

Abstract
Essential to iron homeostasis is the transport of iron by the bilobal protein human serum transferrin (hTF). Each lobe (N- and C-lobe) of hTF forms a deep cleft which binds a single Fe3+. Iron-bearing hTF in the blood binds tightly to the specific transferrin receptor (TFR), a homodimeric transmembrane protein. After undergoing endocytosis, acidification of the endosome initiates the release of Fe3+ from hTF in a TFR-mediated process. Iron-free hTF remains tightly bound to the TFR at acidic pH; following recycling back to the cell surface, it is released to sequester more iron. Efficient delivery of iron is critically dependent on hTF/TFR interactions. Therefore, identification of the pH-specific contacts between hTF and the TFR is crucial. Recombinant protein production has enabled deconvolution of this complex system. The studies reviewed herein support a model in which pH-induced interrelated events control receptor-stimulated iron release from each lobe of hTF.
1. IRON
As a transition element, iron can assume a number of different oxidation states, from −2 to +6. However, the ferrous (Fe2+) and ferric (Fe3+) states are most common and are easily exchangeable through the transfer of one electron (Aisen, Enns, & Wessling-Resnick, 2001). Due to its inherent redox properties, iron is critical to a number of biological processes, including oxygen and electron transport, making it essential for most life on Earth (Aisen et al., 2001). Only two organisms have been identified that do not require iron: Borrelia burgdorferi, the bacteria responsible for Lyme disease, and a bacterium found in soil, Lactobacillus plantarum (Posey & Gherardini, 2000). These organisms substitute other metals, such as manganese, for iron to maintain activity of biologically important molecules. The same redox properties that provide biological versatility also make iron potentially dangerous. In oxygen-rich environments, Fe3+ forms extremely insoluble iron oxides (i.e. rust), while Fe2+ catalyzed Fenton reactions that can be toxic to living cells (Fenton, 1876, 1893). Specifically, reduction of O2 by Fe2+ generates superoxide, which can ultimately lead to the formation of the hydroxyl radical, a powerful oxidant known to damage DNA, proteins and lipids (Park, Bacon, Brittenham, & Tavill, 1987). In an evolutionary conundrum, aerobic life on Earth requires iron while the oxygen-rich environment of the planet maintains most of it in a chemically inert state (Fe3+). However, it must be remembered that oxygen conditions on Earth have changed. When the first single-cell organisms began to appear on Earth, the atmospheric composition was drastically different, with very little dissolved O2. The ready availability of Fe2+ at such low oxygen concentrations allowed iron to procure a strong foothold in essential biological processes. Hence, organisms that had evolved biological pathways utilizing Fe2+ were presented with a dilemma when high oxygen conditions limited iron bioavailability. Life has adapted to the ever-changing conditions of the atmosphere by manipulating the metallome in a manner that allows for the endless redox cycling that takes place in modern day organisms.
2. IRON HOMEOSTASIS
2.1. Iron Uptake
It is estimated that the adult human body contains ~3.5 g of iron (Theil & Goss, 2009). To prevent detrimental effects due to the redox potential of the Fe2+/Fe3+ pair, iron must be tightly regulated within the body. Since no physiological excretion mechanism exists (McCance & Widdowson, 1938), uptake of iron into the body is strictly controlled. Erythroid precursor cells are the largest consumer of bodily iron, utilizing nearly 1000 million iron atoms a day (ca. two third of the total body iron content) to maintain hemoglobin levels (Andrews & Schmidt, 2007; Theil & Goss, 2009). A large portion of the iron contained in hemoglobin is recycled from aged and damaged erythrocytes that are phagocytosed by tissue macrophages in the spleen (Andrews & Schmidt, 2007). However, 1–2 mg of iron is lost daily due to normal sloughing of epithelial cells (from skin, gastrointestinal and urinary tracts). Therefore, this same amount (~1 to 2 mg) must be offset by iron acquired from the diet every day.
The diet of omnivorous humans generally contains both heme and non-heme iron. Of the non-heme sources, only ferrous iron can be directly absorbed at the apical surface of duodenal enterocytes. Therefore, any Fe3+ in the diet must first be reduced by the ferric reductase, duodenal cytochrome b, before being transported into the enterocyte by the divalent metal transporter 1 (DMT1) (Fleming et al., 1997; Gunshin et al., 1997, 2005). An energy-dependent transporter, DMT1, transports a number of other divalent metals and protons as well as Fe2+ (Gunshin et al., 1997). Although the majority of the iron in most diets is non-heme iron (~90 to 95%), it is absorbed rather inefficiently (only ~2 to 20%) (Sharp & Srai, 2007). On the other hand, heme iron accounts for only 5–10% of the iron in the diet, but is absorbed more efficiently (~20 to 30%) (Hallberg, Bjorn-Rasmussen, Howard, & Rossander, 1979). While the mechanism of non-heme iron absorption is fairly well characterized, far less is understood about the mechanism of heme transport into duodenal enterocytes. It is thought that the intact porphyrin of heme is absorbed. Although a number of different potential heme transporters have been suggested (Sharp, 2010), only one, heme carrier protein-1 (HCP1), has been identified in the duodenum (Shayeghi et al., 2005). However, more recent studies have revealed that HCP1 displays ~100-fold lower Km for folate than heme (Qiu et al., 2006), suggesting HCP1 is actually a specific cell membrane transporter for folate, not heme. Thus, the mechanism of heme absorption in the duodenal enterocyte remains unknown. Iron absorbed as heme is freed within the enterocytic endosomes by the enzyme heme oxygenase 1 (Raffin, Woo, Roost, Price, & Schmid, 1974). Iron acquired from either the heme or non-heme pathway becomes part of the same intracellular iron pool, where it can either be stored in the enterocyte by ferritin (see below) or trafficked to the basolateral surface of the enterocyte for transport into the blood.
The transfer of Fe2+ from the basolateral membrane of the enterocyte into the circulation is mediated by ferroportin (FPN) (Donovan et al., 2000), the only transporter involved in iron efflux from the enterocyte identified to date (Sharp, 2010). The iron released from the enterocytes of the duodenum via FPN is in the more dangerous ferrous form. To avoid Fenton chemistry and the production of damaging free radicals, iron must be oxidized to the ferric form. This oxidation is accomplished by the membrane-bound multi-copper-containing ferroxidase hephaestin (Vulpe et al., 1999), located on the basolateral surface of the enterocyte. A homologue of hephaestin is the soluble ferroxidase, ceruloplasmin (Gubler, Lahey, Chase, Cartwright, & Wintrobe, 1952; Lahey, Gubler, Chase, Cartwright, & Wintrobe, 1952), which circulates in the serum. Critically, following the degradation of senescent red blood cells, Fe2+ released from macrophages (via FPN) is oxidized by ceruloplasmin to Fe3+. Following oxidation, Fe3+ is carefully chaperoned through the body by the bilobal iron binding and transport protein human serum transferrin (hTF) (Kaplan & O’Halloran, 1996).
3. TRANSFERRIN
Synthesized in the liver and secreted into the blood plasma, hTF is an ~80 kDa bilobal (N- and C-lobes) glycoprotein that binds Fe3+ very tightly (Kd ~1022/M) (Aisen, Leibman, & Zweier, 1978), yet reversibly. Since hTF can bind iron in both lobes, four different hTF species may circulate in the blood, differing only in iron content. The ~25 to 50 μM (~2.5 mg/ml) hTF in the serum (Sun, Li, & Sadler, 1999) appears to be nonrandomly distributed among diferric (Fe2hTF, ~11 to 27%), monoferric N-lobe hTF (FeNhTF, ~21 to 23%), monoferric C-lobe hTF (FeChTF, ~11 to 17%) and iron-free hTF (apohTF, ~40 to 51%) (Makey & Seal, 1976; Williams & Moreton, 1980) (see below). At any given time only ~30% of the hTF in circulation is saturated with iron although even within an individual the range is considerable with variables including diet and time of day. Importantly, the iron-binding sites in the unsaturated 70% of hTF remain available and provide significant buffering capacity in times of iron influx or overload. Interestingly, although the C-lobe has a greater affinity for Fe3+ at neutral pH than the N-lobe (Cannon & Chasteen, 1975; Lestas, 1976; Princiotto & Zapolski, 1975), there appears to be a somewhat greaterpercentage of FeNhTF in the serum than FeChTF (Williams & Moreton, 1980). The source of the unequal distribution between lobes (if it exists, see for example Huebers, Josephson, Huebers, Csiba, & Finch, 1984) could include either incomplete loading of hTF at the level of uptake from the enterocytes and/or incomplete iron removal within cells. Although the distribution appears to be nonrandom and to favor the kinetically weaker N-lobe, several observations should be considered: 1) Using electrophoretic measurements, it is challenging to determine the distribution in fresh serum without disturbing the equilibrium. Most studies employ urea gel analysis alone or in combination with other techniques (Leibman & Aisen, 1979); 2) The small number of individual serum samples in combination with the large differences in hTF saturation may belie the generality of the conclusions that are derived from such data; 3) The substantial concentration of hTF in the serum, in addition to the difference in the binding affinity of Fe2hTF in comparison to either monoferric species, is probably sufficient to guarantee that binding of Fe2hTF will be favored. Thus, the concentration of the monoferric species may be somewhat irrelevant.
