

Abstract
Since the original descriptions of post-concussive pathophysiology, there has been a significant increase in interest and ongoing research to study the biological underpinnings of concussion. The initial ionic flux and glutamate release result in significant energy demands and a period of metabolic crisis for the injured brain. These physiological perturbations can now be linked to clinical characteristics of concussion, including migrainous symptoms, vulnerability to repeat injury and cognitive impairment. Furthermore, advanced neuroimaging now allows a research window to monitor post-concussion pathophysiology in humans noninvasively. There is also increasing concern about the risk for chronic or even progressive neurobehavioral impairment after concussion/mild TBI. Critical studies are underway to better link the acute pathobiology of concussion with potential mechanisms of chronic cell death, dysfunction and neurodegeneration. This “new and improved” paper summarizes in a translational fashion and updates what is known about the acute neurometabolic changes after concussive brain injury. Furthermore, new connections are proposed between this neurobiology and early clinical symptoms as well as to cellular processes that may underlie long term impairment.
Introduction
One of the hallmarks of concussion is that neurological signs and symptoms are imparted after biomechanical force to the brain in the absence of macroscopic neural damage 1. In general, this has been interpreted as a result of predominantly functional or microstructural injury to neural tissue. Functional injury can refer to perturbations of cellular or physiological function including but not limited to ionic shifts, metabolic changes or impaired neurotransmission. Microstructural injury refers to physical changes not readily evident on CT scanning, but now detectable through advanced imaging (such as diffusion tensor imaging – DTI – for delineating signs of axonal injury). An important additional characteristic of sports-related concussion is the potential for repeated mild traumatic brain injuries (TBI) over the course of an athletic event, a season or even a lifetime. The basic neurobiology of concussion/mild TBI has been elucidated in animal models, is increasingly corroborated in human studies and has been described as a neurometabolic cascade of events that involves bioenergetic challenges, cytoskeletal and axonal alterations, impairments in neurotransmission and vulnerability to delayed cell death and chronic dysfunction 2,3. This review will use a translational approach to describe advances in our understanding of the underlying neurobiology of concussive injuries, with particular emphasis on linking the neurometabolic cascade to clinical characteristics as well as on new connections being made between acute post-concussion pathophysiology, long-term biological changes and chronic sequelae.
Acute Pathophysiology (Figures 1 and 2)
Figure 1.
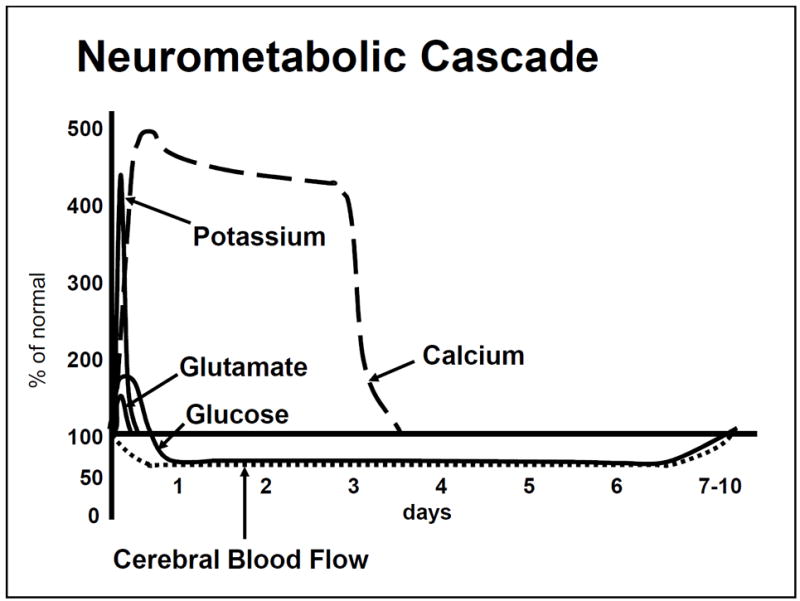
Time course of the neurometabolic cascade of concussion.
Figure 2.
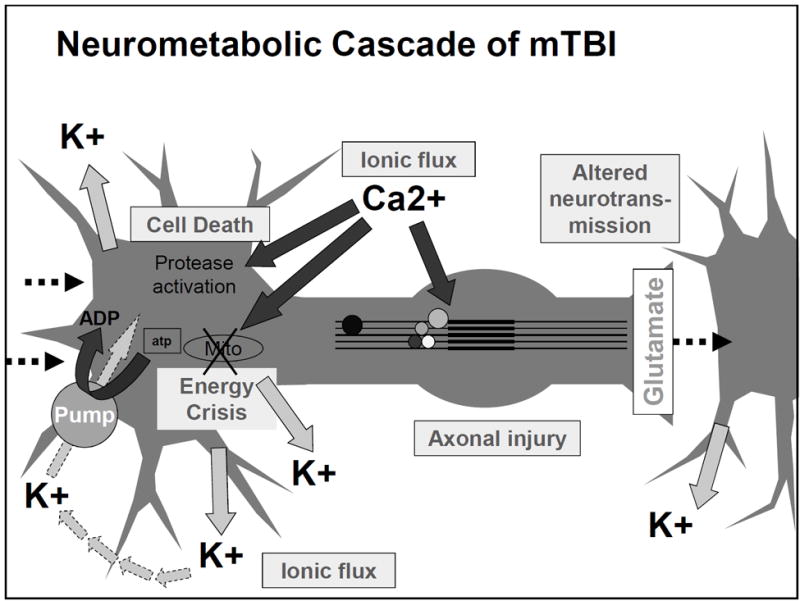
Diagram of the acute cellular biological processes occurring after concussion/mild TBI.
Ionic flux and glutamate release
Early studies demonstrated that biomechanical injury results in ionic flux and hyperacute indiscriminate glutamate release 4,5. Potassium efflux, and sodium and calcium influx, occur due to mechanoporation of lipid membranes (creation of sublethal defects via traumatic insult) at the cellular level. Initial ionic flux and depolarization can then trigger voltage- or ligand-gated ion channels, creating a diffuse ‘spreading depression-like’ state that may be the biological substrate for very acute post-concussive impairments.
Energy crisis
In an effort to restore ionic and cellular homeostasis, ATP-requiring membrane ionic pumps shift into overdrive, causing hyperglycoloysis, relative depletion of intracellular energy reserves and increase in ADP 6. In very early phases, this increased demand for energy occurs in a setting of normal or reduced cerebral blood flow, resulting in an uncoupling, or mismatch, between energy supply and demand.
Intracellular calcium flux, which occurs early and may persist longer than other ionic disturbances, is accommodated by sequestration of calcium into mitochondria. However, this short-term solution can then result in mitochondrial dysfunction, exacerbate problems with oxidative metabolism and worsen the cellular energy crisis.
In addition to the acute energy perturbations, intracellular redox state is altered. This puts additional stress on the system by generative damaging free radicals and shifting metabolic pathways that can trigger longer-lasting impairments and set the stage for vulnerability to repeated injury, which is particularly relevant for the clinical setting of sports-related concussion.
After an initial period of hyperglycolysis and metabolic uncoupling, glucose metabolic rates go into a state of impaired metabolism that can last up to 7-10 days in adult animals, and is associated with behavioral impairments in spatial learning 6,7. The duration of this hypometabolic period appears to vary with age, with younger animals showing shorter periods (3 days) of impairment 8. Post-traumatic changes in metabolism may be mediated by altered gene expression 9 and enzyme/transporter regulation 10.
