walsh [24] was used:
where N is the total number of animals, and NAABB, NAABb, NAaBB, and NAaBb are the corresponding number of individuals in each genotypic category (AABB, AABb, AaBB, and AaBb). Another commonly used pair-wise measure of LD is D’ [25]. The reason for using r2 rather than D’ is that r2 is less sensitive to allele frequency and small sample size [26]. Values range from 0 (no LD) to 1 (complete LD) between two markers. If we consider the r2 between a bi-allelic marker and an (unobserved) bi-allelic QTL, r2 is the proportion of variation caused by the alleles at a QTL that is explained by the markers [27].
We calculated r2 for each pair of loci on each chromosome to determine the LD between adjacent SNPs, and the LD decay over different distances. To examine the decay of LD with physical distance, SNP pairs on the autosomes were sorted into bins based on pair-wise marker distance and the average of each bin was calculated. We defined 20 distance bins: lower than 0.02 Mb, from 0.02 until 0.1 defined every 0.01 Mb from 0.1 to 1 Mb defined every 0.1 Mb from 1 to 1.2 Mb and greater than 1.2 Mb.
Consistency of gametic phase
The consistency of gametic phase was defined by the Pearson correlation of signed r values between two breeds. For each marker pair with a measure of r2 the signed r value was determined by taking the square root of the r2 value and assigning the appropriate sign based on the calculated disequilibrium (D) value. Data was sorted into bins based on pair-wise marker distance to determine the breakdown in the consistency of gametic phase across distances and to assess the consistency of gametic phase at the smallest distances possible, given the number of genotyped SNPs. For each distance bin, the signed r values were then correlated between all 36 pairs of breeds using the CORR procedure in SAS (SAS Institute Inc., Cary, USA).
Ancestral effective population size
The r2 measures combined with markers distance can be used to estimate the approximate effective population size (Ne) at a given point in the past time. The Ne in each generation was determined based on the expectation of r2 in different distances and assuming a model without mutation as described by Sved [15]: , in which, c is the distance in Morgans between the SNPs. Ne is the effective population size and r2 is the average r2 value at a given distance. Each genetic distance (c) corresponds to a value of t generations in the past. This value was calculated as t = 1/(2c) as suggested by Hayes et al. [17].
The ancestral Ne was investigated at 21 time points from 5 until 1500 generations in the past. The distances (c) were taken as the middle of a range and the average r2 value was estimated at that distance. Ne was then calculated at each distance using that specific average r2.
Admixture analysis
In order to have an insight about the evolutionary history of the breeds included in this study we performed an admixture analysis. The same genotype quality control presented in Table 1 was applied to the admixture analysis. We used the ADMIXTURE software [28] to determine the level of admixture of each animal. This software applies a model based on a clustering algorithm that identifies subgroups that have distinctive allele frequencies. It places individuals into k predefined clusters.
The choice of an appropriate value for k is a notoriously difficult statistical problem. It seems that this choice should be guided by knowledge of a population’s history [28]. In this study we evaluated k from 6 to 10 as it would be a more representative value of the expected number of subpopulations in our data set. Two out of nine populations were from the same breed (Australian and Canadian Boer populations). Furthermore, it is known that the Rangeland is a composite breed population. So only results for k = 7 were shown, which have a more reasonable biological interpretation, as suggested by Pritchard et al. [29].
Principal component analysis (PCA)
In order to better assess the breed composition of the animals and for graphically display the results we also performed a principal component analysis. Principal components were calculated from the genomic relationship matrix (G) using prcomp function of R [30]. The G matrix was calculated using the method described by VanRaden [31]:
where M is a matrix of counts of the alleles “A” (with dimensions equal to the number of animals by number of SNPs), pi is the frequency of allele “A” of the ith SNP, P is a matrix (with dimensions equal to the number of animals by number of SNPs) with each row containing the pi values, I is the identity matrix (of size equal to the number of animals). Missing values in M were replaced by 2 times the frequency of allele “A” in the breed.
Results
SNP frequency and distribution
The level of genetic diversity present within and between the goat populations can be measured by the number of polymorphic loci and their allele frequencies distributions. Table 1 indicates that the Rangeland Alpine and Saanen breeds had the highest number of loci remaining after filtering based on MAF, HWE and other metrics. Fig. 1 presents the distribution of SNP by MAF range, and shows that Rangeland goats had the highest rate of high MAF loci, where more than 90 % of SNPs displayed MAF in excess of 0.15. Conversely, the Nubian and Toggenburg breeds had 67.41 and 68.68 % of loci with MAF > 0.15. Only one animal from the Rangeland breed was excluded due to low call rate (<0.90). Alpine and Saanen breeds presented very similar SNPs distribution for all MAF ranges. Canadian Boer population presented a higher proportion of SNPs with MAF < 0.15 compared to the Australian Boer population.
Fig. 1.
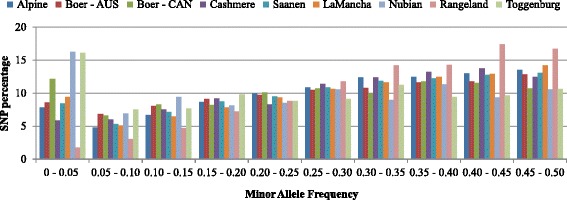
Distribution of SNPs by MAF ranges and breed. AUS: Australia, CAN: Canada
A descriptive summary of chromosomes and SNPs for the Alpine breed (larger sample size) is shown in Table 2. Diploid cells of Capra hircus contain 29 homologous autosomal pairs (CHI) and one pair of sex chromosomes. The total autosomal genome length was 2402.526 Mb with the shortest CHI being 41.478 Mb (CHI25) and the longest CHI being 154.929 Mb (CHI1).
Table 2.