3.1. The Transferrin Cycle
At the pH of the serum (~7.4) iron-bearing hTF binds with nanomolar affinity to the specific transferrin receptor (TFR, also referred to as TFR1), located on the cell surface of all iron-requiring cells. It is important to note that at pH 7.4, although Fe2hTF binds to the TFR with the highest affinity (Kd ~4 nM), the two monoferric hTFs (FeNhTF and FeChTF) also form a high-affinity stable complex with the TFR (Kd ~36 nM and Kd ~32 nM, respectively) (Mason et al., 2009) while apohTF binds very weakly at this pH. Following clathrin-dependent endocytosis of the hTF/TFR complex (Morgan & Appleton, 1969), the pH within the endosome is lowered through the action of ATP-dependent H+ pumps (Morgan, 1981). Salt and a currently unidentified chelator, along with the significantly lower pH (~5.6) within the endosome, initiate receptor-stimulated iron release from hTF. Although the TFR purportedly influences the redox potential of Fe3+ bound to hTF (Dhungana et al., 2004), the recent discovery of an endosomal ferrireductase (Steap3) in erythroid cells suggests that the reduction of Fe3+ to Fe2+ is accomplished only after Fe3+ is released from hTF within the endosome (Ohgami et al., 2005). Critical to the hTF endocytic cycle, apohTF remains bound to the TFR with high affinity at endosomal pH. This allows the apohTF/TFR complex to return to the cell surface instead of being targeted to late endosomes and avoid degradation within the lysosome. Upon returning to the cell surface, apohTF is released into the serum, either through dissociation from the TFR or displacement by an iron-containing hTF (Leverence, Mason, & Kaltashov, 2010), where it is free to bind more Fe3+. The process of internalization and recycling of the hTF/TFR complex has been so extensively studied that it has become the classic example of clathrin-dependent receptor-mediated endocytosis and often serves as a positive control for other systems (Grant & Donaldson, 2009).
3.2. Cellular Iron Utilization
Once inside the cell, iron is compartmentalized in a number of different ways. These include incorporation into heme, iron–sulfur clusters, and other iron-requiring enzymes/proteins, formation of an intracellular labile iron pool or storage in the iron storage protein ferritin. The majority of heme biosynthesis occurs within the mitochondria, with a few intermediary steps taking place in the cytosol (Andrews & Schmidt, 2007). The molecular pathway utilized to traffic iron from the endosome to the mitochondria has been a subject of debate for many years. One model proposes that after leaving the endosome via DMT1, Fe2+ enters a cytosolic pool of iron composed of low molecular weight iron–chelate complexes (Greenberg & Wintrobe, 1946; Jacobs, 1977). A combination of pulse-chase experiments (Ponka, Borova, Neuwirt, & Fuchs, 1979) and cytosolic fractionation studies (Weaver & Pollack, 1989) were first utilized to suggest that such a chelatable intracellular pool of iron might exist. However, a major drawback is that these studies required cell lysis that can often lead to decompartmentalization and iron release from organelles such as the mitochondria. More recently, cell permeable fluorescent iron chelators have detected low (~2.5 to 5 μM), but significant, levels of free cytosolic iron (Kakhlon & Cabantchik, 2002). The concept of a labile iron pool is very controversial given the redox-active chemical properties of the Fe2+/Fe3+ pair. Another theory, supported by experiments in the Ponka laboratory and termed the “kiss and run” hypothesis (Sheftel, Mason, & Ponka, 2012) proposes that, at least in reticulocytes, iron bypasses the cytosol and is instead directly transferred from hTF-laden endosomes to the mitochondria through direct interaction of the two organelles. Given the considerable iron requirements of reticu-locytes, alternative iron trafficking pathways may be utilized in other cell types (Sheftel et al., 2012). Further study is required to delineate exactly how iron is trafficked once inside the cell.
Iron not utilized in the formation of heme or other iron-containing proteins is stored in the cytosolic iron storage protein ferritin. First identified and crystallized from horse spleen (Laufberger, 1937), 24 subunits (H- and L-chains) form the large cage-like structure (~80 Å diameter) of ferritin, within which up to 4500 Fe3+ atoms may be stored (Harrison & Arosio, 1996). Ferritin also possesses ferroxidase activity that is critical to consuming Fe2+ released by endosomes via DMT1 and storing the less toxic Fe3+ (Arosio & Levi, 2010). However, the bioavailability of the iron stored in ferritin remains questionable (Sheftel et al., 2012). Some suggest that the ferric iron is readily bioavailable via eight hydrophilic channels in the protein shell of ferritin (Bou-Abdallah, McNally, Liu, & Melman, 2011), while other evidence suggests that at least partial degradation of the protein shell must occur before iron can be released from ferritin (Sheftel et al., 2012). Although the mechanism of iron release from ferritin remains unclear, its biological importance is emphasized by its conservation among various life forms, e.g. some form of ferritin has been identified in nearly every biological classification, except for yeast which stores excess iron in vacuoles (Arosio & Levi, 2010).
3.3. Transferrin Structural Overview
As mentioned previously, hTF is a bilobal protein comprised of 679 amino acids. Connected by a seven amino acid bridge (residues 332–338), the N-and C-lobes are ~40% identical and probably arose from gene duplication followed by fusion (Park et al., 1985). Each lobe folds into two subdomains (termed N1 and N2 and C1 and C2, Fig. 1). The N1 and C1 subdomains are discontinuous in primary sequence (N1: residues 1–93 and 247–331, C1: 339–425 and 573–679), while the N2 and C2 subdomains are continuous (N2: residues 94–246, C2: 426–572). The Fe3+ binds in a deep cleft formed between the two subdomains of a lobe. The subdomains are comprised of similar α/β folds centered around a β-sheet (Hall et al., 2002; Noinaj et al., 2012). A characteristic structural feature of most transferrins (except insect transferrins) is a classical γ turn in the N1 and C1 subdomains (Lambert, Perri, Halbrooks, & Mason, 2005). The strain on the Leu–Leu–Phe sequence (which forms the γ turn) places the second leucine residue in the disallowed region of the Ramachandran plot and may help to correctly position pH-sensitive elements critical to iron release from each lobe (Eckenroth, Mason, McDevitt, Lambert, & Everse, 2010). Iron release from each lobe is accompanied by large rigid body movements of the subdomains (opening of ~50°) (Hall et al., 2002; Wally et al., 2006). A “hinge” region, consisting of two β-strands connecting the subdomains of each lobe, mediates opening of the cleft (Jeffrey et al., 1998; MacGillivray et al., 1998; Wally et al., 2006). Additional stability is provided by the participation of all 38 cysteine residues in hTF in the formation of 19 disulfide bonds (eight in the N-lobe and 11 in the C-lobe, Fig. 2). Native hTF is a glycoprotein with two N-linked biantennary glycans in the C-lobe (at residues Asn413 and Asn611). While glycosylation appears to have no effect on TFR binding or iron uptake or release by hTF (Mason et al., 1993), it has been suggested that loss of the terminal sialic acids over time may play a role in clearance of hTF from the blood by asialo receptors in the liver (Regoeczi, Bolyos, & Chindemi, 1989).
Figure 1.

hTF. Structures of diferric hTF (Left, PDB ID: 3V83) and apohTF (Right, PDB ID: 2HAU). Individual subdomains are colored accordingly: N1-blue, N2-red, C1-green, C2-yellow. The two Fe3+ atoms in the diferric transferrin are shown as orange spheres. Figure generated using PyMOL (Delano, 2002). See the color plate.
Figure 2.

Disulfide bonds of hTF. The 19 disulfide bonds formed within hTF are shown numbered according to primary sequence. Equivalent disulfide bonds from each lobe are listed on the same line. Individual subdomains are colored accordingly: N1, blue; N2, red; C1, green; C2, yellow. See the color plate.
3.4. Transferrin Iron-Binding Ligands
For many years, the identity of the iron-binding residues of hTF was unknown and the subject of much conjecture. Based on various biochemical studies, histidine, tyrosine and tryptophan residues were suggested as candidates. It was not until the first crystal structure of the transferrin family member lactoferrin (LTF) was determined by the Baker laboratory (Anderson et al., 1987) that the iron-binding ligands were finally identified. The Fe3+ is coordinated by identical ligands in both lobes of hTF: two tyrosine residues, one aspartic acid and one histidine residue (Tyr95, Tyr188, Asp63 and His249 in the N-lobe; Tyr426, Tyr517, Asp392 and His585 in the C-lobe, hTF numbering) (Fig. 3). The remaining two ligands which complete the distorted octahedral coordination of the iron are provided by carbonate, which is anchored in place by a conserved arginine residue (Arg124 in the N-lobe and Arg456 in the C-lobe). Interestingly, synergistic anion binding is an absolute requirement for high-affinity Fe3+ binding by hTF (Price & Gibson, 1972b). The initial binding of carbonate may be necessary to bring the remaining iron-binding ligands of hTF, which are very distant in the primary sequence of the protein, into proximity (Gaber, Miskowski, & Spiro, 1974).
Figure 3.

Close-up of ligands and second-shell residues in the hTF N- and C-lobes of Fe2hTF (PDB 3V83). The ligands are colored orange. The second-shell residues are depicted in pink. The conserved arginine is in green. The figure was produced by Dr Stephen J. Everse using PyMOL. See the color plate.
Given its relatively high concentration in the serum, carbonate is the physiologically relevant synergistic anion (Schade, Reinhart, & Levy, 1949); however, other molecules (oxalate, glycolate, malonate, etc.) can substitute for carbonate to promote high-affinity Fe3+ binding to hTF (Schlabach & Bates, 1975). All functional synergistic anions appear to follow an interlocking sites model (Schlabach & Bates, 1975) in which the carboxylate group of the anion is available to bind the anchoring arginine residue in each lobe of hTF (Arg124 and Arg456), as well as a proximal electron donor group 1–2 carbon atoms away to complete the distorted octahedral coordination of Fe3+(Baker, 1994).
The coordination of Fe3+ by the two tyrosine residues in each lobe of hTF not only produces an intense ligand-to-metal charge transfer band but also disrupts the π–π★ transition of the two tyrosines. This causes an increase in the UV absorbance at 280 nm that overlaps with and strongly quenches the tryptophan fluorescence through what is thought to be radiationless transfer of excited-state energy (Lehrer, 1969; Patch & Carrano, 1981). Importantly, the overall effect is that Fe3+ binding by hTF significantly quenches the intrinsic tryptophan fluorescence of the protein (Lehrer, 1969) such that upon transitioning from Fe2hTF to apohTF a 368% increase in the intrinsic tryptophan fluorescence is observed (Fig. 4) (Byrne & Mason, 2009). This spectral property has been invaluable in the study of the kinetics of iron release from hTF.