Cytoskeletal damage
Biomechanical forces imparted onto neurons and glia can damage the delicate and complex microstructural components including dendritic arbors, axons and astrocytic processes. Following intra-axonal calcium flux, neurofilament side-arms can be phosphorylated and collapse, resulting in loss of structural integrity in axons 11. Microtubule disruption due to axonal stretch can interfere with bi-directional axonal transport, potentially isolating the synapse, diminishing normal neurotransmission and in severe cases, result in axonal disconnection. Intra-axonal calcium flux results in proteolytic damage to subaxolemmal spectrin and other cytoskeletal components 12. More recent studies have indicated that cytoskeletal anchor points in the cell membrane, mediated through proteins such as integrins, may represent a primary molecular target of traumatic injury in both neurons and vascular cells 13,14.
Axonal dysfunction
Axons prove to be particularly vulnerable to biomechanical stretch. Early studies showed increased axolemmal permeability after experimental TBI 15. As mentioned above, damage to the neurofilaments and microtubules leads to axonal dysfunction and potential for disconnection. While axonal disconnection was once thought to invariably lead to demise of the neuronal cell body, in vivo animal studies have shown that perisomatic axotomy can occur with atrophy and shrinkage of the neuron, without cell death 16. It is assumed that this type of severely damaged neuron is not capable of normal function. Some studies have suggested that dietary supplementation of particular fatty acids may mitigate, or at least alter the timeline of, axonal damage after experimental TBI 6. In vitro stretch injury models demonstrate post-stretch axonal undulations and beading – in some cases these would recover, but in others the axonal damage was lasting 17,18.
In addition, recent studies using fluid percussion injury centered over the white matter tracts of the corpus callosum indicate that unmyelinated axons are more vulnerable to injury (and show greater resultant impairment of electrophysiological function) than myelinated fibers 19. This finding, although not replicated in a comparative study of immature animals, has developmental implications, as it is known that myelination of axonal tracts is one of the ongoing maturational changes that occur in the young brain.
Recent studies of repeat mild TBI in the immature brain have demonstrated white matter damage and associated cognitive impairments 20. This is consistent with studies in adult animals that have also shown more prominent axonal injury with repeated mild TBI 21.
Altered neurotransmission
Changes in ligand-gated excitatory and inhibitory neurotransmission have been reported after in vivo experimental TBI. Alterations in glutamate (N-methyl-D-aspartate NMDA) receptor subunit composition and function have been reported following TBI in both the immature 22 and mature brain 23-25. Functional consequences of NMDAR subunit change in developing rat pups have been described as interfering with normal developmental plasticity, electrophysiology and memory 26. In adult rats, NMDAR changes have also been associated with functional alterations including differential patterns of calcium flux 25, immediate early gene activation and phosphorylation/activation of downstream signal transduction molecules such as CaMKII (calcium/calmodulin-dependent protein kinase II), ERK (extracellular signal-related kinase), CREB (cAMP response element binding protein) and BDNF (brain derived neurotrophic factor) 23,27.
Post-injury excitatory-inhibitory balance can also be upset by changes in inhibitory neurotransmission involving GABA and its receptors. Studies have consistently shown dropout of hilar GABAergic interneurons following lateral fluid percussion injury (FPI) in adult rats 28,29. In a recent study of post-TBI augmentation of fear-based learning, a putative model for post-TBI vulnerability to develop anxiety disorders and/or PTSD, decreased levels of GAD67, the biosynthetic enzyme for GABA, have been reported in the amygdala 30. The amygdala is a critical structure implicated in the development of aberrant fear-based learning in animal models 31 but also in some human studies 32.
Inflammation
Somewhat overlooked in acute studies of mild TBI, changes in inflammatory markers have been well-reported in more severe TBI for some time 33. Following TBI there is an activation and infiltration of microglia, particularly in cortical impact models but also after less severe models such as fluid percussion 34.
More recent studies suggest that inflammatory changes are also triggered by mild TBI. In both adults and immature rats, microarray studies report extensive upregulation of cytokine and inflammatory genes after TBI 9,35. Microglial activation after adult FPI, with or without exposure to the pesticide paraquat, has been associated with damage to the substantia nigra and implicated in the increased risk for parkinsonism after TBI 36. Similar neuroinflammatory changes have also been reported subacutely in the absence of behavioral deficits in a different study 37. A theory relating glutamate release and activation of immune receptors to oxidative stress and potentially later cellular injury has been proposed and termed “immunoexcitotoxicity” 38.
Cell death
Models of mild TBI have generally shown little cell death, even in settings where measurable cognitive impairments are described 20,39,40. However, the consequences of repeated mild TBI are less well described – functional impairments appear to be greater than single TBI in most models 21,41,42, raising the possibility that there may be longer term structural changes, although studies looking at chronic axonal injury and/or atrophy are lacking.
It is unclear whether chronic structural changes may evolve over longer time courses after even a single mild injury. However, following a single moderate/severe FPI in adult rats, ongoing cerebral and hippocampal atrophy have been described 43, as have chronic losses in dopaminergic neurons in the substantia nigra 36. Chronic evolution of injury has also been described after a single controlled cortical impact (CCI) injury in adult animals 44,45. Immature (P7) mice show late appearance of cognitive deficits and tissue loss after a single CCI 46, but other studies using a single FPI in developing rats (P19) showed no significant hippocampal or cortical neuronal loss 39.
Pathophysiology Meets Symptoms (Table 1)
Table 1.
Physiological perturbations after concussion and proposed clinical correlates.
| Post-TBI pathophysiology | Acute symptom / clinical correlate |
|---|---|
| Ionic flux | Migraine headache, photophobia, phonophobia |
| Energy crisis | Vulnerability to second injury |
| Axonal injury | Impaired cognition, slowed processing, slowed reaction time |
| Impaired neurotransmission | Impaired cognition, slowed processing, slowed reaction time |
| Protease activation, altered cytoskeletal proteins, cell death | Chronic atrophy, development of persistent impairments |
While studies proving associations between the following pathophysiological changes after concussion/mild TBI remain to be conducted, it is tempting to speculate on how acute neurobiological change results in the clinical symptoms associated with concussion. Several reasonable connections can be made, and are presented in Table 1.
Ionic flux and migraine
The widespread ionic flux described after FPI has often been described as ‘spreading depression-like’, and the original description of the spreading depression of Leao was in the context of migraine 47. In fact, DC potential shifts have been described in both experimental TBI 48,49 and with direct electrocorticography after more severe human TBI 50,51.
An additional intriguing connection that suggests a relationship between ionic compensatory mechanisms and cerebral responses to concussion is the description of significant mental status change and evidence for cerebral edema after mild TBI in the pedigree of a family with an ion channelopathy normally associated with familial hemiplegic migraine 52. In this description, the CACNA1A mutation was associated with several clinical phenotypes, including an exaggerated response to mild TBI in some family members. There are an increasing number of ion channelopathies being identified, and it is interesting to speculate whether genetic factors such as these may underlie the ‘glass jaw’ phenomenon whereby some athletes are more readily symptomatic after mTBI, or whether this type of genetic vulnerability underlies the malignant cerebral edema seen in cases of ‘second impact syndrome’.
Finally, there is considerable overlap of typical post-concussion symptoms with those commonly described in migraine, particularly headache, nausea, vomiting, photophobia, phonophobia and dizziness. In fact, some studies indicate the reporting of headache symptoms or a prior history of migraine as potential risk factors for prolonged recovery or more severe symptoms after concussion 53.
