

Abstract
The first-line treatment of hyperuricemia, which causes gout, is allopurinol. The allopurinol response is highly variable, with many users failing to achieve target serum uric acid (SUA) levels. No genome-wide association study (GWAS) has examined the genetic factors affecting allopurinol effectiveness. Using 2,027 subjects in Kaiser Permanente’s Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort, we conducted a GWAS of allopurinol-related SUA reduction, first in the largest ethnic group, non-Hispanic white (NHW) subjects, and then in a stratified transethnic meta-analysis. ABCG2, encoding the efflux pump BCRP, was associated with SUA reduction in NHW subjects (P = 2 × 10−8), and a missense allele (rs2231142) was associated with a reduced response (P = 3 × 10−7) in the meta-analysis. Isotopic uptake studies in cells demonstrated that BCRP transports allopurinol and genetic variants in ABCG2 affect this transport. Collectively, this first GWAS of allopurinol response demonstrates that ABCG2 is a key determinant of response to the drug.
Introduction
Gout is a painful condition caused by deposition of uric acid (UA) crystals in the joints. This leads to an inflammatory arthritis with joint pain and swelling. The main cause of gout is hyperuricemia, which can be influenced by many factors including genetics, diet, medications, and renal failure (UA is mainly eliminated by the kidney with much lower elimination by the intestine).[1] Gout remains the most common form of inflammatory arthritis in Western countries, and is on the rise worldwide in countries such as China, New Zealand, and Taiwan, with a particularly high prevalence (~10%) in the aboriginal populations of these latter two countries.[1, 2] In the UK, a recent study found that the prevalence of gout had increased to 2.5% by 2012.[3, 4] Importantly, less than half of UK patients with gout are treated with UA-lowering drugs.[4] If left untreated, hyperuricemia can lead to nephrolithiasis and nephropathy and has also been associated with an increased risk of cardiovascular events, notably heart failure.[2]
Currently, allopurinol is the first-line medication for gout prevention.[5] Allopurinol is a purine analog that is metabolized into oxypurinol. Both compounds act by inhibiting xanthine oxidase, the enzyme that converts xanthine into UA, thereby lowering serum uric acid (SUA) levels through the inhibition of its formation.
While a number of genetic risk factors for gout and hyperuricemia have been well established through multiple large genome-wide association studies (GWAS) and meta-analyses,[6, 7] only a few GWAS have focused on allopurinol, and all of them have centered on identifying genetic influences on its rare, life-threatening skin toxicities.[8, 9] No GWAS for allopurinol-related SUA reduction have been performed despite the fact that only 42% of patients on allopurinol are estimated to achieve the recommended SUA target of ≤6 mg/dl.[10]Here we describe the first pharmacogenomic GWAS of allopurinol to identify genetic factors for response to the drug in a large multiethnic cohort of patients with comprehensive electronic pharmacy, laboratory, and clinical records. As confirmation, we followed up with laboratory experiments to verify some of the associations observed in the GWAS and explore the mechanism through which they affect response.
RESULTS
Overall, 2,027 patients in the GERA cohort met study inclusion criteria (Table 1). The resulting group of subjects had an average age of 68 at the time of treatment and were mainly male (75%), consistent with the higher prevalence of gout in men.[2] As expected, this group had an elevated mean baseline SUA of 8.9 mg/dL, which is indicative of hyperuricemia and consistent with values normally associated with uncontrolled gout. This is much greater than allopurinol’s therapeutic goal of 6 mg/dL.[5] Most members of the study group were non-Hispanic white (NHW) (N = 1,607), with East Asians comprising the second largest group (N = 238).
Table 1.
Demographic, laboratory, and treatment characteristics of the study sample
| Number | Percent | ||
|---|---|---|---|
| Gender | Male | 1,515 | 74.7 |
| Female | 512 | 25.3 | |
| Race-ethnicity | Non-Hispanic whites | 1,607 | 79.3 |
| East Asians | 238 | 11.7 | |
| African Americans | 84 | 4.1 | |
| Latinos | 85 | 4.2 | |
| Other | 13 | 0.6 | |
| Measurements | Mean | SD | |
| Age at treatment | 67.9 | 10.4 | |
| Baseline SUA | 8.9 mg/dL | 2.2 | |
| Median post-SUA | 6.3 mg/dL | 1.6 | |
| SUA change | −2.6 mg/dL | 2.2 | |
| Allopurinol dose | 201.3 mg/day | 101.4 |
Values are over all individuals who have a pre- and a post-serum uric acid (SUA). Median post-SUA is calculated over all post SUA values for the first allopurinol prescription that has both a pre- and a post-SUA measurement. SUA change is calculated as the difference between the median post-SUA and the pre-SUA. Allopurinol dose is the dose at the time of the median post-SUA measurement.