Figure 4.

Steady-state tryptophan fluorescence of Fe2hTF. Steady-state emission spectra Fe2hTF before (iron containing 100 mM HEPES buffer (4-(2-hydroxyethyl)-1-piperazineethansulfonic acid), pH 7.4) and after treatment with iron removal buffer (apohTF 100 mM MES (2-(N-morpholino)ethanesulfonic acid), pH 5.6, containing 300 mM KCl and 4 mM EDTA for 15 min) following constant excitation at 280 nm. For color version of this figure, the reader is referred to the online version of this book.
3.5. Second-Shell Residues
Although the iron-binding ligands are completely conserved in each lobe of hTF, the mechanism of iron release from each lobe is distinctly different, largely because of differences in “second-shell” residues (these residues are not directly involved in the coordination of the Fe3+ but form an intricate hydrogen bonding network with the primary Fe3+-binding ligands of hTF). Notably, the two lobes differ in their response to pH (Baldwin & de Sousa, 1981; Lestas, 1976; Princiotto & Zapolski, 1975), anions (Baldwin, 1980; Baldwin & de Sousa, 1981), the TFR (Bali & Aisen, 1991; Byrne, Chasteen, Steere, & Mason, 2010; Zak & Aisen, 2003), and the conformation of the other lobe in part due to differences in the composition of the second-shell residues in each lobe.
The first suggestion of a potential iron release mechanism from the N-lobe of hTF was provided by the crystal structure of the N-lobe of hen ovotransferrin (Dewan, Mikami, Hirose, & Sacchettini, 1993). Two second-shell lysine residues (one from each subdomain) in the N-lobe, Lys206 and Lys296, are close enough to share a hydrogen bond (3.0 Å) and constitute what is referred to as the dilysine trigger (Dewan et al., 1993) (Fig. 3). The formation of this hydrogen bond is attributed to an unusually low pKa value of either one or both of the lysine residues due to their location within a hydrophobic environment. Protonation of one of the lysine residues due to a reduction in pH causes the positively charged lysines to repel each other (moving at least 9 Å apart in the apo N-lobe structure; Jeffrey et al., 1998), and literally triggers cleft opening to promote iron release. Mutation of either Lys206 or Lys296 to a glutamate or alanine drastically slows the rate of iron removal, validating the critical nature of the dilysine trigger in the mechanism of iron release from the N-lobe of hTF (He & Mason, 2002).
Until recently, the mechanism of iron release from the C-lobe has been unclear. As described above, crystal structures of iron-bound hen oTF and apohTF helped clarify the action of the dilysine trigger in the N-lobe. However, no crystal structure of an iron-containing C-lobe of hTF was available. Using sequence alignments and an unpublished crystal structure of FeChTF (Zuccola, 1993), Dewan et al. (1993) proposed a slightly different mechanism of iron release from the C-lobe in which the dilysine trigger is replaced by a triad of residues (Lys534, Arg632 and Asp634) (Fig. 3). Similar to the dilysine trigger, Lys534 and Arg632 may share a hydrogen bond that is stabilized by Asp634, which upon protonation would trigger iron release from the C-lobe; however, in the structure of porcine transferrin, the NZ and NE group of the homologous lysine (Lys543) and arginine (Arg641) are ~4.1 Å apart, seemingly too far to share a hydrogen bond (Hall et al., 2002). Lacking a crystal structure of an iron-containing C-lobe of hTF, the precise mechanism by which the triad might trigger iron release from the C-lobe remains unclear. However, it has been shown that mutation of Lys534 or Arg632 to an alanine severely retards iron release from that lobe, essentially locking iron in the C-lobe (Halbrooks et al., 2003). It has recently become clear that the triad is less relevant to the mechanism of iron release from the C-lobe in the presence of the TFR (Eckenroth, Steere, Chasteen, Everse, & Mason, 2011; Steere, Byrne, et al., 2010) (see discussion of hTF residue His349 below).
3.6. Kinetically Significant Anion-Binding Sites
Along with synergistic anion-binding, non-synergistic anions are known to bind to hTF as well. By definition non-synergistic anions bind to sites on hTF but do not facilitate high-affinity Fe3+ binding. As originally detailed by Folajtar and Chasteen (1982) the allosteric binding of non-synergistic anions to hTF follows the lyotropic series ( ) and is thought to induce structural changes in hTF that perturb the Fe3+-binding center and thereby influence iron release. Furthermore, as clearly demonstrated by Kretchmar and Raymond (1988), binding of non-synergistic anions to hTF is absolutely required for iron release: iron release from hTF ceases as the ionic strength is extrapolated to zero at pH 7.4. In order to emphasize their important allosteric effect on iron release from hTF, these non-synergistic anion-binding sites were designated “kinetically significant anion-binding” or KISAB sites by Egan et al. (Egan, Ross, Purves, & Adams, 1992; Egan, Zak, & Aisen, 1993; Marques, Egan, & Pattrick, 1990; Marques, Watson, & Egan, 1991).
In the past, many different spectroscopic techniques, including electron paramagnetic resonance spectroscopy (Chasteen, 1983; Folajtar & Chasteen, 1982; Grady, Mason, Woodworth, & Chasteen, 1995; Price & Gibson, 1972a; Thompson, McCarty, & Chasteen, 1986), nuclear magnetic resonance spectroscopy (Kubal, Mason, Patel, Sadler, & Woodworth, 1993) and UV-difference spectra (Harris, 1985; Harris & Bali, 1988; Harris, Cafferty, Abdollahi, & Trankler, 1998; Harris, Nesset-Tollefson, Stenback, & Mohamed-Hani, 1990; Pecoraro, Harris, Carrano, & Raymond, 1981) have been employed to detect anion binding to hTF. It was clearly demonstrated (Williams, Chasteen, & Moreton, 1982) that at pH 7.4, the presence of salt retards the rate of iron release from the N-lobe while facilitating iron release from the C-lobe. The UV-difference technique applied by the Harris laboratory provided an estimate of the binding strength of various anions to apohTF (Bali & Harris, 1990; Harris, 1985; Harris et al., 1990). In addition, the Sadler group published a review summarizing the equilibrium binding constants for 13 non-synergistic anions to hTF (Sun et al., 1999).
Importantly, many of these earlier studies monitoring anion binding to hTF were performed at neutral pH (~7.4), and in the absence of the TFR. Our laboratory has suggested that in order to be designated as kinetically significant, non-synergistic anion binding must correlate with low pH; i.e. anion binding to a KISAB site must exert maximal effect at endosomal pH (~5.6) (Byrne, Steere, Chasteen, & Mason, 2010). Although it is well documented that non-synergistic anions are required for iron release from hTF, the specific identification of these binding sites has remained elusive. Obvious choices for potential KISAB sites are positively charged (lysine, arginine and histidine) residues. The most likely KISAB sites were long thought to be iron liganding residues, second-shell residues and residues in the hinging β-strands that facilitate opening and closing of each lobe of hTF (Egan et al., 1993; Grady et al., 1995; Harris & Bali, 1988). Although anions may bind to these residues (normally buried within the cleft of each lobe when Fe3+ is bound), it is likely that this only occurs after iron is released. Thus, residues within these buried regions of hTF do not qualify as legitimate KISAB sites, but may function to stabilize the apo conformation of each lobe.
4. THE TFR
In the 1960s, it was proposed that a membrane-bound receptor for transferrin might exist ( Jandl & Katz, 1963). However, it was not until 1977 that (Leibman and Aisen 1977) verified the presence of the TFR1 in a detergent-solubilized fraction of lysed rabbit reticulocytes that had been previously incubated with 125I-labeled transferrin. Soon after, (Seligman, Schleicher, and Allen 1979) were the first to isolate and characterize the TFR from human placental tissue. In 1984, two laboratories published the primary amino acid sequence of the TFR1 based on the messenger RNA (mRNA) sequence (Kuhn, McClelland, & Ruddle, 1984; McClelland, Kuhn, & Ruddle, 1984; Schneider, Owen, Banville, & Williams, 1984). Intriguingly, the human TFR gene is located on chromosome 3, as are the genes for hTF and melanotransferrin. The significance of the proximity of these related genes within the genome remains unclear, but raises some interesting evolutionary questions. The ubiquitously expressed TFR1 is the main route of entry for iron into most cells. Importantly, TFR1 expression is controlled at the translational level by five iron responsive element (IREs) in the 3′ UTR of the mRNA, which allows the quantity of TFR1 at the cell surface to be modulated by intracellular iron concentrations (Subramaniam, Summerville, & Wallace, 2002). Due to neurological defects and impaired erythropoiesis, TFR1 knockout mice do not survive past embryonic day 12.5, emphasizing the essential nature of the TFR1 (Levy, Jin, Fujiwara, Kuo, & Andrews, 1999).
TFR1 is a type II transmembrane homodimeric receptor. Two ~90 kDa monomers form the TFR homodimer and are comprised of a short cytoplasmic tail (residues 1–67) with an internalization motif, a membrane-spanning portion (residues 68–88), a stalk region (residues 89–120) which contains intermolecular two disulfide bonds (Cys89 and Cys98), which covalently link the two TFR monomers and a large extracellular ectodomain (residues 121-760) (McClelland et al., 1984). The ectodomain is further subdivided into three domains: the protease-like domain (residues 121–188 and 384–606), the apical domain (residues 189–383) and the helical domain (residues 607–760) responsible for dimerization (Fig. 5) (Lawrence et al., 1999). Each TFR monomer binds one hTF molecule such that a 2:2 complex is formed (Enns & Sussman, 1981).
Figure 5.