Energy crisis and vulnerability
Clinically it is known that a history of prior concussion/mTBI is a strong risk factor for subsequent concussion, and that in prospective studies, this risk appears greatest in the first 10 days post-injury 54-56. There are many factors that may play into this increased clinical risk, but biological vulnerability due to ongoing energy crisis is one that can be addressed by the current practice of delaying return to contact risk. The challenge here is identifying the duration of this period of metabolic vulnerability, as it likely varies between concussed individuals.
Human studies using magnetic resonance spectroscopy (MRS) have shown reductions in the metabolite N-acetylaspartate (NAA) in frontal white matter of concussed adults compared to normal controls 57,58. Over 30 days, NAA levels returned to normal, except in individuals who sustained a second concussion, in whom NAA did not recover fully until 45 days after the initial injury 58. Such findings support the concept of metabolic vulnerability after concussion. However, in these studies, the depressed NAA levels did not correlate well with reported symptoms, and thus the clinical significance of post-concussion NAA remains uncertain.
Animal studies have shed some light on this. In a weight drop model in adult rats, the greatest perturbations of neurochemical markers of metabolic stress occurred when injuries were separated by 3 days 10,59. Repeated concussive injuries in adult mice showed worsening of neurocognitive function and traumatic axonal injury when injuries were spaced out by 3 or 5 days, but not when injuries were separated by 7 days 21.
In a closed head impact model in juvenile rats, the period of reduced glucose metabolism post-injury was associated with impairments in working memory and generally lasted about 3 days. When a second injury occurred within this period of impaired glucose metabolism (post-injury day 1), the severity of both hypometabolism and memory impairment was greater. If the second injury occurred after full metabolic recovery from the first injury (post-injury day 5), the two injuries acted like single, separate injuries 42.
Overall, these investigations indicate that the timing of repeated injuries may have significant consequences on the severity of post-concussion pathophysiology and subsequent cognitive disturbances. They also indirectly support the current clinical management protocols of removing concussed athletes from contact risk immediately to allow time for biological recovery. Further work is needed to identify what biomarker can be used to determine the time of recovery for individual patients – symptoms, cognition, balance, reaction time and neuroimaging have all been proposed. It is likely that the duration of time for recovery in humans will be longer than that described in the animal models.
Axonal dysfunction and slowed cognition
White matter injury and damage to neural networks has been associated with cognitive impairments after moderate and severe TBI. Diffuse axonal injury (DAI) is traditionally described as a pathological finding of damage to white matter tracts associated with frank axonal disconnection and microhemorrhages, often seen post-mortem after severe TBI 60. Furthermore, it is recognized that after TBI, axonal injury also occurs on a microscale, with mechanoporation, stretching and beading of individual axons, disruption of axonal transport and axonal blebbing, as well as disconnection. The complete spectrum of axonal pathophysiology is sometimes termed traumatic axonal injury (TAI), to distinguish it from the more classical pathological description of DAI 61.
For mild TBI and concussion in humans, there is little pathological data 62, but advanced MRI using diffusion tensor imaging (DTI) has been used to measure changes in directionality of water diffusion that may be indicative of white matter disruption. Fractional anisotropy (FA) one such measure, with higher FA values indicating more water diffusion in a particular direction and lower FA values indicating less directionality. Increased FA values have been reported in corpus callosum early after mild TBI (6 days) in adolescents, and these values were correlated with a validated symptom scale 63W. Early increases in FA were also reported in a cohort of adolescents after concussion but no correlation was found between the DTI changes and cognitive function 64. Recent studies have reported changes in DTI measures and cognitive testing after a period contact/collision sports participation in individuals with no diagnosed concussion 65,66.
For more severe TBI, damage to the white matter strongly correlates to burden of neurocognitive impairment. FA generally is decreased in major fiber tracts (corpus callosum being most studied), and white matter disruption is associated with burden of neuropsychological deficits using standard testing 67,68.
Overall, data from DTI studies of mild TBI has been more variable, with some acute studies showing increased FA 64,69-71 and some showing decreased FA 65,72,73. It is likely that multiple parameters can affect the results of DTI studies, including timing after injury, age of subjects, severity of injury, region of brain analyzed as well as the specifics of the methods used 74. Thus, while DTI appears to be a very sensitive imaging modality to study concussion/mTBI noninvasively, its use for routine clinical management of individual patients is still premature.
Again, animal studies provide some additional perspective. Using a mouse model of more severe TBI - controlled cortical impact - correlations between DTI measures and histopathology were elegantly described across a time range from hours up to one month post TBI 75. These studies show that DTI is more sensitive than conventional MRI at detecting white matter injury after TBI. They also shed some light into the changing nature of DTI signal as the TBI evolves over time. Early changes (<24 hrs) included maximal staining of axonal varicosities with amyloid precursor protein (APP) and neurofilament light chain markers, while later time points (4 days to 1 month) showed maximal staining for gliosis (GFAP - glial fibrillary acidic protein).
Rodent models of mild repeated TBI have shown impairments in spatial learning and memory in the absence of overt histological damage or cell death 41. Increased APP was most prominent in animals with repeat closed head injury separated by 3 days, and these animals also showed the greatest impairment in motor function and spatial learning, when compared to single injury animals or those with repeat TBI separated by 1 week 21. Graded deficits in working memory after mild and repeat closed head injury (24 hours apart) have been reported along with increases in APP staining in corpus callosum and subcortical white matter 20.
Taken together, these studies show that cognitive deficits occur after mild TBI in the absence of overt cell death. Graded degrees of axonal injury/dysfunction occur after mTBI and repeat mTBI, and can be associated with graded levels of neurobehavioral impairment. However, in mild TBI, the linkage between imaging measures of axonal damage and clinically measurable cognitive problems is still in evolution.
Altered neurotransmission, slowed cognition and reaction time
Neurotransmitter and synaptic alterations are other mechanisms of neural dysfunction that occur in the absence of overt cell death and may contribute to acute and transient neurobehavioral impairments after concussion. Clinical studies show early post-concussion problems in attention and memory that generally recover over time; these have been measured using simple sideline questions 76, the Standardized Assessment of Concussion (SAC) 77, paper and pencil neuropsychological testing 78 and computerized cognitive testing 79. Slowed reaction time has been measured in conjunction with computerized tests as well as with a simple ‘stick-drop’ device 80.
Determining the physiological correlate of impaired post-concussion cognition has been attempted through the use of functional MRI (fMRI). Functional MRI measures changes in blood oxygen level dependent (BOLD) signal driven by increases in neural activity associated with specific cognitive tasks. While there remains debate about the physiology of the BOLD signal 81, evidence from animal models shows that up to 50% of the BOLD signal may be driven by excitatory glutamatergic neurotransmission 82.
Human studies of fMRI activation following concussion have often used cognitive measures of working memory. Abnormal patterns of increased BOLD activation have been reported days and weeks after concussion/mTBI in adults 83 and older teens (average age 16.6y) 84. These changes may serve as a biomarker of ongoing neural dysfunction or in one study, as a marker for prolonged recovery 84. A more recent study of slightly younger teens (average age 14.5y) after concussion showed worse working memory associated with significantly reduced task-related BOLD activity 85.