Summary of analyzed single nucleotide polymorphism (SNP) markers for each Capra hircus autosome (CHI) for the Alpine breed
| CHI | Length (Mb) | SNP (n) | Mean distance (Mb) | Longest gap (Mb) | Mean r2 |
|---|---|---|---|---|---|
| 1 | 154.929 | 2,939 | 0.0527 ± 0.0273 | 0.2724 | 0.156 |
| 2 | 135.409 | 2,578 | 0.0525 ± 0.0292 | 0.3905 | 0.142 |
| 3 | 116.773 | 2,129 | 0.0548 ± 0.0296 | 0.2931 | 0.138 |
| 4 | 115.925 | 2,192 | 0.0529 ± 0.0296 | 0.4332 | 0.151 |
| 5 | 110.992 | 2,049 | 0.0540 ± 0.0316 | 0.4313 | 0.133 |
| 6 | 114.319 | 2,176 | 0.0525 ± 0.0290 | 0.3011 | 0.145 |
| 7 | 106.469 | 1,972 | 0.0540 ± 0.0304 | 0.3245 | 0.160 |
| 8 | 110.894 | 2,134 | 0.0519 ± 0.0264 | 0.2591 | 0.146 |
| 9 | 90.267 | 1,725 | 0.0523 ± 0.0262 | 0.2640 | 0.135 |
| 10 | 99.104 | 1,894 | 0.0522 ± 0.0264 | 0.2678 | 0.169 |
| 11 | 105.280 | 1,964 | 0.0535 ± 0.0284 | 0.2998 | 0.143 |
| 12 | 83.588 | 1,545 | 0.0541 ± 0.0344 | 0.7093 | 0.160 |
| 13 | 80.605 | 1,491 | 0.0540 ± 0.0319 | 0.3693 | 0.149 |
| 14 | 92.298 | 1,717 | 0.0537 ± 0.0300 | 0.2954 | 0.164 |
| 15 | 78.973 | 1,504 | 0.0525 ± 0.0298 | 0.4343 | 0.139 |
| 16 | 77.625 | 1,468 | 0.0528 ± 0.0267 | 0.2214 | 0.153 |
| 17 | 71.841 | 1,334 | 0.0537 ± 0.0322 | 0.4750 | 0.135 |
| 18 | 60.990 | 1,161 | 0.0524 ± 0.0291 | 0.3044 | 0.149 |
| 19 | 62.124 | 1,124 | 0.0553 ± 0.0312 | 0.3173 | 0.116 |
| 20 | 71.198 | 1,341 | 0.0530 ± 0.0267 | 0.2238 | 0.144 |
| 21 | 66.714 | 1,307 | 0.0510 ± 0.0253 | 0.2654 | 0.137 |
| 22 | 57.863 | 1,076 | 0.0537 ± 0.0303 | 0.3036 | 0.142 |
| 23 | 49.385 | 950 | 0.0519 ± 0.0257 | 0.2341 | 0.140 |
| 24 | 61.693 | 1,211 | 0.0508 ± 0.0269 | 0.2536 | 0.135 |
| 25 | 41.478 | 787 | 0.0526 ± 0.0296 | 0.2421 | 0.127 |
| 26 | 50.153 | 941 | 0.0531 ± 0.0287 | 0.2997 | 0.139 |
| 27 | 44.099 | 840 | 0.0524 ± 0.0302 | 0.3827 | 0.142 |
| 28 | 43.204 | 841 | 0.0513 ± 0.0273 | 0.2589 | 0.120 |
| 29 | 48.332 | 878 | 0.0550 ± 0.0331 | 0.4650 | 0.122 |
| Overall | 2,402.526 | 45,268 | 0.0530 | 0.7093 | 0.144 |
After application of quality control filters to remove low quality data the 50 k SNP panel showed good coverage of the genome with an average gap size between adjacent SNP varying from 0.05 to 0.07 Mb. Additional file 1: Tables S1.a and S1.b shows the largest intervals by chromosome and breed. The largest gaps were observed on CHI12 (0.7093 Mb), CHI17 (1.1399 Mb), CHI3 (1.9366 Mb), CHI12 (0.6780 Mb), CHI12 (0.7093 Mb), CHI7 (1.6214 Mb), CHI22 (1.0613 Mb), CHI29 (0.4990 Mb), CHI25 (1.1201 Mb) for Alpine, Boer (Australian population), Boer (Canadian population), Cashmere, Saanen, LaMancha, Nubian, Rangeland and Toggenburg animals, respectively. The chromosomes that presented larger gaps in most breeds were: CHI12, CHI17 and CHI29. Most of the breeds with a smaller number of animals had the largest average gap size between adjacent SNPs, due to the exclusion of SNPs with minor allele frequency (MAF) lower than 0.15, while for Alpine and Saanen breeds, MAF threshold was 0.05. However, for the Rangeland breed, even considering a MAF threshold of 0.15, the number of excluded SNPs was similar with those from Alpine and Saanen breeds.
Additional file 2: Table S2 presents the distribution of SNPs by chromosome for each breed. The greater range in the number of SNPs/Mb was observed for the Boer breed (Australian population) from 13.62 (CHI13) to 17.31 (CHI28) SNPs/Mb and the shorter range was observed for the Alpine breed and it varied from 18.09 (CHI19) to 19.63 (CHI19) SNPs/Mb.
Extent of linkage disequilibrium within goat breeds
Linkage disequilibrium was estimated separately within each of 9 goat populations using r2. The average linkage disequilibrium (r2) between adjacent SNPs by breed and average distance between adjacent SNPs (Mb) are presented in Table 3. Average r2 between adjacent SNP pairs was highest within the two geographically distinct populations of Boer goats (0.287 and 0.289) and lowest for the Rangeland and Alpine populations (0.110 and 0.144). The average r2 appears to reflect breed diversity whereby genetically diverse populations have generally lower average LD between adjacent loci. LD was also compared between chromosomes, revealing some variation (Additional file 3). The chromosomes that presented higher levels of LD were not the same for most breeds, except for Canadian and Australian Boer populations that presented more similar LD estimates.
Table 3.
Average linkage disequilibrium (r2) and average distance (Mb) between adjacent SNPs by breed
| Breed | r2 | Distance (Mb) |
|---|---|---|
| Alpine1 | 0.144 | 0.0530 |
| Saanen1 | 0.153 | 0.0534 |
| LaMancha1 | 0.194 | 0.0621 |
| Nubian1 | 0.276 | 0.0733 |
| Toggenburg1 | 0.248 | 0.0731 |
| Boer1 | 0.287 | 0.0674 |
| Boer2 | 0.289 | 0.0648 |
| Cashmere2 | 0.182 | 0.0620 |
| Rangeland2 | 0.110 | 0.0545 |
1Canada; 2Australia
LD is expected to decline as the recombination and physical distance between the markers increases. Fig. 2 displays the average LD values at given distance ranges for each breed (see also Additional file 4). High LD values were observed only at small distances between pairs of SNPs. For all the breeds LD decays rapidly as distance between the two SNPs increases. The average r2 estimates for the Rangeland population were the lowest values across all distances. It was followed by Alpine and Saanen. Alpine and Saanen breeds showed similar pattern of LD, which could be explained by their common ancestral origin [14].
Fig. 2.
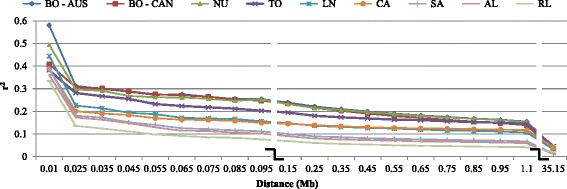
Average r2 values at given distances (Mb) for Australian and Canadian goats. BO-AUS: Australia Boer, BO-CAN: Canadian Boer, NU: Nubian, TO: Toggenburg, LN: LaMancha, CA: Cashmere, SA: Saanen, AL: Alpine, RL: Rangeland
It is important to note that the number of animals varied between groups (Table 1) and this has the potential to influence the observed LD. A correction for sampling error was applied that accounts for the number of haplotypes observed per population. Corrected r2 was calculated as (r2 – 1/N)/(1 – 1/N), where N is the number of haplotypes or twice the number of individuals [23]. The Additional file 5 presents the estimated and corrected r2 values for all the populations included in this study. However, the differences were small and all the results presented in this paper are based on non-corrected r2 estimates.