To obtain a more homogenous sample for initial analysis, we first analyzed the sample of 1,492 NHW that were run on the NHW array (note that 115 NHW subjects were run on other arrays). Among the nongenetic factors associated with allopurinol-related SUA change, the strongest effects were observed for baseline SUA, cumulative dose, and dose at the time of measurement (Table 2). The GWAS found a genomic inflation factor of 1.0048 and revealed a strong association at the genome-wide level significance for ABCG2 (P = 2.0 × 10−8) (Figure 1a, Table 3, Supplemental Table S1). ABCG2 encodes the transporter Breast Cancer Resistance Protein (BCRP), and is a known UA transporter,[11] but has not previously been associated with allopurinol transport or response. Included among the top associated single nucleotide polymorphisms (SNPs) was rs2231142 (P = 1.9 × 10−6), a nonsynonymous reduced function allele coding the Q141K variant[11] and not in high linkage disequilibrium (LD) with the top SNP (r[2] = 0.078 in our NHW sample). We found that the K allele of Q141K was associated with a poorer response. When compared to the residuals after adjusting for nongenetic factors, this variant could account for 1.1% of the unexplained variance in NHW. In light of the BCRP association, we also examined the results for other genes/SNPs previously reported to be associated with gout and/or baseline uric acid.[6] None of these genes/SNPs were found to be associated with allopurinol response (Supplemental Table S2), suggesting a unique association with BCRP.
Table 2.
Nongenetic factors associated with uric acid change in response to allopurinol
| Nongenetic factor | r | P-value |
|---|---|---|
| Age | −0.010 | 0.65 |
| Baseline SUA | −0.71 | <2.2×10−16 |
| BMI | 0.012 | 0.59 |
| Dose | −0.086 | 0.00013 |
| Cumulative dose | −0.18 | 2.6×10−16 |
| First principal component | 0.042 | 0.072 |
| Gender | −0.033 | 0.14 |
| Concomitant medications | 0.019 | 0.41 |
Correlations are calculated comparing the nongenetic factor to the change between the pre-SUA residual and post-SUA residual found after correcting the pre- and post-SUA values for gender, age, BMI, and concomitant medications.
Figure 1.

Manhattan plot of association of SNPs with change in uric acid levels in response to allopurinol in (a) 1,492 patients with European ancestries or (b) all patients using a stratified transethnic meta-analysis. Meta-analysis was conducted combining individual associations of each of the four ethnic groups. Overall, this includes 1,607 non-Hispanic whites, 238 Asians, 84 African Americans, and 85 Hispanics. Gray dots are imputed SNPs. P-values > 0.05 were not plotted.
Table 3.
Top association results in non-Hispanic whites
| RSID | Chr | Position | Beta | SE | P-value | Gene |
|---|---|---|---|---|---|---|
| rs10011796 | 4 | 89090877 | 0.2799 | 0.04959 | 1.99E-08 | ABCG2 |
| rs17211056 | 18 | 73183461 | 0.6488 | 0.1274 | 4.02E-07 | SMIM21 |
| rs3114020 | 4 | 89083666 | 0.2433 | 0.0504 | 1.54E-06 | ABCG2 |
| rs11152031 | 18 | 52533038 | −0.2519 | 0.0522 | 1.54E-06 | CCDC68,RAB27B |
| rs3762427 | 1 | 151867560 | 0.4158 | 0.08677 | 1.82E-06 | THEM4,THEM5 |
| rs2231142 | 4 | 89052323 | 0.323 | 0.06748 | 1.86E-06 | ABCG2 |
| rs2199936 | 4 | 89264355 | 0.3038 | 0.06551 | 3.84E-06 | ABCG2,PKD2 |
| rs12922040 | 16 | 15869466 | 0.2415 | 0.05267 | 4.93E-06 | MYH11,NDE1 |
All SNPs associated with uric acid change in response to allopurinol with P < 5 × 10−6 in a cohort of 1,492 patients of European ancestry. Annotated genes are any gene found within 50 kb of the SNP. Chr, Chromosome. Beta, Beta-coefficient. A positive beta coefficient means that the minor allele is associated with smaller change in SUA from baseline and hence poorer response, whereas a negative beta-coefficient means that the effect allele is associated with larger change in SUA from baseline and hence better response. SE, standard error.