As mentioned above, animal studies have demonstrated TBI-induced deficits in excitatory neurotransmission, often involving the NMDAR 22-25. Interestingly, in the immature brain, these impairments are associated with a loss of experience-dependent plasticity that results in lasting learning and memory deficits 86,87. Even if a period of ‘recovery’ is allowed before stimulating experience-dependent plasticity, restoration of memory functions is only partial 88. Ongoing studies are aimed at developing mechanism-based interventions to help restore these impairments.
It is difficult to entirely tease out the contributions of axonal injury vs impaired neurotransmission to post-concussion cognitive and behavioral deficits. Quite likely both are involved, and determining the predominant mechanism in a given individual may depend on continued study into biomarkers to help identify those at risk for persistent impairments, and to determine interventions most likely to be effective.
Interestingly, as mentioned above, while post-traumatic metabolic changes in the immature brain appear to be shorter lasting than in adults 8, axonal vulnerability 19,20 and impairments in plasticity 86,87 may be more prominent in the young brain. Additional work is necessary to determine on balance whether the young brain is more or less vulnerable to mild TBI, although some clinical evidence suggests, at least for concussion, recovery takes longer in high school athletes compared to collegiate or adult professional athletes.
How much is too much? Acute-to-Chronic Pathobiology (Figure 2)
Evidence for the chronic sequelae of repeated mild TBI/concussion has long been associated with pathological studies of boxers 89,90. More recently, case studies of contact sport athletes have shown both gross and microscopic pathology that has been attributed to repeated exposure to concussive blows 91,92. In these more recent cases, deposition of aggregated tau protein has been reported as the pathological hallmark of the condition now termed chronic traumatic encephalopathy (CTE). However, the waters surrounding our current understanding of CTE have been muddied by selection bias inherent to any case report study, an uncertain dose relationship between mTBI and pathology, concomitant associations between repeat concussion and other neurodegenerative conditions and the lack of a unifying biological mechanism to translate the acute (but generally recoverable) pathophysiology of concussion into a chronically progressive disorder.
Energy crisis, protease activation and cell death
Acute impairments of cellular metabolism have described problems in both glycolytic and oxidative pathways (minutes to days, see above). Excess calcium flux is sequestered in mitochondria but then results in diminution of oxidative capacity that can spill over into the subacute period (days-weeks). Contact/collision sports and military duty, among other professions, are distinct from most accidental mTBI because of the inherent goal of returning to contact risk activity. The occurrence of repeated mTBI prior to full recovery from a first mTBI may set in play more lasting metabolic perturbations that may then trigger activation of intracellular proteases and the cascade that leads to apoptotic cell death.
Human studies have not definitively demonstrated this progression, which can only be done using longitudinal study designs. However, it is well known that gross pathology seen in the brains of boxers and others exposed to repetitive concussions includes cortical and hippocampal atrophy, ventriculomegaly, cavum septum pellucidum and other findings strongly suggestive of chronic cell death 93. More recent studies are beginning to use advances in quantitative MRI and longitudinal study designs to examine progressive atrophy within subject after mild TBI 94.
Animal studies support the concept that more severe TBI can result in chronic evolution of injury that results in ongoing cell death and atrophy out to 1 year 45,95. Even in a pediatric model of moderate-to-severe experimental TBI, progressive cell death and volume loss, associated with emergence of additional cognitive decline, has been reported over 1-3 months post-TBI 46. Longitudinal studies of brain atrophy after experimental repeat mild TBI/concussive injury remain to be done.
Altered protein degradation and toxic accumulation
In addition to the effects of chronic energy impairment as a trigger to protease activation and apoptotic cell death, it is well known that normal cellular protein homeostasis depends upon a functioning system of protein degradation. This system requires energy, ubiquitin and a protein complex called the proteasome 96. There are many examples in neurodegenerative disease of cellular oxidative stress leading to oxidatively damaged proteins that can affect metabolic enzymes and/or the ubiquitin-proteasome system. This could then result in the accumulation of abnormal/toxic proteins 97. These mechanisms have been implicated in Alzheimer disease, tauopathies, synucleinopathies (Parkinson disease) and ALS, to name a few, and it is not surprising that these links are now being made in TBI. Early and prolonged proteasomal activation has been proposed based upon immunohistochemical findings after severe human TBI 98.
Some experimental studies have identified molecular and functional changes in cortical and hippocampal proteasomes after TBI 99. This suggests proteasome dysfunction is a viable mechanism as a precursor to post-traumatic neurodegeneration, although these studies did not specifically measure for accumulation of toxic or abnormal proteins. A more recent study linked abnormalities in proteasomal molecules with enhanced apoptosis after TBI 100.
Accumulation of abnormal proteins has also been reported in animal models of TBI under multiple circumstances. Wild-type rodents show accumulation of oligomeric and phosphorylated tau after parasagittal fluid percussion injury 101 and blast neurotrauma 102. Interestingly, deposition of amyloid or tau was not seen at chronic time points in a study of repetitive mild TBI, even though the animals showed cognitive impairments and astrogliosis 103. Transgenic animals (3xTG-ApoE4) have also been reported to show increased accumulation of abnormal proteins after TBI 104,105.
Aggregation and progression of toxic molecules has recently gained traction in a series of articles suggesting that damaged tau oligomers, not neurofibrillary tangles, are the triggering molecules for spread of tau pathology in Alzheimer disease 106,107 (for review see 108). Elevated levels of extracellular tau have been reported after severe human TBI, and may hypothetically initiate the process of progressive tauopathy 109.
Based upon these data, the mechanism of impaired protein homeostasis and degradation after TBI is plausible. Confirmatory studies remain to be conducted. Such a mechanism may likely involve oxidative damage to metabolic or proteasome molecules. There is evidence that proteins such as amyloid and tau, which accumulate in known neurodegenerative disorders, also show perturbations after both clinical and experimental TBI.
Chronic axonal dysfunction
As discussed above, axonal dysfunction can result in impaired cognitive functioning and slowed reaction times, either through slowed conduction, damage to cerebral networks or deficits in neurotransmission. These mechanisms may be detected early after mTBI using DTI and fMRI 64,83.
DTI has also been used to detect white matter abnormalities subacutely and chronically after mTBI. One recent study used DTI to examine military personnel evacuated from Afghanistan and Iraq for TBI, and found persistent changes in multiple regions 110. Even in a civilian youth cohort with concussions, changes in DTI were detected early after injury while the subjects were symptomatic, but then showed similar changes at 4 months post-injury, a time when most were asymptomatic. The clinical significance of these DTI signal changes in a subacute/chronic setting is only beginning to be understood, but these white matter alterations have been associated with post-traumatic stress disorder 111 and depression 112 in different cohorts. Perhaps equally concerning are recent reports of abnormal DTI measures in athletes subjected to repeated mild head blows (heading in soccer) even without a diagnosis of concussion 65.
While axonal disconnection after severe TBI can certainly result in cognitive impairment, it is uncertain how much post-concussion white matter pathology is due to disconnection vs alterations in axonal physiology 18. Furthermore, the idea that axotomy results in the inevitable death of the neuron has been challenged by evidence showing some neurons survive axonal disconnection but may be rendered dysfunctional 16. What also remains uncertain is the ability of the damage axon to recover, or the time frame of events that dictates recovery vs disconnection. Recent evidence has focused on the possibility for persistent axonal degeneration following TBI, perhaps even a single TBI 17,113. As mentioned earlier, myelination also appears to protect axons from trauma, with unmyelinated axons more vulnerable 19. These mechanisms of chronic axonal damage may then be particularly problematic in settings of repeat mTBI without adequate time for recovery between injuries, immature myelination or individuals with genetic vulnerabilities.