Boer (Australian and Canadian populations) and Nubian had the highest levels of LD across all distances. The r2 values for Canadian and Australian Boer animals were very similar for short distances bins except for distances up to 0.02 Mb. The r2 similarities could be indicating that they were managed together until few generations ago (around 5 generations ago). The Australian Boer goats presented higher estimates at long distances compared to Canadian Boer goats.
Trends across distances were very similar (Fig. 2) for all breeds and the LD level decayed at a very similar rate. The extent of LD decreased substantially from the first (up to 0.02 Mb) to the second range of distances (between 0.02 and 0.03 Mb). The number of SNP pairs at distances < 0.02 Mb was quite small though, which is also indicated by the high standard deviation values (Additional file 4: Tables S4.a and S4.b). The mean r2 decreased more slowly with increasing distance. LD levels were smaller than 0.05 at distances greater than 1.20 Mb for all breeds. The low level of long range LD may indicate that these breeds have not been under intense selection or have had large effective population size in the recent past.
Admixture and principal components analyses
Breed composition for each animal was calculated using the admixture model as described by Alexander et al. [28]. This determines the proportion of a given genome originated from each of k ancestral clusters defined as seven in this study. Fig. 7 and Table 4 show the proportion of each cluster, averaged across individuals within population. Some breeds have distinct genotypes (less clusters) based on SNP50 genotypes, such as the Boer, Cashmere and Nubian populations. The other groups share higher genome proportions with each other, indicating higher admixture and a more diverse genetic composition. The Rangeland population contains the highest rate of admixture, consistent with it being an unmanaged feral population founded by mixing of a number of breeds [21]. The admixture analysis presented here indicates contributions of mainly Cashmere, Nubian and Boer breeds into the Rangeland population. However, there is also a contribution from some dairy breeds. On average, around 13, 27 and 50 % of the Rangeland goat genome was in common to that found in Nubian, Boer and Cashmere, respectively (Table 4). LaMancha breed presents a contribution of Alpine and Nubian breeds (8 and 5 %, respectively). Saanen breed shares a higher proportion of the genome with Alpine (12 %), followed by Toggenburg (6 %) and LaMancha (4 %). The Saanen and Alpine breeds were managed together until few decades ago. However, the Saanen breed appears to be more mixed compared to Alpine breed. Australian and Canadian Boer populations were grouped together with an average of 95 % of their genome in common.
Fig. 7.
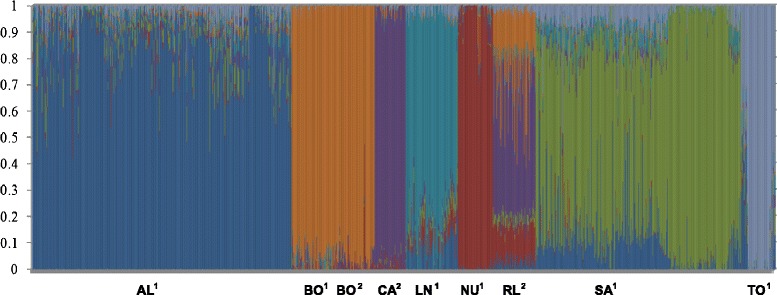
Breed composition per animal using ADMIXTURE software. 1: Canada, 2:Australia, AL = Alpine, BO = Boer, CA = Cashmere, LN = LaMancha, NU = Nubian, RL = Rangeland, SA = Saanen and TO = Toggenburg
Table 4.
Average breed composition of 9 goat populations given 7 clusters estimated by ADMIXTURE software
| Breed1 | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | Cluster 6 | Cluster 7 |
|---|---|---|---|---|---|---|---|
| Alpine1 | 0.8689 ± 0.1250 | 0.0109 ± 0.0314 | 0.0383 ± 0.0445 | 0.0160 ± 0.0203 | 0.0222 ± 0.0214 | 0.0080 ± 0.0102 | 0.0354 ± 0.0446 |
| Saanen1 | 0.1169 ± 0.1324 | 0.0069 ± 0.0239 | 0.7603 ± 0.1967 | 0.0145 ± 0.0182 | 0.0411 ± 0.0326 | 0.0040 ± 0.0079 | 0.0561 ± 0.0536 |
| LaMancha1 | 0.0755 ± 0.0573 | 0.0516 ± 0.0421 | 0.0344 ± 0.0682 | 0.0192 ± 0.0174 | 0.7880 ± 0.1666 | 0.0042 ± 0.0062 | 0.0269 ± 0.0211 |
| Toggenburg1 | 0.0767 ± 0.1389 | 0.0049 ± 0.0126 | 0.0256 ± 0.0391 | 0.0103 ± 0.0169 | 0.0193 ± 0.0300 | 0.0027 ± 0.0049 | 0.8601 ± 0.2218 |
| Nubian1 | 0.0033 ± 0.0072 | 0.9546 ± 0.0724 | 0.0026 ± 0.0046 | 0.0141 ± 0.0198 | 0.0093 ± 0.0131 | 0.0132 ± 0.0438 | 0.0027 ± 0.0048 |
| Boer1 | 0.0029 ± 0.0125 | 0.0204 ± 0.0630 | 0.0011 ± 0.0036 | 0.0214 ± 0.0205 | 0.0021 ± 0.0040 | 0.9506 ± 0.0719 | 0.0011 ± 0.0028 |
| Boer2 | 0.0014 ± 0.0041 | 0.0032 ± 0.0069 | 0.0039 ± 0.0071 | 0.0189 ± 0.0256 | 0.0187 ± 0.0226 | 0.9481 ± 0.0443 | 0.0054 ± 0.0114 |
| Rangeland2 | 0.0289 ± 0.0204 | 0.1337 ± 0.0279 | 0.0276 ± 0.0188 | 0.4971 ± 0.1015 | 0.0218 ± 0.0161 | 0.2704 ± 0.1509 | 0.0203 ± 0.0115 |
| Cashmere2 | 0.0023 ± 0.0064 | 0.0164 ± 0.0171 | 0.0054 ± 0.0098 | 0.9539 ± 0.0519 | 0.0046 ± 0.0071 | 0.0121 ± 0.0165 | 0.0050 ± 0.0085 |
1Canada; 2Australia
Figure 8 presents the first and second principal components calculated based on the G matrix. It shows that some breeds present clear clusters while others are genetically closer to each other. Australian and Canadian Boer were clustered together. The dairy breeds were clustered apart from the dual purpose/fibre/meat breeds. The Rangeland animals were clustered close to Nubian, Cashmere and Boer, what was also observed in the admixture analysis.
Fig. 8.