The results of the meta-analysis across race/ethnicity groups using the imputed genotype data showed that many additional suggestive SNPs within the ABCG2 locus were associated with SUA change, at P-values less than 1.4 × 10−7 that did not reach genome-wide significance (Figure 1b). The previous strongest associated SNP, rs10011796, weakened in the meta-analysis due to differing results in other ethnic groups (from P = 2.0 × 10−8 to P = 6.9 × 10−4). However, the previously noted functional SNP rs2231142 had a consistent direction of effect in all ethnicities, and became more significant (from P = 1.9 × 10−6 to P = 3.4 × 10−7) (Figure 1b, Supplemental Table S3).
To verify our results and determine the mechanism by which ABCG2 variants associate with allopurinol response, we tested the hypothesis that BCRP transports allopurinol and oxypurinol, and that rs2231142 affects transport. Although not necessarily the only causal SNP, we chose to focus on rs2231142 since it is a known functional SNP and its significance strengthened in the meta-analysis. We used stably transfected BCRP-expressing cells, and used a cytotoxicity assay with mitoxantrone, a known BCRP substrate, as a positive control. As expected, cells expressing BCRP reference were the most resistant, followed by those with the Q141K variant, and followed by empty vector (Figure 2). In cell accumulation studies, BCRP-expressing cells had significantly lower levels of allopurinol and oxypurinol in comparison to empty vector transfected cells, consistent with an efflux role for both compounds (Figure 3a,b). Furthermore, addition of a specific BCRP inhibitor, Ko-143, caused significantly greater drug accumulation in BCRP expressing cells (Figure 3a,b). When cells were transfected with the rs2231142 (Q141K) variant, a significantly higher accumulation of both allopurinol and oxypurinol occurred as compared with reference BCRP transfected cells, consistent with reduced BCRP function (Figure 3c). Allopurinol and oxypurinol were not inhibitors of BCRP-mediated efflux of the model substrate pitavastatin (Figure 3d),[12, 13] suggesting that the drugs do not inhibit uric acid secretion via BCRP.
Figure 2.
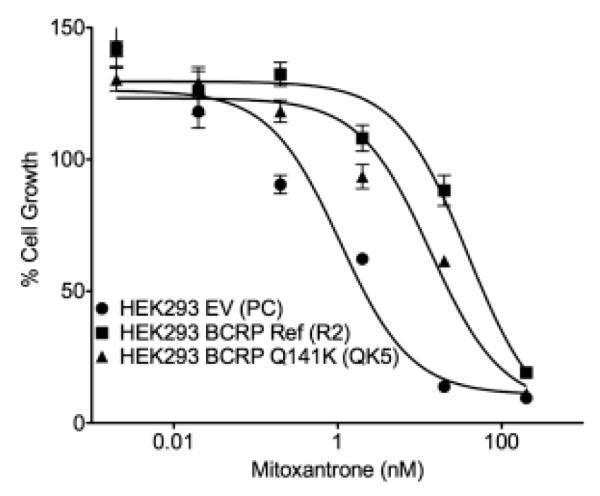
Cell growth of HEK293 cells after treatment with mitoxantrone for 72 hours to test transporter function. Cells were stably transfected with either empty vector, reference ABCG2, or ABCG2 containing the Q141K variant.
Figure 3.

BCRP-mediated transport of allopurinol, oxypurinol, and pitavastatin. Cells stably transfected with empty vector or reference ABCG2 were incubated with (a) [3H]-allopurinol or (b) [3H]-oxypurinol with or without Ko-143, a selective BCRP inhibitor. BCRP is an efflux transporter, so BCRP transport would lead to a lower accumulation of allopurinol in the HEK293 cells. (c) rs2231142 affects BCRP transport of allopurinol and oxypurinol. Cells stably transfected with reference ABCG2 or the ABCG2 Q141K variant were incubated with [3H]-allopurinol or [3H]-oxypurinol. (d) Allopurinol and oxypurinol did not affect BCRP transport of pitavastatin. Cells stably transfected with reference ABCG2 were incubated with [3H]-pitavastatin with or without Ko143, allopurinol (0.5, 1, and 2 mM), or oxypurinol (0.5, 1, and 2 mM). Using Dunnett’s multiple comparison test, only cells incubated with Ko-143 (10 μM) was statistically significant from cells incubated with vehicle (P < 0.0001). Values +/− standard errors are shown in these figures.