Summary
Our understanding of the pathophysiology of concussion has expanded significantly in the last decade. Many of the acute physiological changes originally described have received additional support in newer models of mild repetitive TBI and with the use of advanced noninvasive neuroimaging. The contributions of oxidative stress, impaired axonal transport and altered neurotransmission are now clearly linked to the initial ionic fluxes, indiscriminate glutamate release and metabolic uncoupling. Furthermore, it is increasingly possible to see links between the pathophysiology of concussion and the early clinical signs and symptoms; there is great potential that, as the time course of these cellular events is better elucidated, this information will translate to a clearer idea of the timing of increased risk for repeated concussion in patients. Other accelerated research efforts into the ‘new’ neurometabolic cascade of concussion include studies examining causal linkages between the acute pathobiology and chronic progressive changes that may result in lasting impairments. While still early, untangling the connections between acute and chronic pathophysiology of concussion holds the promise for better prevention of repeated injury and mechanism-based therapies to interrupt the progression to persistent deficits or neurodegeneration.
Figure 3.
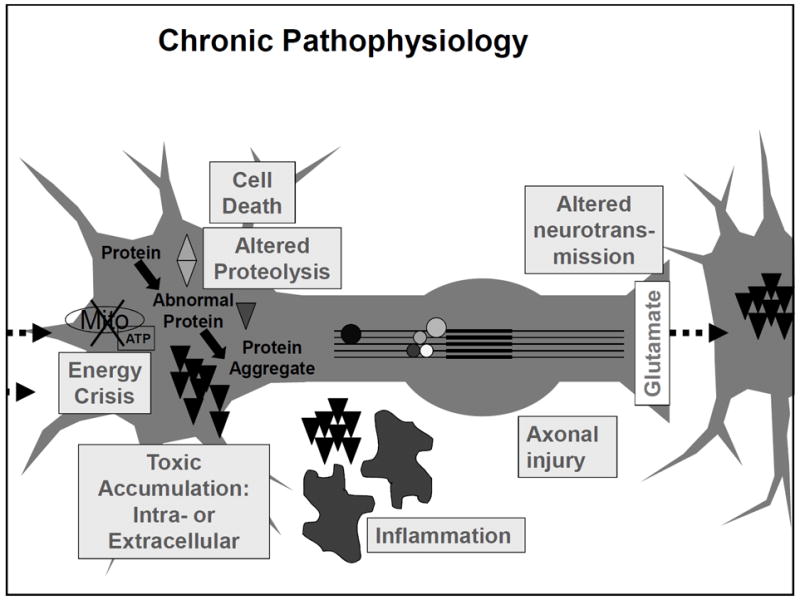
Diagram of linkages between acute post-concussion pathophysiology and mechanisms for chronic impairment and possibly neurodegeneration.
References
- 1.McCrory P, Meeuwisse WH, Aubry M, et al. Consensus statement on concussion in sport. Br J Sports Med; the 4th International Conference on Concussion in Sport held in Zurich; November 2012; 2013. pp. 250–8. [DOI] [PubMed] [Google Scholar]
- 2.Barkhoudarian G, Hovda DA, Giza CC. The Molecular Pathophysiology of Concussive Brain Injury. ClinSports Med. 2011;30(1):33–48. doi: 10.1016/j.csm.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Giza CCC, Hovda DA. The Neurometabolic Cascade of Concussion. [June 25, 2014];J Athl Train. 2001 36(3):228–235. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/pmc155411/ [PMC free article] [PubMed] [Google Scholar]
- 4.Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889–900. doi: 10.3171/jns.1990.73.6.0889. 0022-3085 SB - A SB - M SB - X. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi H, Manaka S, Sano K. Changes in extracellular potassium concentration in cortex and brain stem during the acute phase of experimental closed head injury. J Neurosurg. 1981;55:708–717. doi: 10.3171/jns.1981.55.5.0708. 0022-3085 SB - A SB - M. [DOI] [PubMed] [Google Scholar]
- 6.Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats : evidence of a hyper- and subsequent hypometabolic state. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]
- 7.Hovda DA. Metabolic Dysfunction. In: Narayan RK, Wilberger JE, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1996. pp. 1459–1478. [Google Scholar]
- 8.Thomas S, Prins ML, Samii M, Hovda DA. Cerebral metabolic response to traumatic brain injury sustained early in development: a 2-deoxy-D-glucose autoradiographic study. J Neurotrauma. 2000;17:649–665. doi: 10.1089/089771500415409. 0897-7151. [DOI] [PubMed] [Google Scholar]
- 9.Li HH, Lee SM, Cai Y, Sutton RL, Hovda Da. Differential gene expression in hippocampus following experimental brain trauma reveals distinct features of moderate and severe injuries. J Neurotrauma. 2004;21(9):1141–53. doi: 10.1089/neu.2004.21.1141. [DOI] [PubMed] [Google Scholar]
- 10.Tavazzi B, Vagnozzi R, Signoretti S, et al. Temporal window of metabolic brain vulnerability to concussions: oxidative and nitrosative stresses--part II. Neurosurgery. 2007;61:390–395. doi: 10.1227/01.neu.0000255525.34956.3f. 1524-4040 (Electronic) [DOI] [PubMed] [Google Scholar]
- 11.Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. 0006-8993 SB - M. [DOI] [PubMed] [Google Scholar]
- 12.Büki a, Povlishock JT. All roads lead to disconnection?--Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148(2):181–93. doi: 10.1007/s00701-005-0674-4. discussion 193-4. [DOI] [PubMed] [Google Scholar]
- 13.Alford PW, Dabiri BE, Goss JA, Hemphill MA, Brigham MD, Parker KK. Blast-induced phenotypic switching in cerebral vasospasm. ProcNatlAcadSciUSA. 2011;108:12705–12710. doi: 10.1073/pnas.1105860108. 1091-6490 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemphill Ma, Dabiri BE, Gabriele S, et al. A possible role for integrin signaling in diffuse axonal injury. PLoS One. 2011;6(7):e22899. doi: 10.1371/journal.pone.0022899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettus EH, Christman CW, Giebel ML, Povlishock JT. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11:507–522. doi: 10.1089/neu.1994.11.507. 0897-7151 SB - M. [DOI] [PubMed] [Google Scholar]
- 16.Singleton RH, Zhu J, Stone JR, Povlishock JT. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791–802. doi: 10.1523/JNEUROSCI.22-03-00791.2002. 1529-2401 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang-schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol. 2012;233(1):364–372. doi: 10.1016/j.expneurol.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. 2005 doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Prins ML, Hales A, Reger M, Giza CC, Hovda DA. Repeat Traumatic Brain Injury in the Juvenile Rat Is Associated with Increased Axonal Injury and Cognitive Impairments. DevNeurosci. 2010;32(5-6):510–518. doi: 10.1159/000316800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Longhi L, Saatman KE, Fujimoto S, et al. Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery. 2005;56(2):364–374. doi: 10.1227/01.NEU.0000149008.73513.44. [DOI] [PubMed] [Google Scholar]
- 22.Giza CC, Maria NSS, Hovda Da. N -Methyl- D -Aspartate Receptor Subunit Changes after Traumatic Injury to the Developing Brain. J Neurotrauma. 2006;23(6):950–961. doi: 10.1089/neu.2006.23.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atkins CM, Falo MC, Alonso OF, Bramlett HM, Dietrich WD. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett. 2009;459(2):52–6. doi: 10.1016/j.neulet.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar A, Zou L, Yuan X, Long Y, Yang K. N-Methyl-D-Aspartate Receptors : Transient Loss of NR1 / NR2A / NR2B Subunits After Traumatic Brain Injury in a Rodent Model. 2002;786(October 2001):781–786. doi: 10.1002/jnr.10181. [DOI] [PubMed] [Google Scholar]
- 25.Osteen CL, Giza CC, Hovda DA. Injury-Induced Changes in NMDA Receptor Subunit Composition Contribute to Prolonged Calcium-45 Accumulation in Intact Cortex. J Neurotrauma. 2002;19(10):1384. [Google Scholar]