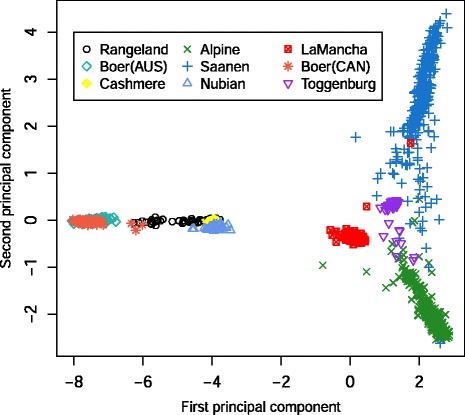
Plot of the first 2 principal components of the genomic relationship matrix for all animals, colored by breed
Linkage phase
The strength of consistency in gametic phase between breeds has implications for the design of successful genomic prediction programs. Specifically it influences what (if any) breed combinations can be merged to form a single training set to estimate SNP effects. Figs. 3 and 4 present the consistency of gametic phase (Pearson correlation between signed r values) between some breed pairs, while Table 5 presents the Pearson correlations between gametic phase of all breeds over distances smaller than 0.20 Mb (above diagonal) and between 0.02 and 0.03 Mb (below diagonal). The estimates for other distances were not presented as they were small. However, it is shown in the Additional file 6 for all distances and breed pairs. The highest consistency of gametic phase was found between Australian and Canadian Boer. This is expected given the two geographically distinct populations were drawn from the same breed. Other groups that presented higher correlations were: Alpine and Saanen, Alpine and LaMancha, Canadian Boer and Rangeland, Australian Boer and Rangeland, and Cashmere and Rangeland. The estimated correlations for all the breed pairs except Canadian and Australian Boer, were lower than 0.70 for all distances greater than 0.02 Mb. The correlations between Australian and Canadian Boer and Cashmere or Rangeland were very similar, suggesting a high relatedness between Australian and Canadian Boer populations.
Fig. 3.
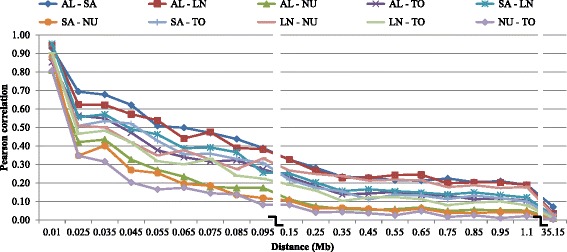
Consistency of gametic phase (Pearson correlations of signed r values) at given distances for 10 selected breed pairs. AL: Alpine, LN: LaMancha, NU: Nubian, SA: Saanen and TO: Toggenburg
Fig. 4.
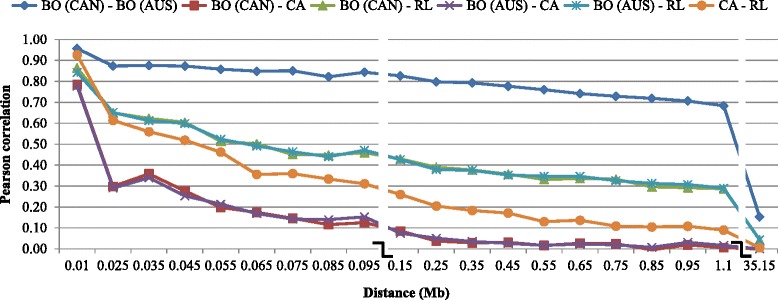
Consistency of gametic phase (Pearson correlations of signed r values) at given distances for 6 selected breed pairs. BO (AUS): Australia Boer, BO (CAN): Canadian Boer, CA: Cashmere and RL: Rangeland
Table 5.
Pearson correlations between gametic phase of all breeds for the distances pairs smaller than 0.2 Mb (above diagonal) and between 0.02 and 0.03 Mb (below diagonal)
| Alpine1 | Saanen1 | LaMancha1 | Toggenburg1 | Nubian1 | Boer1 | Boer2 | Cashmere2 | Rangeland2 | |
|---|---|---|---|---|---|---|---|---|---|
| Alpine1 | 0.93 | 0.94 | 0.85 | 0.81 | 0.75 | 0.74 | 0.83 | 0.87 | |
| Saanen1 | 0.69 | 0.95 | 0.88 | 0.88 | 0.79 | 0.78 | 0.84 | 0.87 | |
| LaMancha1 | 0.62 | 0.56 | 0.90 | 0.87 | 0.74 | 0.76 | 0.82 | 0.82 | |
| Toggenburg1 | 0.56 | 0.51 | 0.47 | 0.81 | 0.70 | 0.75 | 0.78 | 0.77 | |
| Nubian1 | 0.42 | 0.35 | 0.51 | 0.35 | 0.76 | 0.80 | 0.87 | 0.88 | |
| Boer1 | 0.35 | 0.36 | 0.41 | 0.29 | 0.34 | 0.96 | 0.78 | 0.86 | |
| Boer2 | 0.38 | 0.38 | 0.44 | 0.34 | 0.32 | 0.87 | 0.78 | 0.84 | |
| Cashmere2 | 0.42 | 0.37 | 0.38 | 0.33 | 0.36 | 0.30 | 0.29 | 0.93 | |
| Rangeland2 | 0.50 | 0.45 | 0.50 | 0.39 | 0.45 | 0.65 | 0.65 | 0.61 |
1Canada; 2Australia
Ancestral effective population size estimations
A graphical representation of the Ne values at each time point from 1500 to five generations ago is given in Figs. 5 and 6. Looking at the Ne in the distant past (1500 generations ago), effective populations were found to be ~ 5325, 3309, 3057, 3030, 2742, 1967, 1803, 1743, and 1741 animals for Rangeland Alpine, Saanen, Cashmere, LaMancha, Toggenburg, Nubian, Australian Boer and Canadian Boer populations, respectively. It corresponds to the closest measured time to the goat domestication, which occurred around 10,000 years ago [32]. Based on an average generation interval of 4 years [33], they would have been domesticated around 2500 generations ago. However, there were no enough SNP pairs to accurately estimate Ne for more than 1500 generations ago.
Fig. 5.
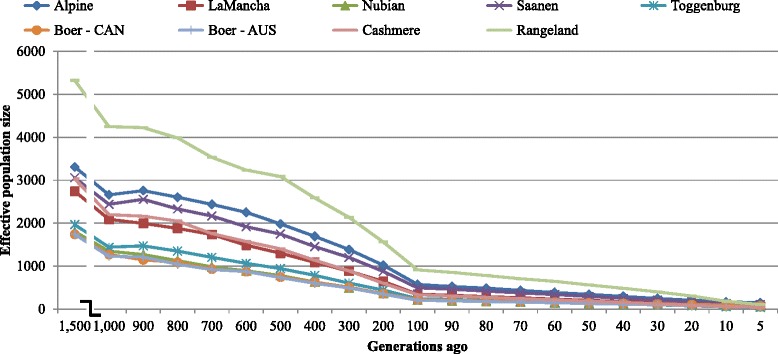
Past effective population size (Ne) over generations based on linkage disequilibrium calculations from 29 autosomes. AUS: Australia, CAN: Canada
Fig. 6.