DISCUSSION
Our finding of a significant effect of genetic variation in ABCG2, and specifically the amino acid substitution Q141K, provides an avenue for understanding the response to allopurinol. SNPs in ABCG2 that have been previously associated with baseline SUA in many studies in multiple ethnic groups.[6] Because BCRP is known to transport UA and various synthetic and naturally occurring purine derivatives,[14] we hypothesized that allopurinol and oxypurinol, both purine analogs, are substrates of the transporter. In vitro studies in stably transfected cells supported our hypothesis, indicating that both compounds are BCRP substrates, and that the Q141K variant can directly modulate BCRP-mediated allopurinol and oxypurinol efflux (Figure 2). This variant has been found to destabilize BCRP and lower its expression on the plasma membrane.[11]
Paradoxically, the K allele of BCRP-Q141K, rs2231142, was associated with a smaller reduction in SUA in response to allopurinol treatment. A reduced function variant of BCRP, which acts as a secretory transporter in the kidney and intestine, should lead to decreased elimination and higher systemic plasma levels of the drug. Thus, a greater SUA reduction would be predicted. Since plasma levels of allopurinol and oxypurinol were not available, the precise mechanism by which the variant causes a reduced response to allopurinol cannot be determined. Although speculative, it is possible that allopurinol and oxypurinol have effects on SUA in addition to xanthine oxidase inhibition. For example, they could act as uricosurics, increasing the renal excretion of UA. In fact, hyperuricemic mice treated with allopurinol have an increased fractional excretion of UA, consistent with uricosuric action.[15] Further, studies in Xenopus laevis oocytes demonstrate that oxypurinol is a potent inhibitor of human SLC2A9, a UA reabsorptive transporter associated with hyperuricemia.[16]Alternatively, oxypurinol may competitively inhibit UA reabsorption via URAT1, which transports both UA and oxypurinol.[17] Thus, oxypurinol in the tubule fluid might inhibit SLC2A9 or URAT1, leading to reduced reabsorption and increased renal excretion of UA, and ultimately to enhanced pharmacologic response. A reduced function variant of BCRP would then lead to decreased drug concentrations in the tubule fluid and thus reduced inhibition of UA reabsorptive transporters and diminished response. If true, either of these possibilities suggests an important mechanism of action for allopurinol, distinct from xanthine oxidase inhibition, and a way of modulating that mechanism. That is, oxypurinol and allopurinol inhibit UA reabsorptive transporters, and BCRP can control their ability to do so. Clearly, further research, including the study of rare reduced function variants in ABCG2, will enhance our understanding of the effects of ABCG2 in modulating the response to allopurinol.
One complication in interpreting our findings is that BCRP could have simply been affecting SUA through effects on baseline values. However, we included baseline SUA as a covariate in our linear regression model, and also baseline SUA was not correlated with Q141K (P = 0.8079). Of note, none of the other known UA transporters, some of which are known to have a strong effect on baseline SUA, were associated with allopurinol response, suggesting a key role of BCRP in allopurinol pharmacoresponse.
Other genes of potential interest in our analysis that were suggestive but did not reach genome-wide statistical significance include RAB27B, THEM4, MYH11, and ANO2 (Supplemental Table 1). Of these, ANO2 is noteworthy, as it is a chloride channel. Chloride is known to be important in the function of many transporters involved in UA clearance, including URAT1, SLC17A1, and SLC2A9.[18-20] While these transporters were not significant in our analysis, it’s still possible that they may transport allopurinol or oxypurinol due to their similarity to UA, and just were not detected in our population. While it is possible that these are false positives, ANO2 and other genes in Supplemental Table 1 should be investigated further if additional evidence supports a possible role in allopurinol response.