- 26.NS S, Harris NG, R ML, Buen F, et al. Glutamatergic mechanisms underlie a deficit in hippocampal activation and neural plasticity following developmental traumatic brain injury. prep. 2014 [Google Scholar]
- 27.Griesbach GS, Gómez-Pinilla F, Hovda Da. Time window for voluntary exercise-induced increases in hippocampal neuroplasticity molecules after traumatic brain injury is severity dependent. J Neurotrauma. 2007;24(7):1161–71. doi: 10.1089/neu.2006.0255. [DOI] [PubMed] [Google Scholar]
- 28.Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992;12(12):4846–53. doi: 10.1523/JNEUROSCI.12-12-04846.1992. Available at: http://www.ncbi.nlm.nih.gov/pubmed/1464770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zanier ER, Lee SM, Vespa PM. Increased Hippocampal CA3 Vulnerability to Low-Level Kainic Acid following Lateral Fluid Percussion Injury. 2003;20(5):409–420. doi: 10.1089/089771503765355496. [DOI] [PubMed] [Google Scholar]
- 30.Reger ML, Poulos AM, Buen F, Giza CC, Hovda DA, Fanselow MS. Concussive Brain Injury Enhances Fear Learning and Excitatory Processes in the Amygdala. BPS. 2011;71(4):335–343. doi: 10.1016/j.biopsych.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fendt M, Fanselow MS. The neuroanatomical and neurochemical basis of conditioned fear. NeurosciBiobehavRev. 1999;23:743–760. doi: 10.1016/s0149-7634(99)00016-0. 0149-7634 (Print) [DOI] [PubMed] [Google Scholar]
- 32.Koenigs M, Huey ED, Raymont V, et al. Focal brain damage protects against post-traumatic stress disorder in combat veterans. Nat Neurosci. 2008;11(2):232–7. doi: 10.1038/nn2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Israelsson C, Bengtsson H, Kylberg A, et al. Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. J Neurotrauma. 2008;25:959–974. doi: 10.1089/neu.2008.0562. 0897-7151 (Print) [DOI] [PubMed] [Google Scholar]
- 34.Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. JNeuropatholExpNeurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. 0022-3069 (Print) [DOI] [PubMed] [Google Scholar]
- 35.Giza CC, Prins ML. Is being plastic fantastic? Mechanisms of altered plasticity after developmental traumatic brain injury. Dev Neurosci. 2006;28(4-5):364–379. doi: 10.1159/000094163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet M-F. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28(9):1783–801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shultz SR, MacFabe DF, Foley KA, Taylor R, Cain DP. Sub-concussive brain injury in the Long-Evans rat induces acute neuroinflammation in the absence of behavioral impairments. Behav Brain Res. 2012;229:145–152. doi: 10.1016/j.bbr.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 38.Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surg Neurol Int. 2011;2:107. doi: 10.4103/2152-7806.83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurkoff GG, Giza CC, Hovda DA. Lateral fluid percussion injury in the developing rat causes an acute, mild behavioral dysfunction in the absence of significant cell death. Brain Res. 2006;1077(1):24–36. doi: 10.1016/j.brainres.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Lyeth BG, Jenkins LW, Hamm RJ, et al. Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. 0006-8993 SB - IM. [DOI] [PubMed] [Google Scholar]
- 41.DeFord SM, Wilson MS, Rice AC, et al. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma. 2002;19(4):427–38. doi: 10.1089/08977150252932389. [DOI] [PubMed] [Google Scholar]
- 42.Prins ML, Alexander D, Giza CC, Hovda DA. Repeated mild traumatic brain injury: mechanisms of cerebral vulnerability. J Neurotrauma. 2013;30:30–38. doi: 10.1089/neu.2012.2399. 1557-9042 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith DH, Chen X, Pierce JES, et al. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14(10):715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 44.Dixon CE, Kochanek PM, Yan HQ, et al. One-year study of spatial memory performance, brain morphology, and cholinergic markers after moderate controlled cortical impact in rats. J Neurotrauma. 1999;16:109–122. doi: 10.1089/neu.1999.16.109. 0897-7151 (Print) [DOI] [PubMed] [Google Scholar]
- 45.Kochanek PM, Hendrich KS, Dixon CE, Schiding JK, Williams DS, Ho C. Cerebral blood flow at one year after controlled cortical impact in rats: assessment by magnetic resonance imaging. J Neurotrauma. 2002;19(9):1029–37. doi: 10.1089/089771502760341947. [DOI] [PubMed] [Google Scholar]
- 46.Pullela R, Raber J, Pfankuch T, et al. Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. DevNeurosci. 2006;28:396–409. doi: 10.1159/000094166. 0378-5866 (Print) [DOI] [PubMed] [Google Scholar]
- 47.Leao AA. Further observations on the spreading depression of activity in the cerebral cortex. JNeurophysiol. 1947;10:409–414. doi: 10.1152/jn.1947.10.6.409. 0022-3077 (Print) [DOI] [PubMed] [Google Scholar]
- 48.Kubota M, Nakamura T, Sunami K, et al. Changes of local cerebral glucose utilization, DC potential and extracellular potassium concentration in experimental head injury of varying severity. NeurosurgRev. 1989;12(Suppl 1):393–399. doi: 10.1007/BF01790681. 0344-5607 SB - IM. [DOI] [PubMed] [Google Scholar]
- 49.Rogatsky G, Mayevsky A, Zarchin N, Doron A. Continuous multiparametric monitoring of brain activities following fluid-percussion injury in rats: preliminary results. JBasic ClinPhysiol Pharmacol. 1996;7:23–43. doi: 10.1515/jbcpp.1996.7.1.23. 0792-6855 SB - IM. [DOI] [PubMed] [Google Scholar]
- 50.Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738–2743. doi: 10.1161/01.str.0000043073.69602.09. 1524-4628 (Electronic) [DOI] [PubMed] [Google Scholar]
- 51.Hartings JA, Strong AJ, Fabricius M, et al. Spreading depolarizations and late secondary insults after traumatic brain injury. J Neurotrauma. 2009;26:1857–1866. doi: 10.1089/neu.2009.0961. 1557-9042 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kors EE, Terwindt GM, Vermeulen FL, et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49(6):753–60. doi: 10.1002/ana.1031. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11409427. [DOI] [PubMed] [Google Scholar]
- 53.Mihalik JP, Stump JE, Collins MW, Lovell MR, Field M, Maroon JC. Posttraumatic migraine characteristics in athletes following sports-related concussion. J Neurosurg. 2005;102(5):850–5. doi: 10.3171/jns.2005.102.5.0850. [DOI] [PubMed] [Google Scholar]
- 54.Giza CC, Kutcher JS, Ashwal S, et al. Summary of evidence-based guideline update: Evaluation and management of concussion in sports. Neurology. 2013;80(24):2250–2257. doi: 10.1212/WNL.0b013e31828d57dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guskiewicz KM, McCrea M, Marshall SW, et al. Cumulative effects associated with recurrent concussion in collegiate football players: the NCAA Concussion Study. JAMA. 2003;290(19):2549–55. doi: 10.1001/jama.290.19.2549. [DOI] [PubMed] [Google Scholar]