Ectodomain of the TFR. Crystal structure of the homodimeric ectodomain of the TFR (PDB ID: 1CX8) is oriented with the cell membrane at the bottom (Lawrence et al., 1999). The domains of each monomer are colored as follows: protease-like domain, blue; apical domain, orange; helical domain, green. Figure created using PyMOL (Delano, 2002). See the color plate
TFR1 is extensively posttranslationally modified. The ectodomain contains three N-linked glycosylation sites (Asn251, Asn317 and Asn727) and one O-linked glycosylation site (Thr104). The N-linked glycosylation is important for proper folding and translocation of the TFR to the cell surface (Hayes, Williams, Lucas, & Enns, 1997). However, mutagenesis studies performed in our laboratory determined that only N-linked glycosylation at Asn317 was critical to TFR function. The N317D TFR mutation decreased the affinity of the TFR for hTF, affected the rate of iron release from the C-lobe of hTF and decreased the production of the recombinant soluble portion of the TFR (sTFR) (Byrne et al., 2006).
Intracellularly, phosphorylation occurs at residue Ser24 on the TFR. It is thought that phosphorylation at this location may affect endocytic trafficking of the TFR (Daniels, Delgado, Rodriguez, Helguera, & Penichet, 2006); however, the definitive function of this posttranslational modification remains unclear. Additionally, it has been reported that the tetrapeptide sequence YTRF (residues 20–23) serves as an internalization signal for endocytosis of the TFR (Collawn et al., 1993). Palmitoylation occurs at two positions (Cys62and Cys67) within the intracellular portion and may aid in anchoring the TFR to the cell membrane (Aisen, 2004).
5. RECOMBINANT EXPRESSION
5.1. Recombinant Expression and Purification of hTF
Isolation of hTF is economically feasible and relatively simple given the relative abundance in human serum (~25 to 50 μM) (Sun et al., 1999).However, the potential for exposure to blood-borne pathogens and the inability to introduce mutations in serum-derived hTF has prompted the recombinant production of hTF in a number of expression systems. Naturally, one of the first expression platforms utilized to produce recombinant hTF is also one of the most common, Escherichia coli. The numerous advantages of using a bacterial expression system are well established. However, production of hTF in E. coli met with extremely limited success (de Smit et al., 1995; Hershberger et al., 1991; Hoefkens et al., 1996; Ikeda, Bowman, Yang, & Lokey, 1992; Steinlein & Ikeda, 1993). Due to an inability to correctly form 19 disulfide bonds (Fig. 2), attempts to produce hTF in E. coli yielded low amounts of improperly folded protein.
Another common expression system for recombinant proteins is the yeast, Pichia pastoris. Attempts to produce the isolated N-lobe of hTF in this eukaryotic system produced significant amounts (50–250 mg/l) of functional protein (Mason et al., 1996; Steinlein, Graf, & Ikeda, 1995). Although no effect on function was observed, analysis of the purified protein by electrospray mass spectrometry identified an O-linked glycosylation on residue Ser32, not present in the naturally derived protein (Mason et al., 1996). While the P. pastoris expression system offers several advantages (high yields and low cost), until recently no significant amount of full-length hTF has been produced in this system (Mizutani et al., 2010). Recent efforts using common baker’s yeast, Saccharomyces cerevisiae, have produced homogenous non-glycosylated hTF at significantly higher yields (~1.5 g/l) than previously observed in any yeast system (Finnis et al., 2010).
Baculovirus-infected insect cells are another frequently used expression system. Although the production of hTF in suspended insect cells provides the convenience of fast growth rates (as in yeast and bacterial expression systems) and the ability to posttranslationally modify the expressed protein, maximum production of hTF in this system only reached ~20 mg/l (Ali, Joao, Csonga, Hammerschmid, & Steinkasserer, 1996; Retzer, Kabani, Button, Yu, & Schryvers, 1996).
By far, the most well-defined and characterized expression system used to produce recombinant hTF is the mammalian system developed in our laboratory (Funk, MacGillivray, Mason, Brown, & Woodworth, 1990; Mason, Funk, MacGillivray, & Woodworth, 1991; Mason et al., 1993, 2004). This system utilizes baby hamster kidney (BHK) cells transfected with the pNUT expression vector (Palmiter et al., 1987) containing the complementary DNA (cDNA) sequence for hTF. Importantly, the pNUT vector contains an ampicillin resistance gene that allows for propagation in E. coli. The hTF gene expressed in the pNUT vector by our laboratory also possesses a number of key features, the first of which is the hTF 19-amino acid signal peptide. This sequence is critical to the natural secretion of hTF from the liver into the serum. Normally within the body, this signal peptide is cleaved. In order to assure cleavage of the signal peptide, the first four amino acids of hTF (Val-Pro-Asp-Lys) precede the N-terminal hexa-histidine tag (used for purification). Next, our hTF construct contains a factor Xa cleavage sequence (Ile-Glu-Gly-Arg) to allow for facile removal of the hexa-His tag. Finally, the plasmid contains the polynucleotide sequence coding for hTF (Yang et al., 1984). The resulting cDNA is then transfected into BHK cells using a standard calcium phosphate precipitation method (Mason et al., 1991). Of importance, the pNUT vector also includes a mutated dihydrofolate reductase enzyme that allows for rapid selection (1–2 weeks) with methotrexate of only those BHK cells containing the plasmid following transfection (Funk et al., 1990). The use of this recombinant system has provided the means to produce hTFs that are either incapable of binding (mutation of the two liganding Tyr residues to Phe precludes iron binding in one lobe to create authentic FeNhTF or FeChTF constructs) (Mason et al., 2004) or releasing iron from one of the two lobes (mutation of residues in the dilysine trigger or C-lobe triad prevent iron removal to create LockNhTF or LockChTF constructs) (Fig. 6) (Byrne, Chasteen, et al., 2010; Byrne & Mason, 2009; Halbrooks et al., 2003). Although production of the isolated N-lobe of hTF has been very successful and well documented (Funk et al., 1990; Mason et al., 1993), until recently, production of the isolated C-lobe has been far more problematic. Again, bacterial expression of the C-lobe is rendered nearly impossible by the need to correctly form 11 disulfide bonds (MacGillivray & Mason, 2002), although a low yield (~5%) of C-lobe with questionable conformation was reported by one laboratory (Hoefkens et al., 1996). Other attempts to produce the C-lobe of hTF using bacterial, yeast and mammalian systems have met with limited success and poor yields (Hoefkens et al., 1996; Steinlein et al., 1995; Steinlein & Ikeda, 1993). Even previous attempts to express the C-lobe as a recombinant entity using the BHK expression system, although successful, produced limited amounts of protein (Mason et al., 1997). Moreover, the C-lobe produced contained a complex glycosylation pattern at each of the two N-linked glycosylation sites in the C-lobe, resulting in a heterogeneous sample, further exacerbating the purification process. The Aisen laboratory produced a small amount of isolated C-lobe using the BHK cell system and an hTF construct with a factor Xa cleavage site in the bridge between the N- and C-lobes (Zak & Aisen, 2002). Using the BHK system, we followed a similar strategy in which the seven amino acids in the bridge were replaced by the tobacco etch virus (TEV) protease cleavage sequence, allowing utilization of the highly specific TEV protease to produce high yields of the isolated C-lobe (Steere, Roberts, et al., 2010). This particular C-lobe was recently crystallized (Noinaj et al., 2012).
Figure 6.
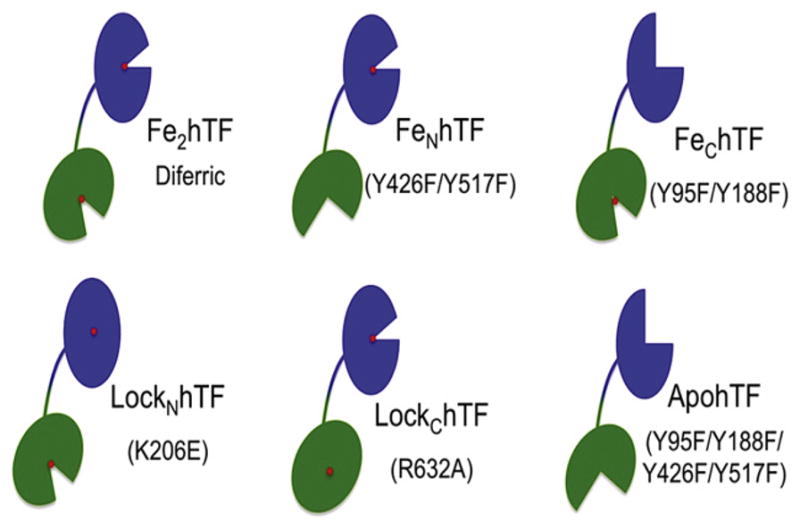
Recombinant hTF constructs. Schematic representing recombinant hTF constructs. Mutations introduced into hTF to prevent iron binding in a lobe (as in the case of the FeNhTF, FeChTF and apohTF constructs) or prevent iron release from one lobe (as in the case of LockNhTF and LockChTF) are indicated. All constructs containing an N-terminal hexahistidine tag are non-glycosylated and have all been previously described (Byrne, Chasteen, et al., 2010; Mason et al., 2004). For color version of this figure, the reader is referred to the online version of this book.
Recently, recombinant production of human proteins in plants has become an appealing alternative expression system and the topic of a great deal of research. Plant sources provide a relatively inexpensive animal-free method to the production of recombinant proteins for biopharmaceutical applications. Plant-derived recombinant proteins eliminate any potential contamination by animal pathogens. Furthermore, edible transgenic plants provide an attractive possibility for therapeutic purposes through direct oral delivery. Thus far, attempts to produce recombinant hTF in tobacco (Nicotiana tabacum) have met with rather limited success (estimated 0.25% total soluble protein) (Brandsma et al., 2010), while the production of hTF in rice (Oryza sativa) is estimated to be ~40% of total soluble protein and is now available commercially under the trade name Optiferrin™ (Zhang et al., 2010). While Optiferrin™ appears to function almost identically to BHK-derived recombinant hTF in a number of aspects, the production of trans-ferrins using this transgenic rice system requires ~1 to 2 years, making it somewhat impractical for the production of mutant transferrins (Steere, A.N., Bobst, C.E., Zhang, D., Pettit, S., Kaltashov, I.A., Huang, N., & Mason, A.B., Journal of Inorganic Biochemistry, In Press.). It seems imperative that all recombinant hTF and TFR samples undergo a rigorous evaluation of their ability to function in a physiologically relevant manner.