Recently, gout treatment has undergone a renaissance in the drug development industry. A newer, more costly alternative exists in febuxostat. In addition, new drugs under development include small molecules (e.g., rilonacept) and monoclonal antibodies (e.g., canakinumab) against interleukin 1 beta and its receptors as well as drugs that target UA reabsorptive transporters in the kidney (e.g., lesinurad).[3] With newer drugs on the market and under development, the decision to move away from the low-cost drug, allopurinol, needs to be both economically and clinically rationalized. For example, lesinurad is under development for use in patients who suffer from allopurinol hypersensitivity reactions, and thus cannot tolerate the drug. In addition, the drug is being combined with allopurinol in patients who are inadequate allopurinol responders. With recent GWAS findings for allopurinol hypersensitivity reactions including Stevens-Johnson syndrome and toxic epidermal necrolysis,[8, 9, 21] as well as the results from the current study, it may be possible to use genetic biomarkers to identify patients who should not be on allopurinol therapy. These patients may represent a smaller fraction of gout patients who would benefit from more costly drug therapies.
METHODS
Study population
Participants were drawn from the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort, a subsample of the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH). A detailed description of the cohort and study design can be found in dbGaP.[22] The GERA cohort comprises a sample of 110,266 adult members of Kaiser Permanente Medical Care Plan, Northern California Region (KPNC) with high-density single nucleotide polymorphism (SNP) markers linked to comprehensive electronic health records (EHR) containing information on pharmacy utilization, laboratory test results, clinical diagnoses, and other clinical utilization. All patients signed broad consent forms for use of their data in research on health and disease. In addition, the Kaiser Permanente Northern California Division of Research’s Institutional Review Board (IRB) reviewed and approved the current study. The average age of the multiethnic GERA cohort was about 63 years at the time of sample collection and includes about 81% self-identified NHW participants, and 19% self-identified ethnic minorities (7% Asian, 3.5% African American, 7% Latinos, and 1.5% Other). The EHRs were examined to identify all subjects with an allopurinol prescription who also had SUA measurements. Allopurinol prescriptions within 0.2 years of each other were combined into a single prescription period while still tracking any changes in prescribed dosage. SUA measurements were classified as being untreated (not taken during or within 0.5 years of the end of an allopurinol prescription), treated (taken during or within 0.3 years of the end of an allopurinol prescription), or neither. In addition, the EHR was queried for body mass index (BMI), gender, age at time of the SUA measurements, allopurinol dose, and UA-affecting concomitant medications, defined as diuretics and urate-lowering drugs.
Participants for this study were selected as individuals having an allopurinol prescription with an untreated SUA obtained at most 1.5 years prior to the prescription, and a treated value for the same prescription. If multiple prescriptions fulfilled the criteria, the earliest prescription was used to try to lessen any possible interactions with worsening health conditions or development of drug tolerance.
Genotyping
Genome-wide genotypes on GERA cohort members were generated at the University of California, San Francisco, and underwent detailed quality control. Saliva samples were obtained from the participants and normalized to 10 ng/μL at Kaiser Permanente. Genotyping was conducted using one of four race/ethnic-specific Affymetrix Axiom arrays (Santa Clara, CA). These were the Axiom_KP_UCSF_EUR, _EAS, _AFR, and the _LAT array and were designed to maximize coverage respectively in NHW, East Asians, African Americans, and Latinos.[23, 24] The resulting calls were evaluated and samples with DashQC values <0.82 or with sample call rates <97% were removed, leaving 103,006 well-genotyped samples. Additional QC steps included filtering of autosomal SNPs with large allele frequency differences between genders or SNPs with poor overall call rates among other criteria. Full details can be found in the GERA dbGaP submission.[22] In total, 670,176 SNPs, 801,830 SNPs, 877,845 SNPs, and 708,134 SNPs in the NHW, Latinos, African Americans, and East Asians, respectively, passed initial QC. For this study, additional filters removed polymorphisms with more than 10% missing genotypes or less than 1% minor allele frequency.
Genetic ancestry
EIGENSOFT4.2[25] was used to compute principal components (PC) separately for individuals on the four different arrays using 144,799 high-quality SNPs common to all arrays. Since the calculations were computationally intensive in NHW, they were instead run on a subset of individuals (N = 20,000) with the remaining individuals projected into the same space. The results were validated in three random sets of NHW from the cohort. Full details can be found in the GERA dbGaP submission.[22] These PCs were used in the GWAS to adjust for genetic ancestry.
Imputation
We performed imputation of subject genotypes separately by array. The genotypes in our cohort were first prephased with Shape-it v2.r727.[26] Subsequently, we imputed variants from the 1000 Genomes Project[27] as a cosmopolitan reference panel with Impute2 v2.3.0.[28-30]We used the quality control r[2] info metric from Impute2, which estimates the correlation of the true genotype to the imputed genotype,[31]and set a cutoff filter to eliminate SNPs with an r[2] < 0.3.