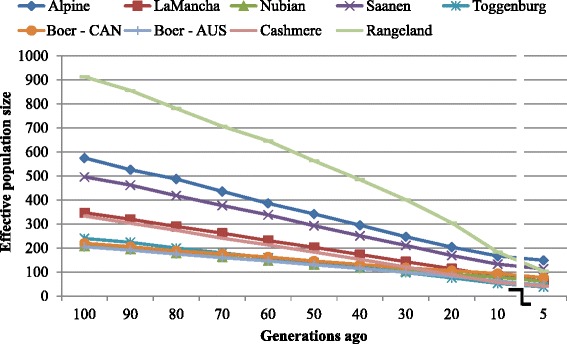
Past effective population size (Ne) from 100 to 5 generations ago based on linkage disequilibrium calculations from 29 autosomes. AUS: Australia, CAN: Canada
The results suggest that Ne has been lower in the recent past compared to the ancient past. The effective population size at five generations ago is calculated to be 104, 149, 113, 41, 62, 38, 61, 46 and 77 for Rangeland, Alpine, Saanen, Cashmere, LaMancha, Toggenburg, Nubian, Australian Boer and Canadian Boer populations, respectively. At the most recent measure of effective population size, five generations ago, Alpine breed presented the highest Ne, followed by Saanen and Rangeland. On the other hand, Toggenburg, Cashmere and Australian Boer populations presented the lowest values. The estimates for Australian and Canadian Boer populations were very similar for most of the measured time, except for the most recent generations studied. The Canadian Boer population presented higher Ne than Australian Boer population and it was particularly low at the most recent generations studied.
Discussion
Genotypic data and levels of LD
For breeds with small number of samples higher MAF threshold was applied and therefore more SNPs were excluded (Table 1). However, for the Rangeland population, even using a 0.15 MAF threshold, it presented a number of excluded SNPs by MAF criteria similar with Alpine and Saanen breeds, indicating high levels of polymorphism in that population. The high diversity level in the Rangeland population was previously discussed by Kijas et al. [21]. From the breeds included in this study, only Alpine, Boer and Saanen were represented within the SNP discovery panel used during the development of the goat SNP50 chip. Even though, all of them presented high levels of polymorphisms. The observed levels of MAF within breeds should provide enough variability for genomic studies such as genome-wide association studies and genomic evaluations.
The amount of SNPs remaining for the Alpine and Saanen breeds were slightly smaller than those attained by Carillier et al. [14] for the same breeds. They applied a call rate threshold of 98 % a MAF greater than 0.01 and Hardy-Weinberg equilibrium test (p-value < 10−6) and validated 46,959 out of 53,347 SNPs. Mucha et al. [34] working with a crossbred population (Alpine, Saanen and Toggenburg), filtered out SNPs that were not in Hard Weinberg equilibrium, had MAF below 0.05, call rate below 0.95 or GC content below 0.6. After the quality control 47,306 markers were available for further analyses. Despite of the number of SNPs excluded in our study, the 50 k panel showed good coverage of the genome.
The number of SNPs excluded due to low SNP call rate (CR) was very similar for the Canadian breeds. The smaller number of SNPs excluded due to low CR for the Australian breeds is due to the pre quality control that was done previously in the Australian dataset in which 1145 markers with call rates lower than 90 % were removed. Greater gaps were observed between SNP pairs in some chromosomes for most of the breeds (e.g. CHI12, CHI17 and CHI29), suggesting that in future development of another SNP panel for goats more SNPs could be included in those chromosomes for better coverage.
In the present study the number of genotyped animals differed considerably across breeds, with the largest number of genotyped animals in Alpine (403) and Saanen (318) breeds (Table 1). The differences in average r2 values for the breeds may be in part due to sampling effects, the low numbers of animals genotyped in some breeds and it could be due to different effective population sizes of those populations, which seems particularly appropriate for some breeds. Bohmanova et al. [26] recommended that for Holstein cattle at least 55 animals should be used to avoid overestimation of r2. In this study for three populations (Cashmere, Nubian and Toggenburg) there were fewer animals genotyped than that value. To address this concern, we applied a correction suggested by Hill and Robertson [23]. However, even for the Cashmere breed (smallest sample size) the highest difference between r2 estimated and corrected were around 0.01 units. Therefore, we decided to present the non-corrected values in the main text.
The average LD estimates in the goat breeds studied were quite variable. For Alpine and Saanen breeds average r2 values at 50 kb were slightly smaller than the values reported by Carillier et al. [14] (0.17 at 50 kb). In a crossbred population (Alpine, Saanen, and Toggenburg) Mucha et al. [34] observed a mean r2 at 50 kb of 0.18. For the other breeds, this was the first study done, which did not allow us to compare the results.
For the breeds Alpine Cashmere, Saanen, and Rangeland, the LD levels appear to be lower than that reported in Holstein dairy cattle (from 0.18 to 0.31, [35, 9, 36, 10]) or pigs (0.36 to 0.46, [6, 5]). The r2 estimates for the Saanen breed were similar with those attained for the Churra breed sheep of 0.152 from 40 to 60 kb [12]. Kijas et al. [13] found average r2 values for five sheep breeds for marker pairs at 70 kb apart varying from 0.08 to 0.22.
There is variation in the published extent of LD because the estimates of LD strongly depend on various factors such as: history and structure of the studied population (evolutionary forces that affected the population) sample size, marker type (microsatellites or SNPs), density and distribution of markers, type of method used for haplotype reconstruction, strictness of SNP filtering (threshold of minor allele frequencies and Hardy-Weinberg equilibrium), use of maternal haplotypes only or both maternal and paternal haplotypes [26].
As pointed out in Hayes et al. [17] LD at small distances reflects Ne in the distant past whereas LD at large distances reflects Ne in the recent past. The r2 values for Canadian and Australian Boer animals were very similar for short distances, except for distances up to 0.02 Mb, which could be indicating that they were managed together until few generations ago. The differences observed for distances up to 0.02 Mb could be explained by the small number of SNP pairs used to estimate r2 for that distance range. The higher r2 estimates at long distances observed in Australian Boer goats compared to the Canadian Boer population could be due to a smaller effective population size in the more recent past in the Australian Boer population compared to the Canadian one or it could be due to the fact that all Australian Boer animals were sampled in the same region and they could be more related than the average of the Australian Boer population. The standard deviations values (SD) for the r2 estimates at given distances (Additional file 4) were quite high, mainly for shorter distances, which may be due to the smaller number of SNP pairs available for the r2 estimations.
The extent of LD decreased substantially from the first (up to 0.02 Mb) to the second range of distances (between 0.02 and 0.03 Mb) (Fig. 2). The low level of long range LD may indicate that these breeds have not been under intense selection or genetic drift.
Alpine and Saanen were the breeds with the largest sample sizes. The higher observed levels of LD at short ranges in some of the other breeds could be due to sampling but they are more likely to be due to smaller effective population size in those breeds, as Rangeland population also presented low r2 values. Therefore, it would be interesting to confirm the LD results obtained in this investigation using a larger number of genotyped animals.