5.2. Recombinant Production and Purification of the sTFR
Initial studies involving the TFR utilized full-length TFR isolated from the placenta. Solubilization of the membrane-bound TFR required challenging purification techniques and the presence of detergent micelles, resulting in relatively low yields of TFR (Bali, Zak, & Aisen, 1991; Turkewitz, Amatruda, Borhani, Harrison, & Schwartz, 1988). The recombinant production of the sTFR (residues 121–706, Fig. 5) has helped overcome the challenges of working with the full-length placental-derived TFR (Byrne et al., 2006; Lawrence et al., 1999; Lebron & Bjorkman, 1999). The use of the sTFR eliminates the need for detergent required in the full-length TFR preparations. It is important to note that although two disulfide bonds in the stalk region (Cys89 and Cys98) are formed as a result of TFR dimerization, the homodimer is maintained even in their absence (as in the sTFR) due to the strong interaction between the helical domains of each monomer.
Production of the sTFR has been successful in a number of different expression systems including Chinese hamster ovary cells (Lawrence et al., 1999), baculovirus-infected insect cells (Giannetti et al., 2005; Giannetti, Snow, Zak, & Bjorkman, 2003; Lebron & Bjorkman, 1999; West et al., 2001) as well as our BHK cell system (Byrne et al., 2006). The expression in BHK cells and purification of the N-terminally hexa-His tagged sTFR in our laboratory is accomplished in much the same way as recombinant hTF and generally produces between 30 and 40 mg of protein per production run (Byrne et al., 2006).
6. KINETICS OF IRON RELEASE
Work from the Aisen laboratory provided some of the first mechanistic insights into the kinetics of iron release from hTF at pH 5.6 (Bali & Aisen, 1991, 1992). Specifically, the initial evidence that the TFR slows iron release from hTF at neutral pH while enhancing iron release from hTF at endosomal pH was provided by the polyethyleneglycol (PEG) precipitation studies of Bali and Aisen (Bali et al., 1991). By binding 59Fe to apohTF, these experiments allowed iron release at various pH values to be monitored by PEG precipitation of the 59Fe2hTF (the chelate–iron complex 59Fe–PPi remains in the soluble fraction).
Over a number of years, the laboratory of el Hage Chahine has reported the results of stopped-flow absorbance and pH jump chemical relaxation studies monitoring iron release from serum-derived diferric hTF and monoferric C-lobe hTF in the presence of detergent-solubilized full-length placental-derived TFR (Chahine & Pakdaman, 1995; Hemadi & Ha-Duong, 2006). Discordant results have been obtained in this system in comparison to those of Aisen (Bali & Aisen, 1992) and our own laboratory, as recently discussed in some detail (Steere, Byrne, Chasteen, & Mason, 2012). Specifically, the detergent-solubilized TFR used by the el Hage Chahine laboratory promotes iron release preferentially from the N-lobe of hTF first. We provide evidence (below) that the sTFR promotes iron release preferentially from the C-lobe of hTF first. The numerous differences in the conditions used, including the absence of a chelator and pH values of <4 in the system of el Hage Chahine, may well account for the discrepancy. It appears that further work is required to completely resolve the substantial discrepancy in the findings. Of course, the use of any in vitro system to model what may be occurring in vivo is always subject to a good deal of uncertainty and questions of physiological relevance.
We suggest that, to date, our laboratory has provided the most comprehensive kinetic analysis of iron release from hTF in the absence and presence of the sTFR (Byrne, Chasteen, et al., 2010). Using an optimized stopped-flow iron removal assay, the increase in intrinsic tryptophan fluorescence (Ex. 280 nm) is measured as hTF, alone or in complex with the sTFR in 300 mM KCl is rapidly mixed with our standard iron removal buffer (200 mM MES (2-(N-morpholino)ethanesulfonic acid), pH 5.6 containing 300 mM KCl and 8 mM EDTA). In combination with the ability to produce recombinant hTF with specific properties of iron binding and/or release, the use of the stopped-flow spectrofluorometer has allowed a comprehensive determination of all kinetic steps involved in iron release from hTF in the presence and absence of the sTFR. With all the proper controls and equations, we can confidently extract accurate kinetic constants that provide important information with regard to the detailed mechanism of iron release from various hTF and TFR mutants. Understanding this complex system, especially with regard to the role of the TFR, is crucial to ultimately discovering how to manipulate it.
6.1. Kinetics of Iron Release from hTF in the Absence of the TFR
As mentioned, it is clear from decades of research that iron release from hTF is complex and involves a number of factors including pH, anion binding, chelator, lobe–lobe cooperativity and binding to the TFR. Under pseudo-first-order conditions, iron release from Fe2hTF can occur via two pathways giving rise to four microscopic rate constants, (k1C, k2N, k1N, and k2C) (Bali & Aisen, 1992; Harris, Bali, & Crowley, 1992). The kinetic assessment of diferric locked constructs (LockNhTF and LockChTF) allows the specific assignment of k1C and k1N, while the use of authentic monoferric constructs (FeNhTF and FeChTF) allows the specific assignment of k2N and k2C, respectively. Curve fitting of Fe2hTF shows that in the absence of the sTFR iron release from the N-lobe is very rapid (17.7 ± 2.2/min) and relatively slow from the C-lobe (0.65 ± 0.06/min). Thus, iron is released from Fe2hTF almost exclusively (~96% of the time) via the k1N → k2C pathway, precluding the need to consider the negligible k1C → k2N pathway in the analysis of Fe2hTF data. The validity of these values is further supported through analysis of the LockChTF and FeChTF constructs which provides independent measures of k1N and k2C.
6.1.1. Cooperativity between hTF Lobes
Given the bilobal nature of hTF, it is critical to examine whether cooperativity exists between the two iron-binding sites. Kinetic analysis of FeNhTF and the isolated N-lobe, along with k1N values obtained from the Fe2hTF and LockChTF constructs, indicates that in the absence of the sTFR, the rate of iron release from the N-lobe (kN) is impacted not only by the presence but also the iron occupancy of the C-lobe. However, the rate of iron release from the C-lobe of hTF (kC) is clearly unaffected by the presence or iron status of the N-lobe. Kinetic cooperativity factors are used as a measure of the cooperative effect of iron release in one lobe on the other lobe (Bali & Aisen, 1992; Byrne, Chasteen, et al., 2010). Given as a ratio of k2/k1 of a respective lobe, a number greater than one indicates positive cooperativity between the two lobes, whereas a number equal to or less than one indicates a lack of cooperativity. Based on these determined kinetic values, the calculated kinetic cooperativity figures (k2N/k1N = 24.8/17.9 = 1.4 and k2C/k1C = 0.79/0.72 = 1.1) indicate that iron release from the N-lobe is accelerated by the absence of iron in the C-lobe as well as by the complete absence of the C-lobe, whereas iron occupancy of the N-lobe has little affect on the kinetics of iron release from the C-lobe.
6.1.2. Anion Binding
A complex relationship exists between anion binding to KISAB sites and iron release from hTF. At pH 5.6, anion concentration most dramatically affects the rate of iron release from the N-lobe (increasing it ca. four-fold from 50 to 600 mM [Cl−]) in the absence of the TFR (Byrne, Chasteen, et al., 2010; Byrne, Steere, et al., 2010), while the rate of iron release from the C-lobe is relatively unaffected (Byrne, Chasteen, et al., 2010). The first authentic KISAB site in the N-lobe, residue Arg143, was not definitively identified until recently (Byrne, Steere, et al., 2010). Mutation of Arg143 to alanine completely eliminates iron release from the N-lobe of hTF at all tested chloride concentrations (50–600 mM) in the absence of the TFR. Thus, binding of anions to Arg143 may (at least in part) account for the salt sensitivity of iron release from the N-lobe of hTF. Additionally, mutation of C-lobe residue Lys569 in the Fe2hTF background was previously found to slow iron release by 15- to 20-fold and inhibit chloride enhancement of iron release from the C-lobe (Zak, Tam, MacGillivray, & Aisen, 1997). Therefore, Lys569 was putatively identified as a KISAB site thought to be essential for iron release from the C-lobe of hTF. However, more recently we demonstrated that mutation of Lys569 to alanine (K569A) has little effect on the rate of iron release from Fe2hTF in the absence or presence of the sTFR (Eckenroth et al., 2011). These data are consistent with the finding that the rate of iron release from the C-lobe is relatively unaffected by anion concentration. The discrepancy between more recent and previously published data again highlights the limitations of the steady-state tryptophan fluorescence technique used in the earlier work.
6.2. Kinetics of Iron Release from hTF in the Presence of the TFR
The TFR plays an integral role in physiological iron release from hTF. Therefore, it is absolutely essential that iron release from hTF also be monitored in the presence of the TFR. In accordance with previous findings (Bali & Aisen, 1991, 1992), under our standard conditions, the sTFR induces a switch in the order of iron release from hTF, such that iron is preferentially removed from the C-lobe first followed by the N-lobe (k1C → k2N) (Byrne, Chasteen, et al., 2010). However, the fits of the kinetic data indicate that this is not the case 100% of the time. The k1N → k2C pathway is reduced from 96% in the absence of the sTFR to only 35% in the presence of the sTFR. It is clear that both pathways are physiologically relevant and must be included in the fits of Fe2hTF/sTFR complex. While analyzing Fe2hTF/sTFR complex kinetic data, in order to limit the number of variables, the values for the other pathway (k1N and k2C) are held constant based on kinetic values obtained from the LockChTF/sTFR and FeChTF/sTFR complexes. Again, the values obtained independently for the k1C → k2N pathway from the LockNhTF/sTFR and FeNhTF/sTFR complexes (k1C = 5.0 ± 1.6 and k2N = 1.7 ± 0.6) corroborate the validity of the Fe2hTF/sTFR complex analysis.