Association
Association analysis was conducted using a linear regression model in PLINK v1.07.[32] Initial analysis focused on NHW subjects, as they formed the largest homogenous subgroup. To calculate the change in SUA (our measure of pharmacoresponse), we selected the untreated value as the baseline measurement; if multiple values were available, we chose the measurement closest to treatment initiation. We then adjusted the baseline measurements for BMI, age, gender, and concomitant medications by linear regression. The same was done for the treated values found in our cohort. We then calculated the difference between the treated SUA residuals and the residuals for the associated baseline value. A strong nonlinear relationship between cumulative dose and SUA difference was observed, so we adjusted for cumulative dose using a spline regression. If multiple treated values were present for a particular patient, we used the median change after the cumulative dose adjustment step for subsequent analyses. In the PLINK linear regression model, covariates were included for baseline SUA, age, current dose, BMI, gender, concomitant medications, and the top population structure principal component, as further principal components were much weaker and not significantly associated with the outcome. As a QA step, top associated SNPs were checked for extreme HWE departures in our samples.
The imputed data were analyzed using a meta-analysis. Association analysis was conducted separately on subjects in each race/ethnicity group (NHW, East Asian, Latino, and African American) using all typed and imputed SNPs and using the same covariates as the original analysis with the appropriate principal components for that ethnic group. The results were then combined in a transethnic meta-analysis in PLINK using a random effects model. As described elsewhere,[22] a small subset of NHW were genotyped on arrays other than the NHW array. They were excluded from the initial GWAS of NHW but included in the meta-analysis using imputed genotypes.
R v2.14.1[33] was used to create Manhattan plots and analyze regressions between risk factors and phenotypes.
Experimental materials
Allopurinol, oxypurinol, mitoxantrone, and Ko-143 were purchased from Sigma-Aldrich (St. Louis, MO). [3H]-Allopurinol was purchased from Moravek Biochemicals (Brea, CA), [3H]-oxypurinol was custom-synthesized by tritiation performed by IsoSciences (King of Prussia, PA), and [3H]-Pitavastatin was purchased from American Radiolabeled Chemicals (St. Louis, MO). Cell culture medium, Hank’s Balanced Salt Solution (HBSS), fetal bovine serum (FBS), penicillin/streptomycin, and glutamine were purchased from University of California San Francisco Cell Culture Facility (San Francisco, CA).
Cell lines and cell culture
Human embryonic kidney (HEK) 293 cells expressing vector only (pcDNA3) or vector expressing myc-tagged ABCG2 construct were provided by Dr. Jian-Ting Zhang (Indiana University, Indianapolis, IN).[34, 35] The cells were maintained in DME-H21, supplemented with 10% FBS, 2 mM glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, and 200 μg/mL G418 at 37°C in 5% CO2. HEK293 cells expressing vector only (pcDNA3.1) and cells expressing ABCG2 or ABCG2-Q141K were provided by Dr. Susan E. Bates (National Institutes of Health, Bethesda, MD).[36] The cells were maintained in DME-H21 as described above and in 2 mg/mL G418.
Cytotoxicity assay
HEK293 cells stably expressing vector only or ABCG2 or ABCG2-Q141K provided by Dr. J.T. Zhang and Dr. S.E. Bates were seeded at 6,000 cells per well in 96-well poly-d-lysine-coated plates (BD Biosciences, San Jose, CA). On the following day, cells were exposed to growth medium containing mitoxantrone (200 nM, 100 nM, 50 nM, 25 nM, 12.5 nM) or vehicle for 72 hours. Cell density was measured using the CellTiter-Glo cell viability kit (Promega, Madison, WI) according to the manufacturer’s instructions. Proliferation of each HEK293 cell line after 72 hours was normalized to the density measured on cells treated with mitoxantrone to vehicle treatment. The concentration of mitoxantrone to inhibit 50% of the cell proliferation (IC50) was computed by fitting the data using GraphPad Prism (v. 5.0, GraphPad Software, San Diego, CA).