A higher level of LD is related to a higher accuracy of genomic estimated breeding values. Some studies (e.g. [1, 37]) have recommended that an r2 value greater than 0.2 would be sufficient for genomic selection. At the average distance between adjacent SNPs in the goat 50 k SNP panel (~0.06 Mb) the breeds LaMancha, Nubian, Toggenburg, and Australian and Canadian Boer exceeded or approached this value. This indicates that, with a large enough training population, genomic selection could potentially be implemented with reasonable accuracy using the current 50 k panel within breed, but the other breeds might benefit from a denser panel. For the Rangeland population, the LD levels were very low even for short distances, suggesting that this breed come from a highly heterogeneous population and a higher density panel might be needed to implement genomic selection in this breed.
Admixture and principal component analyses and linkage phase
The results show that a great number of animals have a significant portion of their genotype coming from another cluster (Fig. 7). Boer, Cashmere and Nubian breeds seem to have a smaller level of admixture compared to the other breeds, indicating that there is less remaining from any other ancestral breed that may have interacted with them.
For the animals that had estimates of breed composition more diverse (less than 75 % of their genes coming from a single breed) it can be assumed that a more recent admixture event could have occurred. According to Larmer et al. [38] this may be useful in identifying locations of certain QTL that are present in only one breed. If an animal has a phenotype that is significantly different from other animals in the breed to which it is registered and chunks coming from other breeds can be identified in the genome we could propose that one or more of those chunks have a QTL for that trait on them.
The higher level of admixture seen in the Saanen breed when compared to the Alpine breed implies that a greater degree of admixture has undergone since these breeds diverged historically. Consistently PCA (Fig. 8) also showed this trend. Animals from Alpine and Saanen breeds showed more spread clusters, indicating a higher breed admixture level among those breeds and other dairy breeds such as LaMancha and Toggenburg. LaMancha and Toggenburg showed clear individual clusters and a smaller genetic variation among animals from within those breeds. The larger degree of admixture seen in the Rangeland population is consistent with its evolutionary history, as the Rangeland goats are largely unmanaged feral goats. The results indicate that Boer, Cashmere and Nubian breeds are likely to have contributed to create the feral population. On average, 50 % of the Rangeland genome was in common to that found in the Cashmere breed. It indicates that the Rangeland population may have been formed by the introgression of mainly Boer and Nubian animals in the Cashmere genetics to develop the Rangeland population. PCA (Fig. 8) also confirmed this relationship, where animals from Nubian, Cashmere, Boer and Rangeland were closely clustered compared to the dairy breeds. This sharing of the gene pool may be due to mixing of the breeds as discussed before, especially for Rangeland population.
Canadian and Australian Boer populations seem to share a great proportion of the genome. The small level of admixture coming from other breeds (clusters) indicates that admixture may have taken place on average, in the distant past. The high degree of genotype sharing among both Boer populations is consistent with their evolutionary history, as the Boer breed was developed in South Africa [39] and exported to Canada and Australia a few decades ago. Furthermore, according to Casey and Van Niekerk [39], the Boer breed was formed with infusion of Indian and European blood, which could explain the admixture contribution, even small, from other breeds. The close relationship between Australian and Canadian Boer populations was also confirmed in the PCA plot (Fig. 8), where animals were clustered together based on the first two principal components.
The PCA analysis showed that the Illumina 50 K goat beadchip was able to discriminate most of the breeds. Some of them were more clearly clustered while others were clustered more closely. However this trend is consistent with the breeds history. Huson et al. [40] reported that the Illumina 50 K goat beadchip can effectively distinguish goat populations, specifically indigenous African goat populations. In a comparison of 14 African goat breeds, New Zealand Boer, three Italian Alpine breeds, and six United States of America breeds, the first principal component generated a continental categorization by Italy, United States of America, and Africa with the second principal component distinguishing the Boer breed.
The consistency of gametic phase between breeds indicates whether or not different breeds could potentially be pooled into one common training population to better estimate SNP effects. For goat genomic evaluation this would be very important due to the fact that there is a small number of genotyped animals in the breeds with small population size. The highest consistency of phase was found between Australian and Canadian Boer populations suggesting a greater level of relatedness between these populations. They may be still connected through exchange of genetic material or have diverged a few generations ago. It was also confirmed in the admixture analysis, where they were always grouped together. According to Malan [19], Boer goats were imported to North America directly from South Africa or via Australia or New Zealand, which is another evidence of their close relationship. The correlation values for them were consistent until greater distances, indicating that both populations could be pooled in a single training population. The other groups that presented higher correlations were: Alpine and Saanen, Alpine and LaMancha, Canadian Boer and Rangeland, Australian Boer and Rangeland and Cashmere and Rangeland. Based on the admixture levels observed for some breeds, it was expected a higher consistency of phase among them. However, even for those breed pairs the consistency of gametic phase between adjacent markers was not high enough to support the pooling of breeds in a training population for genomic selection. The estimated correlations for all the breed pairs, except Canadian and Australian Boer, were lower than 0.70 for distances greater than 0.02 Mb. It indicates that markers and QTL phases might not be strongly associated across those breeds.
Carillier et al. [14] found consistency of gametic phases at 50 kb (i.e. average distance between two SNPs) among French Alpine and Saanen breeds of 0.56. According to them, the two goat breeds (Alpine and Saanen) were genetically close until a couple of generations ago. In dairy cattle, de Roos et al. [41] evaluated the effect of combining multiple populations on the reliability of genomic predictions and concluded that the benefits of combining populations in a training set were higher when the populations have diverged for only a few generations ago, when the marker density was high, and when heritability was low. From the simulation studies reported by these authors, populations that had diverged six generations ago presented a correlation of phase higher than 0.8 for distances up to 0.45 Mb. Therefore, for multi-breed genomic evaluation in goats, a denser SNP panel seems to be required. For implementing genomic selection using the 50 k panel in goat breeds, other ways to increase the training population should be sought, such as genotyping more animals in each breed or collaborate with other countries and share genotypes and phenotypes/EBVs for genomic selection.
Ancestral effective population size
We observed an initial pattern of decreasing Ne with values of over 1740 for Australian and Canadian Boer populations and 5325 for Rangeland population estimated in the distant past (1500 generations ago) and values closer or even smaller than 100 estimated at 5 generations ago (Figs. 5 and 6 and Additional file 7).
The Ne estimates at 5 generations ago found for Alpine and Saanen breeds (149 and 113 respectively) were similar with those reported by Larroque et al. [42] for French Alpine and Saanen breeds, 143 and 120, respectively. Garcia-Gamez et al. [12] have reported a Ne estimate of 128 in the more recent generation studied for Churra breed sheep population. Alpine and Saanen breeds are the most common dairy breeds raised over the world, which is reflected by their highest Ne measures in the most recent time. The similar estimates attained for both Boer populations are another evidence of their relatedness. The differences observed in the most recent past may be due to sampling errors or smaller number of animals in the Canadian population compared to the South African population, where Australian and Canadian Boer animals were probably imported. The high Ne observed in the ancient past for the Rangeland population reflects the great level of admixture observed for this breed. As observed in the admixture analysis, Boer, Cashmere, Nubian and other breeds contributed to its formation.
According to Meuwissen [43] a threshold of Ne = 100 would be necessary to ensure that an animal population is long-term viable in terms of genetic diversity. Our results of current effective population size are above the threshold only for 3 breeds, indicating that care should be taken in this regard to ensure that the effective population size and consequently a reasonable diversity level are maintained.