Previous studies by the Aisen laboratory clearly demonstrated that the TFR enhances the rate of iron release from the C-lobe of hTF at pH 5.6 (Bali & Aisen, 1991; Zak & Aisen, 2003). Using our standard conditions, the sTFR enhances iron release from the C-lobe by ~7- to 11-fold (k1C, complex/k1C, alone and k2C, complex/k2C, alone) and retards iron release from the N-lobe by 6- to 15-fold (k1N, complex/k1N, alone and k2N, complex/k2N, alone) (Byrne, Chasteen, et al., 2010). Thus, not only does binding to the sTFR switch the order of iron release from hTF but also makes the rates of iron release from the two lobes more equivalent than in the absence of the sTFR. Consequently, the calculated cooperativity factors of each lobe are much different in the presence of the sTFR (N-lobealone = 1.4 versus N-lobecomplex = 0.61 and C-lobealone = 1.1 versus C-lobecomplex = 1.5) (Byrne, Chasteen, et al., 2010). Therefore, in the presence of the sTFR, the rate of iron release from the C-lobe is slowed by the presence of iron in the N-lobe, while for the N-lobe the reverse is true: iron occupancy in the C-lobe accelerates iron release from the N-lobe.
6.2.1. pH Effects
The endosomal pH (~5.6) is a critical component of the endocytic cycle and iron delivery by hTF. It is evident from a pH titration of the FeChTF/ sTFR complex that iron release from the hTF/TFR complex has an apparent pKa of ~5.8 to 5.9 (Steere, Byrne, et al., 2010), a value similar to the pKa of histidine residues. As previously mentioned, His349 in the C-lobe of hTF has been a residue of particular interest for some time (Giannetti et al., 2005). Interestingly, mutation of His349 to alanine or lysine completely abolishes the pH dependence of iron release from the FeChTF/ sTFR complex (Steere, Byrne, et al., 2010). This finding led to the proposal that His349 acts as a pH-inducible switch that accelerates iron release from the C-lobe of hTF. More recently, the effect of the H349A mutation in the Fe2hTF background has been investigated. Curiously, the H349A Fe2hTF/sTFR complex displays three kinetic rates (unlike the Fe2hTF/sTFR control where only two rates, k1C and k2N, are obtained) (Eckenroth et al., 2011). The first rate (k1 = 23.7 ± 4.6/min), ascribed to a rapid conformational change, is very similar to the conformational change observed in both monoferric complexes, as discussed above. However, unlike the Fe2hTF/sTFR control complex, iron release from the H349A Fe2hTF/sTFR complex follows only the k1N → k2C pathway. Interestingly, the rate of iron release from the N-lobe of the H349A Fe2hTF/sTFR complex is nearly doubled (k1N = 6.7 ± 0.3/min) in comparison to the Fe2hTF/sTFR complex (k1N = 2.8 ± 0.8/min) while iron release from the C-lobe of the H349A Fe2hTF/sTFR complex is greatly retarded (k2C = 0.61 ± 0.02/min). Hence, the effect of receptor stimulation on iron release from the C-lobe is completely abrogated in the presence of the H349A mutation (Eckenroth et al., 2011), verifying its crucial role in the mechanism of iron release from the C-lobe of the hTF/TFR complex.
6.2.2. Anion Binding
In contrast to the findings of Egan et al. (1993), we find that at pH 5.6 the effect of anion concentration on iron release from hTF in the presence of the sTFR is significantly muted (Byrne, Chasteen, et al., 2010). Therefore, although a slight salt-induced acceleration in iron removal from the hTF/ TFR complex is observed at pH 5.6, the overriding kinetic effect of the TFR appears to outweigh any kinetic anion effects. In accordance with these findings (Byrne, Chasteen, et al., 2010), the effect of the Arg143 mutation (described above) is largely abolished in the presence of the TFR (Byrne, Steere, et al., 2010).
7. STRUCTURE OF THE HTF/TFR COMPLEX
Given that the hTF/TFR interaction promotes conformational changes required for iron release, the molecular details of the interactions between the two proteins is clearly critical. Although transferrin has been studied extensively since the late 1940s and the existence of the TFR was confirmed in the 1970s, the precise structural details of the hTF/TFR interaction, as well as the functional implications of those interactions, have remained unclear. Therefore, identification of the contacts formed between the two proteins is critical. Studying the hTF/TFR interaction in the past was hindered by a lack of structural information on the complex. In 2004, the first model of the TF/sTFR complex was completed. A 7.5-Å resolution cryo-electron microscopy model of the TF/sTFR complex utilized the crystal structure of the unliganded sTFR and individually modeled in the structures of the isolated human N-lobe and rabbit C-lobe (~85% similar to the human C-lobe). The model indicated that the N-lobe is situated between the membrane and the TFR, while the C-lobe makes significant contacts with the helical domain of the TFR (PDB ID: 1SUV) (Cheng, Zak, Aisen, Harrison, & Walz, 2004). However, confidence in the model is limited by the relatively low resolution and the fact that placement of the N-lobe into the model required an ~9 Å shift with respect to the C-lobe (Cheng et al., 2004; Sakajiri, Yamamura, Kikuchi, & Yajima, 2009). More recently, the inclusion of the available structure of the apohTF (Wally et al., 2006) in addition to consideration of various mutagenesis studies of hTF and the TFR led to a new in silico model of hTF bound to the TFR (Sakajiri et al., 2009). This computational model eliminated the need for the 9 Å gap between the N- and C-lobes of hTF and appeared to be consistent with all available mutagenesis data from both the TFR and the hTF (Giannetti et al., 2003; Mason et al., 2009).
The recently published high-resolution (3.22 Å) crystal structure of the FeNhTF/sTFR complex (Eckenroth et al., 2011) provided precise molecular details of the interaction between the two binding partners. Three distinct binding motifs between hTF and the TFR are observed: the hTF N1-TFR motif, the hTF N2-TFR motif and the hTF C1-TFR motif. Moreover, the structure clearly illustrates how binding to the TFR hinders iron release from the N-lobe while promoting iron release from the C-lobe. As in the structure of the hemochromatosis protein (HFE) and the TFR (Bennett, Lebron, & Bjorkman, 2000), the FeNhTF/sTFR complex structure definitively showed that conformational changes in the TFR occur as a result of ligand binding. Importantly, the insight gained from the recent crystal structure has provided new mutagenic targets in both the hTF and the TFR. By assessing various single-point mutants, we have been able to identify residues within both hTF and the sTFR that are critical to the receptor–ligand binding interaction and also influence the kinetics of iron release from hTF.
7.1. Features of the FeNhTF/sTFR Structure
An interesting feature of the FeNhTF/sTFR crystal structure was the presence of a Ca2+ ion bound between the apical and protease-like domains of the sTFR. Highlighting the structural stability provided by this Ca2+ ion, mutation of two of the coordinating amino acids (E465A/E468A) produces an unstable aggregated sTFR (Steere, Chasteen, et al., 2012). A number of intriguing questions remain with regard to this calcium ion in the sTFR. First, although the coordinated metal in sTFR produced in our BHK system was definitively identified as calcium by inductively coupled plasma mass spectrometry (Eckenroth et al., 2011), calcium may not be the physiologically relevant metal. It is possible that this metal-binding site in the sTFR may be occupied by some other metal ion (Zn2+ or Mg2+ for example) in vivo. Additionally, the identity of the physiological chelator of iron within the endosome remains unknown. We have previously shown that decreased pH (~5.6) alone is not sufficient to remove iron from hTF or the hTF/ sTFR complex and that the presence of a chelator is indeed required to promote iron release in a physiologically relevant time frame (Byrne, Chasteen, et al., 2010). Although abundant small molecules such as citrate or PPi have been suggested, to date, the endosomal chelator has not been definitively identified. Given the presence of another chelatable metal ion other than iron, the question of whether this other metal (putatively Ca2+) is chelated within the endosome remains.
One especially interesting feature of the FeNhTF/sTFR structure was a large intersection formed between the apical and the protease-like domains of one TFR monomer, the helical domain of the other TFR monomer and the C1 subdomain of hTF (TFR–TFR′–C1 subdomain intersection) (Eckenroth et al., 2011). The significance of this intersection is emphasized by the large conformational changes in this region of the TFR that take place upon hTF binding. Furthermore, given the pH range experienced by hTF throughout the endocytic cycle (between ~7.5 and 5.5), it is not entirely surprising that histidine residues in both hTF (Eckenroth et al., 2011; Steere, Byrne, et al., 2010) and the sTFR (Steere, Chasteen, et al., 2012) within this TFR–TFR′–C1 subdomain intersection participate in pH-dependent receptor-stimulated iron release from the C-lobe of hTF. Through a series of interrelated pH-induced events involving His residues within the TFR–TFR′–C1 subdomain intersection iron release from the C-lobe of hTF is triggered. Specifically, protonation of sTFR residue His318 promotes a pH-inducible change in the sTFR that is communicated to the nearby C-terminus (residues 757–760) of the other TFR monomer (TFR′). Meanwhile, due to its protonation and a related conformational change in hTF, His349 (hTF) interacts with the C-terminus of the TFR′ and ultimately promotes iron release from the C-lobe of the hTF/sTFR complex (Steere, Chasteen, et al., 2012).
It is well established that highly proliferative cells have dramatically increased iron requirements and therefore an increased number of TFRs, making the TFR a prime candidate for targeted therapeutics (Daniels et al., 2011). Hence, the molecular details of how the TFR interacts with its ligand, hTF, are critical and could be used in order to develop small molecules or toxin conjugates to target the hTF/TFR pathway in cancer cells. Moreover, identification of specific residues that account for the pH-sensitive binding affinity of hTF throughout the endocytic cycle could be essential to delivery of therapeutics inside the cell. We are slowly identifying specific residues and/or structural elements within both the hTF and the TFR that directly correlate to events that occur during iron release.