- 56.McCrea M, Guskiewicz K, Randolph C, et al. Effects of a symptom-free waiting period on clinical outcome and risk of reinjury after sport-related concussion. Neurosurgery. 2009;65:876–882. doi: 10.1227/01.NEU.0000350155.89800.00. 1524-4040 (Electronic) [DOI] [PubMed] [Google Scholar]
- 57.Vagnozzi R, Signoretti S, Cristofori L, et al. Assessment of metabolic brain damage and recovery following mild traumatic brain injury : a multicentre, proton magnetic resonance spectroscopic study in concussed patients. 2010 doi: 10.1093/brain/awq200. [DOI] [PubMed] [Google Scholar]
- 58.Vagnozzi R, Signoretti S, Tavazzi B, et al. Temporal window of metabolic brain vulnerability to concussion: a pilot 1H-magnetic resonance spectroscopic study in concussed athletes--part III. Neurosurgery. 2008;62:1286–1295. doi: 10.1227/01.neu.0000333300.34189.74. 1524-4040 (Electronic) [DOI] [PubMed] [Google Scholar]
- 59.Vagnozzi R, Tavazzi B, Signoretti S, et al. Temporal window of metabolic brain vulnerability to concussions: mitochondrial-related impairment--part I. Neurosurgery. 2007;61:379–388. doi: 10.1227/01.NEU.0000280002.41696.D8. 1524-4040 (Electronic) [DOI] [PubMed] [Google Scholar]
- 60.Adams JH, Jennett B, Murray LS, Teasdale GM, Gennarelli TA, Graham DI. Neuropathological findings in disabled survivors of a head injury. J Neurotrauma. 2011;28:701–709. doi: 10.1089/neu.2010.1733. 1557-9042 (Electronic) [DOI] [PubMed] [Google Scholar]
- 61.Buki A, Povlishock JT. All roads lead to disconnection?--Traumatic axonal injury revisited. Acta Neurochir(Wien) 2006;148:181–193. doi: 10.1007/s00701-005-0674-4. 0001-6268 (Print) [DOI] [PubMed] [Google Scholar]
- 62.Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet. 1994;344:1055–1056. doi: 10.1016/s0140-6736(94)91712-4. 0140-6736 SB - A SB - M SB - X. [DOI] [PubMed] [Google Scholar]
- 63.Oni MB, Wilde Ea, Bigler ED, et al. Diffusion tensor imaging analysis of frontal lobes in pediatric traumatic brain injury. J Child Neurol. 2010;25(8):976–84. doi: 10.1177/0883073809356034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilde EA, McCauley SR, Hunter JV, et al. Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology. 2008;70(12):948–955. doi: 10.1212/01.wnl.0000305961.68029.54. [DOI] [PubMed] [Google Scholar]
- 65.Lipton ML, Kim N, Zimmerman ME, et al. Soccer heading is associated with white matter microstructural and cognitive abnormalities. Radiology. 2013;268:850–857. doi: 10.1148/radiol.13130545. 1527-1315 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McAllister TW, Ford JC, Flashman La, et al. Effect of head impacts on diffusivity measures in a cohort of collegiate contact sport athletes. Neurology. 2014;82(1):63–9. doi: 10.1212/01.wnl.0000438220.16190.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Babikian T, Marion SD, Copeland S, et al. Metabolic levels in the corpus callosum and their structural and behavioral correlates following moderate to severe pediatric TBI. J Neurotrauma. 2009 doi: 10.1089/neu.2009.1058. 1557-9042 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kraus MF, Susmaras T, Caughlin BP, Walker CJ, Sweeney JA, Little DM. White matter integrity and cognition in chronic traumatic brain injury : a diffusion tensor imaging study. 2007:2508–2519. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- 69.Bazarian JJ, Zhong J, Blyth B, Zhu T, Kavcic V, Peterson D. Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. J Neurotrauma. 2007;24(9):1447–59. doi: 10.1089/neu.2007.0241. [DOI] [PubMed] [Google Scholar]
- 70.Henry LC, Tremblay J, Tremblay S, et al. Acute and chronic changes in diffusivity measures after sports concussion. J Neurotrauma. 2011;28:2049–2059. doi: 10.1089/neu.2011.1836. 1557-9042 (Electronic) [DOI] [PubMed] [Google Scholar]
- 71.Mayer AR, Ling JM, Yang Z, Pena A, Yeo RA, Klimaj S. Diffusion abnormalities in pediatric mild traumatic brain injury. J Neurosci. 2012;32:17961–17969. doi: 10.1523/JNEUROSCI.3379-12.2012. 1529-2401 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miles L, Grossman RI, Johnson G, Babb JS, Diller L, Inglese M. Short-term DTI predictors of cognitive dysfunction in mild traumatic brain injury. Brain Inj. 2008;22:115–122. doi: 10.1080/02699050801888816. 0269-9052 (Print) [DOI] [PubMed] [Google Scholar]
- 73.Arfanakis K, Haughton VM, Carew JD, Rogers BP, Dempsey RJ, Meyerand ME. Diffusion tensor MR imaging in diffuse axonal injury. AJNR AmJNeuroradiol. 2002;23:794–802. 0195-6108 (Print) [PMC free article] [PubMed] [Google Scholar]
- 74.Shenton ME, Hamoda HM, Schneiderman JS, et al. A review of magnetic resonance imaging and diffusion tensor imaging findings in mild traumatic brain injury. Brain Imaging Behav. 2012;6:137–192. doi: 10.1007/s11682-012-9156-5. 1931-7565 (Electronic)) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mac Donald CL, Dikranian K, Bayly P, Holtzman D, Brody D. Diffusion tensor imaging reliably detects experimental traumatic axonal injury and indicates approximate time of injury. J Neurosci. 2007;27(44):11869–76. doi: 10.1523/JNEUROSCI.3647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maddocks DL, Dicker GD, Saling MM. The assessment of orientation following concussion in athletes. ClinJSport Med. 1995;5:32–35. doi: 10.1097/00042752-199501000-00006. 1050-642X (Print) [DOI] [PubMed] [Google Scholar]
- 77.McCrea M, Kelly JP, Randolph C, et al. Standardized assessment of concussion (SAC): on-site mental status evaluation of the athlete. JHead Trauma Rehabil. 1998;13:27–35. doi: 10.1097/00001199-199804000-00005. 0885-9701 (Print) [DOI] [PubMed] [Google Scholar]
- 78.Killam C, Cautin RL, Santucci AC. Assessing the enduring residual neuropsychological effects of head trauma in college athletes who participate in contact sports. Arch Clin Neuropsychol. 2005;20(5):599–611. doi: 10.1016/j.acn.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 79.Van Kampen DA, Lovell MR, Pardini JE, Collins MW, Fu FH. The “Value Added ” of Neurocognitive Testing After Sports-Related Concussion. :1630–1635. doi: 10.1177/0363546506288677. [DOI] [PubMed] [Google Scholar]
- 80.Eckner JT, Kutcher JS, Broglio SP, Richardson JK. Effect of sport-related concussion on clinically measured simple reaction time. Br J Sports Med. 2014;48(2):112–8. doi: 10.1136/bjsports-2012-091579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bonvento G, Sibson N, Pellerin L. Does glutamate image your thoughts? Trends Neurosci. 2002;25:359–364. doi: 10.1016/s0166-2236(02)02168-9. 0166-2236 (Print) [DOI] [PubMed] [Google Scholar]