Conclusions
At the average distance between adjacent SNPs in the current 50 k SNP panel (~0.06 Mb) the breeds LaMancha, Nubian, Toggenburg and Australian and Canadian Boer exceeded or approached the level of linkage disequilibrium that is useful (r2 > 0.2) for genomic prediction. This indicates that, with a large enough training population, genomic selection could potentially be implemented within breed with the current 50 k panel, but the breeds Alpine, Saanen, Cashmere and Rangeland might benefit from a denser panel.
The highest consistency of gametic phase was found between Australian and Canadian Boer populations indicating a greater level of relatedness between these two breeds and a possibility of pooling them in a single reference population. However, for the other breeds, the consistency of gametic phase between adjacent markers is not high enough to encourage the pooling of breeds in a single training population for genomic selection. For multi-breed genomic evaluation, a denser SNP panel seems to be required. Therefore, other ways to increase the training population for genomic selection using the 50 k panel should be sought, such as genotyping more animals in each breed and/or collaborating with other countries for sharing genotypes and phenotypes/EBVs.
Acknowledgements
The authors thank the following organizations for providing funds and collaborating within the project: the sector councils of Quebec, Ontario and British-Columbia, who administer the Canadian Agricultural Adaptation Program (CAAP) for Agriculture and Agri-Food Canada; Ontario Goat; Société des éleveurs de chèvres laitières de race du Quebec; GoatGenetics.Ca; and the Brazilian Government through the Science without Borders Program that provides graduate fellowship for the first author. We also thank the International Goat Genome Consortium (IGGC) for developing the goat SNP50 BeadChip and Meat and Livestock Australia (MLA) for support to collect and genotype the three Australian goat populations.
Abbreviations
- AAFC
Agriculture and Agri-Food Canada
- AI
Artificial insemination
- AL
Alpine breed
- AUS
Australia
- BO
Boer breed
- CA
Cashmere breed
- CAN
Canada
- Chr
Chromosome
- CHI
Capra hircus homologous autosomal pairs
- CR
Call Rate
- CSIRO
Commonwealth Scientific and Industrial Research Organisation
- DNA
Deoxyribonucleic acid
- EBV
Estimated Breeding value
- GEBV
Genomic Estimated Breeding Value
- GS
Genomic Selection
- GWAS
Genome-Wide Association Studies
- HWE
Hardy-Weinberg Equilibrium
- IGGC
International Goat Genome Consortium
- kb
kilo base pairs
- LD
Linkage Disequilibrium
- LN
LaMancha breed
- MAF
Minor Allele Frequency
- Mb
Mega base pairs
- Ne
Effective population size
- NU
Nubian breed
- PCA
Principal component analysis
- QTL
Quantitative Trait Loci
- RL
Rangeland population
- SA
Saanen breed
- SD
Standard deviation
- SNP
Single Nucleotide Polymorphism
- TO
Toggenburg breed
Additional files
Largest gaps between adjacent SNPs by chromosome and breed. Showing the largest intervals between adjacent SNPs for each autosome and for all 9 goat populations.
Number of SNPs/Mb by breed for each autosome (CHI). Presenting the distribution of SNPs by chromosome for each breed.
Linkage disequilibrium (r2) estimates by chromosome for each breed. Showing the LD estimates by chromosome for each breed.
Average r2 values (± standard deviation) at a given distance range. Displays the average LD values at given distance ranges for each breed.
Average r2 and corrected r2 at given distances. Presenting the estimated and corrected r2 values for all the populations included in this study.
Pearson correlations between gametic phase of all breeds pairs. Showing the estimates for the Pearson correlations between gametic phase of all breeds pairs, including those there were not presented in the main text.
Effective population size for all studied breeds for a given number of generations ago. Presenting the effective population size for all studied breeds for a given number of generations ago estimated based on the LD levels.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LFB participated in the design of the study, carried out the analyses and results interpretation, was involved in the discussions, prepared and drafted the manuscript. MJ participated in the design of the study, was involved in the discussions and helped to draft the manuscript and in the data acquisition. DAG helped with the analysis, results interpretation and manuscript drafting. JWK and LRPN provided the 3 Australian breeds dataset, were involved in the discussions and gave editorial assistance. RVV helped with the analysis, results interpretation and manuscript drafting. MS developed the SNPPLD software, was involved in the discussions, and helped to draft the manuscript. FSS participated in the design of the study, was involved in the discussions and helped to draft the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Luiz F. Brito, Email: lbrito@uoguelph.ca
Mohsen Jafarikia, Email: mohsen@ccsi.ca.
Daniela A. Grossi, Email: d_agrossi@yahoo.com.br
James W. Kijas, Email: James.Kijas@csiro.au
Laercio R. Porto-Neto, Email: Laercio.Portoneto@csiro.au
Ricardo V. Ventura, Email: rventura@uoguelph.ca
Mehdi Salgorzaei, Email: msargol@uoguelph.ca.