Cell accumulation studies
HEK293 cells expressing vector only or ABCG2 or ABCG2-Q141K cDNA constructs were seeded at 2.5 × 105 cells per well in 24-well poly-d-lysine-coated plates (BD Biosciences). Mixtures containing the following were prepared for cell accumulation studies: HBSS with trace amount of radiolabeled compound ([3H]-allopurinol or [3H]-oxypurinol) and its unlabeled compound, allopurinol (1 μM) or oxypurinol (1 μM), were mixed with ABCG2 inhibitor, Ko-143 (10 μM), or with vehicle (0.2% DMSO). After the cells were washed twice and incubated with HBSS for 15 minutes, HBSS were removed and cells were incubated with the mixtures above for 30 minutes at 37°C. For the studies associated with determining accumulation of ([3H]-pitavastatin in BCRP-expressing cells, allopurinol (0.5, 1 and 2 mM) or oxypurinol (0.5, 1 and 2 mM) or Ko-143 (10 μM as positive control) or vehicle (1.0 % DMSO) were used. The accumulation studies were terminated by washing cells twice with 1.0 mL of ice-cold HBSS, followed by addition of 800 μL of lysis buffer (0.1% SDS v/v, 0.1 N NaOH). Intracellular radioactivity was determined by scintillation counting and normalized per well of protein content as measured by bicinchoninic acid protein assay (Pierce, Rockford, IL). The method for this study was modified from Abla et al.[37] Data were analyzed by two-tailed unpaired t-test.P-values <0.05 were considered statistically significant.
Supplementary Material
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ There have been no GWAS of antihyperuricemic response to allopurinol. Two GWAS[6, 7] examined genetic associations with allopurinol induced Stevens-Johnson syndrome, but not with effectiveness in terms of SUA response. Another article has shown that URAT1 transports oxypurinol.[17]
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ What are the genetic factors that affect SUA response to allopurinol as an antihyperuricemic drug?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ We identified variants in ABCG2 that predict SUA response to allopurinol, including rs2231142. We observed that BCRP, the protein encoded by ABCG2, transports allopurinol and that the rs2231142 variant reduces allopurinol transport. This is the first time BCRP has been associated with allopurinol response or been observed to transport allopurinol.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS
✓ This discovery could lead to changes in the way we view the pharmacologic mechanisms of allopurinol. From the clinical perspective, the results may contribute to identifying poor and nonresponders to allopurinol, who may benefit from newer and more expensive alternative drug therapies.
ACKNOWLEDGMENTS
This work was supported by funds provided by the NIH (GM61390 and DK103729) and UCSF Institute for Human Genetics. The genotyping of the GERA cohort was supported by grant RC2 AG036607 from the National Institutes of Health; development of the RPGEH and GERA cohort was supported by grants from the Robert Wood Johnson Foundation, the Ellison Medical Foundation, the Wayne and Gladys Valley Foundation, and Kaiser Permanente. We also thank the Kaiser Permanente RPGEH for providing the initial data and the UCSF IHG for computational time.
Footnotes
CONFLICT OF INTEREST
Dr Giacomini reports personal fees from Apricity Therapeutics, grants from Pfizer, grants from Sanofi Aventis, grants from Astra Zeneca, and grants from GSK outside the submitted work. Dr Yee reports personal fees from Apricity Therapeutics, outside the submitted work.
AUTHOR CONTRIBUTIONS
CCW, CS, NR, and KMG designed the research. SWY, and XL performed the research. CCW, NR, KMG, TJH, MNK, YB, and EJ analyzed the data. CCW, KMG, and NR wrote the article. All authors reviewed and edited the final report.