- 82.Gsell W, Burke M, Wiedermann D, et al. Differential effects of NMDA and AMPA glutamate receptors on functional magnetic resonance imaging signals and evoked neuronal activity during forepaw stimulation of the rat. J Neurosci. 2006;26(33):8409–16. doi: 10.1523/JNEUROSCI.4615-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McAllister TW, Sparling MB, Flashman La, Guerin SJ, Mamourian aC, Saykin aJ. Differential working memory load effects after mild traumatic brain injury. Neuroimage. 2001;14(5):1004–12. doi: 10.1006/nimg.2001.0899. [DOI] [PubMed] [Google Scholar]
- 84.Lovell MR, Pardini JE, Welling J, et al. Functional brain abnormalities are related to clinical recovery and time to return-to-play in athletes. Neurosurgery. 2007;61:352–359. doi: 10.1227/01.NEU.0000279985.94168.7F. 1524-4040 (Electronic) [DOI] [PubMed] [Google Scholar]
- 85.Keightley ML, Saluja RS, Chen J-K, et al. A functional magnetic resonance imaging study of working memory in youth after sports-related concussion: is it still working? J Neurotrauma. 2014;31(5):437–51. doi: 10.1089/neu.2013.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fineman I, Giza CC, Nahed BV, Lee SM, Hovda Da. Inhibition of neocortical plasticity during development by a moderate concussive brain injury. J Neurotrauma. 2000;17(9):739–49. doi: 10.1089/neu.2000.17.739. Available at: http://www.ncbi.nlm.nih.gov/pubmed/11011814. [DOI] [PubMed] [Google Scholar]
- 87.Ip EY-Y, Giza CC, Griesbach GS, Hovda DA. Effects of enriched environment and fluid percussion injury on dendritic arborization within the cerebral cortex of the developing rat. J Neurotrauma. 2002;19(5):573–85. doi: 10.1089/089771502753754055. [DOI] [PubMed] [Google Scholar]
- 88.Giza CC, Griesbach GS, Hovda Da. Experience-dependent behavioral plasticity is disturbed following traumatic injury to the immature brain. Behav Brain Res. 2005;157(1):11–22. doi: 10.1016/j.bbr.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 89.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. PsycholMed. 1973;3:270–303. doi: 10.1017/s0033291700049588. 0033-2917 (Print) [DOI] [PubMed] [Google Scholar]
- 90.Martland HS. Punch Drunk. JAMA J Am Med Assoc. 1928;91(15):1103–1107. [Google Scholar]
- 91.McKee AC, Stein TD, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2012 doi: 10.1093/brain/aws307. 1460-2156 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. 1524-4040 (Electronic) [DOI] [PubMed] [Google Scholar]
- 93.McKee AC, Stern Ra, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136(Pt 1):43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ross DE, Ochs AL, Seabaugh JM, et al. Progressive brain atrophy in patients with chronic neuropsychiatric symptoms after mild traumatic brain injury: a preliminary study. Brain Inj. 2012;26:1500–1509. doi: 10.3109/02699052.2012.694570. 1362-301X (Electronic) [DOI] [PubMed] [Google Scholar]
- 95.Immonen RJ, Kharatishvili I, Gröhn H, Pitkänen A, Gröhn OHJ. Quantitative MRI predicts long-term structural and functional outcome after experimental traumatic brain injury. Neuroimage. 2009;45(1):1–9. doi: 10.1016/j.neuroimage.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 96.Tanaka K, Mizushima T, Saeki Y. The proteasome: molecular machinery and pathophysiological roles. 2012;393(April):217–234. doi: 10.1515/hsz-2011-0285. [DOI] [PubMed] [Google Scholar]
- 97.Martínez A, Portero-Otin M, Pamplona R, Ferrer I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 2010;20(2):281–97. doi: 10.1111/j.1750-3639.2009.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sakai K, Fukuda T, Iwadate K. Immunohistochemical Analysis of the Ubiquitin Proteasome System and Autophagy Lysosome System Induced After Traumatic Intracranial Injury Association With Time Between the Injury and Death. 2014;35(1):38–44. doi: 10.1097/PAF.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 99.Yao X, Liu J, Mccabe JT. Alterations of cerebral cortex and hippocampal proteasome subunit expression and function in a traumatic brain injury rat model. 2008:353–363. doi: 10.1111/j.1471-4159.2007.04970.x. [DOI] [PubMed] [Google Scholar]
- 100.Wan C, Chen J, Hu B, Zou H, Li A, Guo A. Downregulation of UBE2Q1 Is Associated With Neuronal Apoptosis in Rat Brain Cortex Following Traumatic Brain Injury. 2014;12:1–12. doi: 10.1002/jnr.23305. [DOI] [PubMed] [Google Scholar]
- 101.Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. JBiolChem. 2013;288:17042–17050. doi: 10.1074/jbc.M113.472746. 1083-351X (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goldstein LE, Fisher AM, Tagge Ca, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4(134):134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mannix R, Meehan WP, Mandeville J, et al. Clinical correlates in an experimental model of repetitive mild brain injury. AnnNeurol. 2013;74:65–75. doi: 10.1002/ana.23858. 1531-8249 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tran HT, Sanchez L, Esparza TJ, Brody DL. Distinct temporal and anatomical distributions of amyloid-beta and tau abnormalities following controlled cortical impact in transgenic mice. PLoSOne. 2011;6:e25475. doi: 10.1371/journal.pone.0025475. 1932-6203 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bennett RE, Esparza TJ, Lewis HA, et al. Human apolipoprotein E4 worsens acute axonal pathology but not amyloid-beta immunoreactivity after traumatic brain injury in 3xTG-AD mice. JNeuropatholExpNeurol. 2013;72:396–403. doi: 10.1097/NEN.0b013e31828e24ab. 1554-6578 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lasagna-Reeves Ca, Castillo-Carranza DL, Sengupta U, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science (80-) 2012;336:1511–1513. doi: 10.1126/science.1222951. 1095-9203 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Magnoni S, Esparza TJ, Conte V, et al. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain. 2012;135:1268–1280. doi: 10.1093/brain/awr286. 1460-2156 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mac Donald CL, Johnson AM, Cooper D, et al. Detection of blast-related traumatic brain injury in U.S. military personnel. NEnglJMed. 2011;364:2091–2100. doi: 10.1056/NEJMoa1008069. 1533-4406 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yeh PH, Wang B, Oakes TR, et al. Postconcussional disorder and PTSD symptoms of military-related traumatic brain injury associated with compromised neurocircuitry. HumBrain Mapp. 2013 doi: 10.1002/hbm.22358. 1097-0193 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Strain J, Didehbani N, Cullum CM, et al. Depressive symptoms and white matter dysfunction in retired NFL players with concussion history. Neurology. 2013;81:25–32. doi: 10.1212/WNL.0b013e318299ccf8. 1526-632X (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. 1460-2156 (Electronic) [DOI] [PMC free article] [PubMed] [Google Scholar]