In conclusion, after over a half century of research on transferrin, much has been learned (as detailed in the recent Biochimica Biophysica Acta Special Issue titled Transferrins: Molecular Mechanisms of Iron Transport and Disorders), but many interesting and important questions remain. Although the existence of the TFR was confirmed in the 1970s, the precise structural details of the hTF/TFR interaction, as well as the implications of those interactions on cellular iron delivery, have remained unclear. Collectively, these results help to establish a molecular basis for the pH-induced events that dictate efficient release of iron from each lobe within the endosome in a physiologically relevant time frame. We now enjoy a better understanding of how the TFR is able to influence iron release from each lobe of hTF at endosomal pH. Appreciation that the TFR is more than a passive binding partner and that it trumps other factors is emphasized by recent work.
Acknowledgments
This work was supported by U.S. Public Service Grant R01 DK 21739 to A.B.M. and an American Heart Association Predoctoral Fellowship (10PRE4200010) to A.N.S. We wish to thank Drs N. D. Chasteen, Brian E. Eckenroth, Shaina L. Byrne and Nicholas G. James for their substantive contributions that helped to provide the basis for some of this work.
References
- Aisen P. The International Journal of Biochemistry and Cell Biology. 2004;36:2137–2143. doi: 10.1016/j.biocel.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Aisen P, Enns C, Wessling-Resnick M. The International Journal of Biochemistry and Cell Biology. 2001;33:940–959. doi: 10.1016/s1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- Aisen P, Leibman A, Zweier J. The Journal of Biological Chemistry. 1978;253:1930–1937. [PubMed] [Google Scholar]
- Ali SA, Joao HC, Csonga R, Hammerschmid F, Steinkasserer A. The Biochemical Journal. 1996;319(Pt 1):191–195. doi: 10.1042/bj3190191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson BF, Baker HM, Dodson EJ, Norris GE, Rumball SV, Waters JM, et al. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:1769–1773. doi: 10.1073/pnas.84.7.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews NC, Schmidt PJ. Annual Review of Physiology. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- Arosio P, Levi S. Biochimica et Biophysica Acta. 2010;1800:783–792. doi: 10.1016/j.bbagen.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Baker EN. Advances in Inorganic Chemistry. 1994;41:389–463. [Google Scholar]
- Baldwin DA. Biochimica et Biophysica Acta. 1980;623:183–198. doi: 10.1016/0005-2795(80)90020-3. [DOI] [PubMed] [Google Scholar]
- Baldwin DA, de Sousa DM. Biochemical and Biophysical Research Communications. 1981;99:1101–1107. doi: 10.1016/0006-291x(81)90732-4. [DOI] [PubMed] [Google Scholar]
- Bali PK, Aisen P. Biochemistry. 1991;30:9947–9952. doi: 10.1021/bi00105a019. [DOI] [PubMed] [Google Scholar]
- Bali PK, Aisen P. Biochemistry. 1992;31:3963–3967. doi: 10.1021/bi00131a011. [DOI] [PubMed] [Google Scholar]
- Bali PK, Harris WR. Archives of Biochemistry and Biophysics. 1990;281:251–256. doi: 10.1016/0003-9861(90)90440-a. [DOI] [PubMed] [Google Scholar]
- Bali PK, Zak O, Aisen P. Biochemistry. 1991;30:324–328. doi: 10.1021/bi00216a003. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Lebron JA, Bjorkman PJ. Nature. 2000;403:46–53. doi: 10.1038/47417. [DOI] [PubMed] [Google Scholar]
- Bou-Abdallah F, McNally J, Liu XX, Melman A. Chemical Communications. 2011;47:731–733. doi: 10.1039/c0cc03454a. [DOI] [PubMed] [Google Scholar]
- Brandsma ME, Diao H, Wang X, Kohalmi SE, Jevnikar AM, Ma S. Plant Biotechnology Journal. 2010;8:489–505. doi: 10.1111/j.1467-7652.2010.00499.x. [DOI] [PubMed] [Google Scholar]
- Byrne SL, Chasteen ND, Steere AN, Mason AB. Journal of Molecular Biology. 2010;396:130–140. doi: 10.1016/j.jmb.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne SL, Leverence R, Klein JS, Giannetti AM, Smith VC, MacGillivray RT, et al. Biochemistry. 2006;45:6663–6673. doi: 10.1021/bi0600695. [DOI] [PubMed] [Google Scholar]
- Byrne SL, Mason AB. Journal of Biological Inorganic Chemistry. 2009;14:771–781. doi: 10.1007/s00775-009-0491-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne SL, Steere AN, Chasteen ND, Mason AB. Biochemistry. 2010;49:4200–4207. doi: 10.1021/bi1003519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JC, Chasteen ND. Biochemistry. 1975;14:4573–4577. doi: 10.1021/bi00692a003. [DOI] [PubMed] [Google Scholar]
- Chahine JMEH, Pakdaman R. European Journal of Biochemistry. 1995;230:1102–1110. doi: 10.1111/j.1432-1033.1995.tb20661.x. [DOI] [PubMed] [Google Scholar]
- Chasteen ND. Advances in Inorganic Biochemistry. 1983;5:201–233. [PubMed] [Google Scholar]
- Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- Collawn JF, Lai A, Domingo D, Fitch M, Hatton S, Trowbridge IA. The Journal of Biological Chemistry. 1993;268:21686–21692. [PubMed] [Google Scholar]
- Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, et al. Biochimica et Biophysica Acta. 2011;1820:291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. Clinical Immunology. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Delano WL. The PyMOL Molecular Graphics System. 2002. [Google Scholar]
- de Smit DH, Hoefkens P, de Jong G, van Duin J, van Knippenberg PH, van Eijk HG. The International Journal of Biochemistry and Cell Biology. 1995;27:839–850. doi: 10.1016/1357-2725(95)00040-v. [DOI] [PubMed] [Google Scholar]
- Dewan JC, Mikami B, Hirose M, Sacchettini JC. Biochemistry. 1993;32:11963–11968. doi: 10.1021/bi00096a004. [DOI] [PubMed] [Google Scholar]
- Dhungana S, Taboy CH, Zak O, Larvie M, Crumbliss AL, Aisen P. Biochemistry. 2004;43:205–209. doi: 10.1021/bi0353631. [DOI] [PubMed] [Google Scholar]
- Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, et al. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- Eckenroth BE, Mason AB, McDevitt ME, Lambert LA, Everse SJ. Protein Science. 2010;19:1616–1626. doi: 10.1002/pro.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenroth BE, Steere AN, Chasteen ND, Everse SJ, Mason AB. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13089–13094. doi: 10.1073/pnas.1105786108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TJ, Ross DC, Purves LR, Adams PA. Inorganic Chemistry. 1992;31:1994–1998. [Google Scholar]
- Egan TJ, Zak O, Aisen P. Biochemistry. 1993;32:8162–8167. doi: 10.1021/bi00083a016. [DOI] [PubMed] [Google Scholar]
- Enns CA, Sussman HH. The Journal of Biological Chemistry. 1981;256:9820–9823. [PubMed] [Google Scholar]
- Fenton HJH. Chemical News. 1876;33:190. [Google Scholar]
- Fenton HJH. Chemical Society Proceedings. 1893;9:113. [Google Scholar]
- Finnis CJ, Payne T, Hay J, Dodsworth N, Wilkinson D, Morton P, et al. Microbial Cell Factories. 2010;9:87. doi: 10.1186/1475-2859-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming MD, Trenor CC, 3rd, Su MA, Foernzler D, Beier DR, Di etrich WF, et al. Nature Genetics. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- Folajtar DA, Chasteen ND. Journal of the American Chemical Society. 1982;104:5775–5780. [Google Scholar]
- Funk WD, MacGillivray RT, Mason AB, Brown SA, Woodworth RC. Biochemistry. 1990;29:1654–1660. doi: 10.1021/bi00458a043. [DOI] [PubMed] [Google Scholar]
- Gaber BP, Miskowski V, Spiro TG. Journal of the American Chemical Society. 1974;96:6868–6873. doi: 10.1021/ja00829a010. [DOI] [PubMed] [Google Scholar]
- Giannetti AM, Halbrooks PJ, Mason AB, Vogt TM, Enns CA, Bjorkman PJ. Structure. 2005;13:1613–1623. doi: 10.1016/j.str.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Giannetti AM, Snow PM, Zak O, Bjorkman PJ. PLoS Biology. 2003;1:341–350. doi: 10.1371/journal.pbio.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady JK, Mason AB, Woodworth RC, Chasteen ND. The Biochemical Journal. 1995;309(Pt 2):403–410. doi: 10.1042/bj3090403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD, Donaldson JG. Nature Reviews Molecular Cell Biology. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg GR, Wintrobe MM. The Journal of Biological Chemistry. 1946;165:397. [PubMed] [Google Scholar]
- Gubler CJ, Lahey ME, Chase MS, Cartwright GE, Wintrobe MM. Blood. 1952;7:1075–1092. [PubMed] [Google Scholar]
- Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Robine S, Andrews NC. The Journal of Clinical Investigation. 2005;115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, et al. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- Halbrooks PJ, He QY, Briggs SK, Everse SJ, Smith VC, MacGillivray RT, et al. Biochemistry. 2003;42:3701–3707. doi: 10.1021/bi027071q. [DOI] [PubMed] [Google Scholar]
- Hall DR, Hadden JM, Leonard GA, Bailey S, Neu M, Winn M, et al. Acta Crystallographica Section D, Biological Crystallography. 2002;58:70–80. doi: 10.1107/s0907444901017309. [DOI] [PubMed] [Google Scholar]