References
- 1.Roddy E, Doherty M. Epidemiology of gout. Arthritis Res. Ther. 2010;12:223. doi: 10.1186/ar3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh JA. Racial and gender disparities among patients with gout. Curr. Rheumatol. Rep. 2013;15:307. doi: 10.1007/s11926-012-0307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The Lancet. Gout: A disease of the past, the present, but not the future. Lancet. 2014;383:25–31. doi: 10.1016/S0140-6736(14)60088-X. [DOI] [PubMed] [Google Scholar]
- 4.Kuo CF, Grainge MJ, Mallen C, Zhang W, Doherty M. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Ann. Rheum. Dis. 2014 doi: 10.1136/annrheumdis-2013-204463. doi:10.1136/annrhemdis-2013-204463 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khanna D, et al. American College of Rheumatology 2012 American College of Rheumatology guidelines for management of gout Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012;64:1431–1446. doi: 10.1002/acr.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Köttgen A, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013;45:145–154. doi: 10.1038/ng.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okada Y, et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet. 2012;44:904–909. doi: 10.1038/ng.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Génin E, et al. Genome-wide association study of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe. Orphanet J. Rare Dis. 2011;6:52. doi: 10.1186/1750-1172-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tohkin M, et al. A whole-genome association study of major determinants for allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Pharmacogenomics J. 2013;13:60–69. doi: 10.1038/tpj.2011.41. [DOI] [PubMed] [Google Scholar]
- 10.Becker MA, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res. Ther. 2010;12:R63. doi: 10.1186/ar2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodward OM, et al. Gout-causing Q141K mutation in ACG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5223–5228. doi: 10.1073/pnas.1214530110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirano M, Maeda K, Matsushima S, Nozaki Y, Kusuhara H, Sugiyama Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol. Pharmacol. 2005;68:800–807. doi: 10.1124/mol.105.014019. [DOI] [PubMed] [Google Scholar]
- 13.Fujino H, Saito T, Ogawa S, Kojima J. Transporter-mediated influx and efflux mechanisms of pitavastatin, a new inhibitor of HMG-CoA reductase. J. Pharm. Pharmacol. 2005;57:1305–1311. doi: 10.1211/jpp.57.10.0009. [DOI] [PubMed] [Google Scholar]
- 14.Takenaka K, et al. Substrate overlap between Mrp4 and Abcg2/Bcrp affects purine analogue drug cytotoxicity and tissue distribution. Cancer Res. 2007;67:6965–6972. doi: 10.1158/0008-5472.CAN-06-4720. [DOI] [PubMed] [Google Scholar]
- 15.Hua J, Huang P, Zhu CM, Yuan X, Yu CH. Anti-hyperuricemic and nephroprotective effects of modified Simiao decoction in hyperuricemic mice. J. Ethnopharmacol. 2012;142:248–252. doi: 10.1016/j.jep.2012.04.052. [DOI] [PubMed] [Google Scholar]
- 16.Anzai N, et al. Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J. Biol. Chem. 2008;283:26834–26838. doi: 10.1074/jbc.C800156200. [DOI] [PubMed] [Google Scholar]
- 17.Iwanaga T, Kobayashi D, Hirayama M, Maeda T, Tamai I. Involvement of UA transporter in increased renal clearance of the xanthine oxidase inhibitor oxypurinol induced by a uricosuric agent benzbromarone. Drug Metab. Dispos. 2005;33:1791–1795. doi: 10.1124/dmd.105.006056. [DOI] [PubMed] [Google Scholar]
- 18.Enomoto A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 19.Iharada M, et al. Type 1 sodium-dependent phosphate transporter (SLC17A1 protein) is a CL(−)-dependent urate exporter. J. Biol. Chem. 2010;285:26107–26113. doi: 10.1074/jbc.M110.122721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witkowska K, et al. Human SLC2A9a and SLC2A9b isoforms mediate electrogenic transport of urate with different characteristics in the presence of hexoses. Am. J. Physiol. Renal Physiol. 2012;303:F527–539. doi: 10.1152/ajprenal.00134.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung SI, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4134–4139. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Resource for Genetic Epidemiology Research on Adult Health and Aging (GERA) dbGaP study Accession phs000674.v1.p1.
- 23.Hoffmann TJ, et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics. 2011;98:79–89. doi: 10.1016/j.ygeno.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann TJ, et al. Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics. 2011;98:422–430. doi: 10.1016/j.ygeno.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PloS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2011;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 27.1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011;1:457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 32.Purcell S, et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2014. http://www.R-project.org. [Google Scholar]
- 34.Xu J, Liu Y, Yang Y, Bates S, Zhang JT. Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem. 2004;279:19781–1979. doi: 10.1074/jbc.M310785200. [DOI] [PubMed] [Google Scholar]
- 35.Xu J, Peng H, Chen Q, Liu Y, Dong Z, Zhang JT. Oligomerization domain of the multidrug resistance-associated transporter ABCG2 and its dominant inhibitory activity. Cancer Res. 2007;67:4373–4381. doi: 10.1158/0008-5472.CAN-06-3169. [DOI] [PubMed] [Google Scholar]
- 36.Morisaki K, et al. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother. Pharmacol. 2005;56:161–172. doi: 10.1007/s00280-004-0931-x. [DOI] [PubMed] [Google Scholar]
- 37.Abla N, et al. The human multidrug resistance protein 4 (MRP4 ABCC4): functional analysis of a highly polymorphic gene. J. Pharmacol. Exp. Ther. 2008;325:859–868. doi: 10.1124/jpet.108.136523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.