Flavio S. Schenkel, Email: schenkel@uoguelph.ca
References
- 1.Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157(4):1819–29. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tosser-Klopp G, Bardou P, Bouchez O, Cabau C, Crooijmans R, Dong Y, et al. Design and characterization of a 52K SNP chip for goats. PLoS One. 2014;9(1):e86227. doi: 10.1371/journal.pone.0086227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slatkin M. Linkage disequilibrium–understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 2008;9(6):477–85. doi: 10.1038/nrg2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khatkar MS, Nicholas FW, Collins AR, Zenger KR, Cavanagh JA, Barris W, et al. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics. 2008;9:187. doi: 10.1186/1471-2164-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uimari P, Tapio M. Extent of linkage disequilibrium and effective population size in Finnish Landrace and Finnish Yorkshire pig breeds. J Anim Sci. 2011;89(3):609–14. doi: 10.2527/jas.2010-3249. [DOI] [PubMed] [Google Scholar]
- 6.Badke YM, Bates RO, Ernst CW, Schwab C, Steibel JP. Estimation of linkage disequilibrium in four US pig breeds. BMC Genomics. 2012;13:24. doi: 10.1186/1471-2164-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veroneze R, Lopes PS, Guimaraes SE, Silva FF, Lopes MS, Harlizius B, et al. Linkage disequilibrium and haplotype block structure in six commercial pig lines. J Anim Sci. 2013;91(8):3493–501. doi: 10.2527/jas.2012-6052. [DOI] [PubMed] [Google Scholar]
- 8.Corbin LJ, Blott SC, Swinburne JE, Vaudin M, Bishop SC, Woolliams JA. Linkage disequilibrium and historical effective population size in the Thoroughbred horse. Anim Genet. 2010;41(Suppl 2):8–15. doi: 10.1111/j.1365-2052.2010.02092.x. [DOI] [PubMed] [Google Scholar]
- 9.de Roos AP, Hayes BJ, Spelman RJ, Goddard ME. Linkage disequilibrium and persistence of phase in Holstein-Friesian, Jersey and Angus cattle. Genetics. 2008;179(3):1503–12. doi: 10.1534/genetics.107.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larmer SG, Sargolzaei M, Schenkel FS. Extent of linkage disequilibrium, consistency of gametic phase, and imputation accuracy within and across Canadian dairy breeds. J Dairy Sci. 2014;97(5):3128–41. doi: 10.3168/jds.2013-6826. [DOI] [PubMed] [Google Scholar]
- 11.Porto-Neto LR, Kijas JW, Reverter A. The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet, Selection, Evol: GSE. 2014;46:22. doi: 10.1186/1297-9686-46-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Gamez E, Sahana G, Gutierrez-Gil B, Arranz JJ. Linkage disequilibrium and inbreeding estimation in Spanish Churra sheep. BMC Genet. 2012;13:43. doi: 10.1186/1471-2156-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kijas JW, Porto-Neto L, Dominik S, Reverter A, Bunch R, McCulloch R, et al. Linkage disequilibrium over short physical distances measured in sheep using a high-density SNP chip. Anim Genet. 2014;45(5):754–7. doi: 10.1111/age.12197. [DOI] [PubMed] [Google Scholar]
- 14.Carillier C, Larroque H, Palhiere I, Clement V, Rupp R, Robert-Granie C. A first step toward genomic selection in the multi-breed French dairy goat population. J Dairy Sci. 2013;96(11):7294–305. doi: 10.3168/jds.2013-6789. [DOI] [PubMed] [Google Scholar]
- 15.Sved JA. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol. 1971;2(2):125–41. doi: 10.1016/0040-5809(71)90011-6. [DOI] [PubMed] [Google Scholar]
- 16.Hill WG. Estimation of effective population size from data on linkage disequilibrium. Genet Res. 1981;38(03):209–16. doi: 10.1017/S0016672300020553. [DOI] [Google Scholar]
- 17.Hayes BJ, Visscher PM, McPartlan HC, Goddard ME. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003;13(4):635–43. doi: 10.1101/gr.387103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubeuf J-P, Boyazoglu J. An international panorama of goat selection and breeds. Livest Sci. 2009;120(3):225–31. doi: 10.1016/j.livsci.2008.07.005. [DOI] [Google Scholar]
- 19.Malan SW. The improved Boer goat. Small Rumin Res. 2000;36(2):165–70. doi: 10.1016/S0921-4488(99)00160-1. [DOI] [PubMed] [Google Scholar]
- 20.Canadian Agri-Food Research Council. Recommended Code of Practice for the Care and Handling of Farm Animals - GOATS. 2003. https://www.nfacc.ca/pdfs/codes/goat_code_of_practice.pdf .Accessed 15 May 2015.
- 21.Kijas JW, Ortiz JS, McCulloch R, James A, Brice B, Swain B, et al. Genetic diversity and investigation of polledness in divergent goat populations using 52 088 SNPs. Anim Genet. 2013;44(3):325–35. doi: 10.1111/age.12011. [DOI] [PubMed] [Google Scholar]
- 22.Wiggans G, Sonstegard T, VanRaden P, Matukumalli L, Schnabel R, Taylor J, et al. Selection of single-nucleotide polymorphisms and quality of genotypes used in genomic evaluation of dairy cattle in the United States and Canada. J Dairy Sci. 2009;92(7):3431–6. doi: 10.3168/jds.2008-1758. [DOI] [PubMed] [Google Scholar]
- 23.Hill WG, Robertson A. Linkage disequilibrium in finite populations. TAG Theoretical Applied Genet Theoretische und angewandte Genetik. 1968;38(6):226–31. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- 24.Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland, Mass: Sinauer; 1998. [Google Scholar]
- 25.Lewontin RC. The interaction of selection and linkage. I. General considerations; Heterotic models. Genetics. 1964;49(1):49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohmanova J, Sargolzaei M, Schenkel FS. Characteristics of linkage disequilibrium in North American Holsteins. BMC Genomics. 2010;11:421. doi: 10.1186/1471-2164-11-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayes B, Bowman P, Chamberlain A, Goddard M. Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci. 2009;92(2):433–43. doi: 10.3168/jds.2008-1646. [DOI] [PubMed] [Google Scholar]
- 28.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Team RCR. A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- 31.VanRaden P. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91(11):4414–23. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 32.Zeder MA, Hesse B. The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science. 2000;287(5461):2254–7. doi: 10.1126/science.287.5461.2254. [DOI] [PubMed] [Google Scholar]
- 33.Danchin-Burge C, editor. Bilan de variabilité génétique de 9 races de petits ruminants laitiers et à toison. Compte rendu; 2011.
- 34.Mucha S, Mrode R, Coffey M, Conington J. Estimation of genomic breeding values for milk yield in UK dairy goats. 10th world congress on genetics applied to livestock production. Canada: Vancouver; 2014. [Google Scholar]
- 35.Sargolzaei M, Schenkel FS, Jansen GB, Schaeffer LR. Extent of linkage disequilibrium in Holstein cattle in North America. J Dairy Sci. 2008;91(5):2106–17. doi: 10.3168/jds.2007-0553. [DOI] [PubMed] [Google Scholar]
- 36.Habier D, Tetens J, Seefried FR, Lichtner P, Thaller G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet, Selection, Evolution : GSE. 2010;42:5. doi: 10.1186/1297-9686-42-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calus MP, Meuwissen TH, de Roos AP, Veerkamp RF. Accuracy of genomic selection using different methods to define haplotypes. Genetics. 2008;178(1):553–61. doi: 10.1534/genetics.107.080838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larmer S, Ventura R, Buzanskas ME, Sargolzaei M, Schenkel FS. Assessing admixture by quantifying breed composition to gain historical perspective on dairy cattle in Canada. Vancouver, Canada: 10th World Congress on Genetics Applied to Livestock Production; 2014. [Google Scholar]
- 39.Casey N, Van Niekerk W. The Boer goat. I. Origin, adaptability, performance testing, reproduction and milk production. Small Rumin Res. 1988;1(3):291–302. doi: 10.1016/0921-4488(88)90056-9. [DOI] [Google Scholar]
- 40.Huson H, Sonstegard T, Silverstein J, Woodward-Greene M, Masiga C, Muchadeyi F, et al. Genetic and phenotypic characterization of African goat populations to prioritize conservation and production efforts for small-holder farmers in Sub-Saharan africa. Vancouver, Canada: 10th World Congress on Genetics Applied to Livestock Production; 2014. [Google Scholar]
- 41.de Roos AP, Hayes BJ, Goddard ME. Reliability of genomic predictions across multiple populations. Genetics. 2009;183(4):1545–53. doi: 10.1534/genetics.109.104935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larroque H, Barillet F, Baloche G, Astruc J, Buisson D, Shumbusho F, et al. Toward genomic breeding programs in French dairy sheep and goats. Vancouver, Canada: 10th World Congress on Genetics Applied to Livestock Production; 2014. [Google Scholar]
- 43.Meuwissen T. Genetic management of small populations: a review. Acta Agriculturae Scand Section A. 2009;59(2):71–9. [Google Scholar]