- Hallberg L, Bjorn-Rasmussen E, Howard L, Rossander L. Scandinavian Journal of Gastroenterology. 1979;14:769–779. doi: 10.3109/00365527909181403. [DOI] [PubMed] [Google Scholar]
- Harris WR. Biochemistry. 1985;24:7412–7418. doi: 10.1021/bi00346a057. [DOI] [PubMed] [Google Scholar]
- Harris WR, Bali PK. Inorganic Chemistry. 1988;27:2687–2691. [Google Scholar]
- Harris WR, Bali PK, Crowley MM. Inorganic Chemistry. 1992;31:2700–2705. [Google Scholar]
- Harris WR, Cafferty AM, Abdollahi S, Trankler K. Binding of monovalent anions to human serum transferrin. Biochimica et Biophysica Acta. 1998;1383:197–210. doi: 10.1016/s0167-4838(97)00152-0. [DOI] [PubMed] [Google Scholar]
- Harris WR, Nesset-Tollefson D, Stenback JZ, Mohamed-Hani N. Journal of Inorganic Biochemistry. 1990;38:175–183. doi: 10.1016/0162-0134(90)84011-d. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Arosio P. Biochimica et Biophysica Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Hayes GR, Williams AM, Lucas JJ, Enns CA. Biochemistry. 1997;36:5276–5284. doi: 10.1021/bi962884y. [DOI] [PubMed] [Google Scholar]
- He Q-Y, Mason AB. In: Templeton DM, editor. New York: Marcel Dekker, Inc; 2002. pp. 95–124. [Google Scholar]
- Hemadi M, Ha-Duong NT, el Hage Chahine JM. Journal of Molecular Biology. 2006;358:1125–1136. doi: 10.1016/j.jmb.2006.02.055. [DOI] [PubMed] [Google Scholar]
- Hershberger CL, Larson JL, Arnold B, Rosteck PR, Jr, Williams P, DeHoff B, et al. Annals of the New York Academy of Sciences. 1991;646:140–154. doi: 10.1111/j.1749-6632.1991.tb18573.x. [DOI] [PubMed] [Google Scholar]
- Hoefkens P, de Smit MH, de Jeu-Jaspars NM, Huijskes-Heins MI, de Jong G, van Eijk HG. The International Journal of Biochemistry and Cell Biology. 1996;28:975–982. doi: 10.1016/1357-2725(96)00057-x. [DOI] [PubMed] [Google Scholar]
- Huebers HA, Josephson B, Huebers E, Csiba E, Finch CA. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:4326–4330. doi: 10.1073/pnas.81.14.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda RA, Bowman BH, Yang F, Lokey LK. Gene. 1992;117:265–269. doi: 10.1016/0378-1119(92)90737-a. [DOI] [PubMed] [Google Scholar]
- Jacobs A. Blood. 1977;50:433–439. [PubMed] [Google Scholar]
- Jandl JH, Katz JH. The Journal of Clinical Investigation. 1963;42:314–326. doi: 10.1172/JCI104718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey PD, Bewley MC, MacGillivray RT, Mason AB, Woodworth RC, Baker EN. Biochemistry. 1998;37:13978–13986. doi: 10.1021/bi9812064. [DOI] [PubMed] [Google Scholar]
- Kakhlon O, Cabantchik ZI. Free Radical Biology and Medicine. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- Kaplan J, O’Halloran TV. Science. 1996;271:1510–1512. doi: 10.1126/science.271.5255.1510. [DOI] [PubMed] [Google Scholar]
- Kretchmar SA, Raymond KN. Inorganic Chemistry. 1988;27:1436–1441. [Google Scholar]
- Kubal G, Mason AB, Patel SU, Sadler PJ, Woodworth RC. Biochemistry. 1993;32:3387–3395. doi: 10.1021/bi00064a024. [DOI] [PubMed] [Google Scholar]
- Kuhn LC, McClelland A, Ruddle FH. Cell. 1984;37:95–103. doi: 10.1016/0092-8674(84)90304-0. [DOI] [PubMed] [Google Scholar]
- Lahey ME, Gubler CJ, Chase MS, Cartwright GE, Wintrobe MM. Blood. 1952;7:1053–1074. [PubMed] [Google Scholar]
- Lambert LA, Perri H, Halbrooks PJ, Mason AB. Comparative Biochemistry and Physiology Part B, Biochemistry and Molecular Biology. 2005;142:129–141. doi: 10.1016/j.cbpb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Laufberger V. Bulletin de la Société de Chimie Biologique. 1937. pp. 1575–1582. [Google Scholar]
- Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC. Science. 1999;286:779–782. doi: 10.1126/science.286.5440.779. [DOI] [PubMed] [Google Scholar]
- Lebron JA, Bjorkman PJ. Journal of Molecular Biology. 1999;289:1109–1118. doi: 10.1006/jmbi.1999.2842. [DOI] [PubMed] [Google Scholar]
- Lehrer SS. The Journal of Biological Chemistry. 1969;244:3613–3617. [PubMed] [Google Scholar]
- Leibman A, Aisen P. Biochemistry. 1977;16:1268–1272. doi: 10.1021/bi00626a004. [DOI] [PubMed] [Google Scholar]
- Leibman A, Aisen P. Blood. 1979;53:1058–1065. [PubMed] [Google Scholar]
- Lestas AN. British Journal of Haematology. 1976;32:341–350. doi: 10.1111/j.1365-2141.1976.tb00937.x. [DOI] [PubMed] [Google Scholar]
- Leverence R, Mason AB, Kaltashov IA. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8123–8128. doi: 10.1073/pnas.0914898107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Nature Genetics. 1999;21:396–399. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- MacGillivray RT, Moore SA, Chen J, Anderson BF, Baker H, Luo Y, et al. Biochemistry. 1998;37:7919–7928. doi: 10.1021/bi980355j. [DOI] [PubMed] [Google Scholar]
- MacGillivray RTA, Mason AB, Templeton DM. New York: Marcel Dekker, Inc; 2002. pp. 41–69. [Google Scholar]
- Makey DG, Seal US. Biochimica et Biophysica Acta. 1976;453:250–256. doi: 10.1016/0005-2795(76)90270-1. [DOI] [PubMed] [Google Scholar]
- Marques HM, Egan TJ, Pattrick G. South African Journal of Science. 1990;86:21–24. [Google Scholar]
- Marques HM, Watson DL, Egan TJ. Inorganic Chemistry. 1991;30:3758–3762. [Google Scholar]
- Mason AB, Byrne SL, Everse SJ, Roberts SE, Chasteen ND, Smith VC, et al. Journal of Molecular Recognition. 2009;22:521–529. doi: 10.1002/jmr.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason AB, Funk WD, MacGillivray RT, Woodworth RC. Protein Expression and Purification. 1991;2:214–220. doi: 10.1016/1046-5928(91)90074-s. [DOI] [PubMed] [Google Scholar]
- Mason AB, Halbrooks PJ, Larouche JR, Briggs SK, Moffett ML, Ramsey JE, et al. Protein Expression and Purification. 2004;36:318–326. doi: 10.1016/j.pep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Mason AB, Miller MK, Funk WD, Banfield DK, Savage KJ, Oliver RW, et al. Biochemistry. 1993;32:5472–5479. doi: 10.1021/bi00071a025. [DOI] [PubMed] [Google Scholar]
- Mason AB, Tam BM, Woodworth RC, Oliver RW, Green BN, Lin LN, et al. The Biochemical Journal. 1997;326(Pt 1):77–85. doi: 10.1042/bj3260077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason AB, Woodworth RC, Oliver RW, Green BN, Lin LN, Brandts JF, et al. Protein Expression and Purification. 1996;8:119–125. doi: 10.1006/prep.1996.0081. [DOI] [PubMed] [Google Scholar]
- McCance RA, Widdowson EM. Journal of Physiology. 1938;94:148–154. doi: 10.1113/jphysiol.1938.sp003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland A, Kuhn LC, Ruddle FH. Cell. 1984;39:267–274. doi: 10.1016/0092-8674(84)90004-7. [DOI] [PubMed] [Google Scholar]
- Mizutani K, Hashimoto K, Takahashi N, Hirose M, Aibara S, Mikami B. Bioscience, Biotechnology and Biochemistry. 2010;74:309–315. doi: 10.1271/bbb.90635. [DOI] [PubMed] [Google Scholar]
- Morgan EH. Biochimica et Biophysica Acta. 1981;642:119–134. doi: 10.1016/0005-2736(81)90143-7. [DOI] [PubMed] [Google Scholar]
- Morgan EH, Appleton TC. Nature. 1969;223:1371–1372. doi: 10.1038/2231371a0. [DOI] [PubMed] [Google Scholar]
- Noinaj N, Easley NC, Oke M, Mizuno N, Gumbart J, Boura E, et al. Nature. 2012;483:53–58. doi: 10.1038/nature10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, et al. Nature Genetics. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD, Behringer RR, Quaife CJ, Maxwell F, Maxwell IH, Brinster RL. Cell. 1987;50:435–443. doi: 10.1016/0092-8674(87)90497-1. [DOI] [PubMed] [Google Scholar]
- Park CH, Bacon BR, Brittenham GM, Tavill AS. Laboratory Investigation. 1987;57:555–563. [PubMed] [Google Scholar]
- Park I, Schaeffer E, Sidoli A, Baralle FE, Cohen GN, Zakin MM. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3149–3153. doi: 10.1073/pnas.82.10.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch MG, Carrano CJ. Inorganica Chimica Acta. 1981;56:L71–L73. [Google Scholar]
- Pecoraro VL, Harris WR, Carrano CJ, Raymond KN. Biochemistry. 1981;20:7033–7039. doi: 10.1021/bi00527a040. [DOI] [PubMed] [Google Scholar]
- Ponka P, Borova J, Neuwirt J, Fuchs O. FEBS Letters. 1979;97:317–321. doi: 10.1016/0014-5793(79)80111-8. [DOI] [PubMed] [Google Scholar]
- Posey JE, Gherardini FC. Science. 2000;288:1651–1653. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- Price EM, Gibson JF. The Journal of Biological Chemistry. 1972a;247:8031–8035. [PubMed] [Google Scholar]
- Price EM, Gibson JF. Biochemical and Biophysical Research Communications. 1972b;46:646–651. doi: 10.1016/s0006-291x(72)80189-x. [DOI] [PubMed] [Google Scholar]
- Princiotto JV, Zapolski EJ. Nature. 1975;255:87–88. doi: 10.1038/255087a0. [DOI] [PubMed] [Google Scholar]
- Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, et al. Cell. 2006;127:917–928. doi: 10.1016/j.cell.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Raffin SB, Woo CH, Roost KT, Price DC, Schmid R. The Journal of Clinical Investigation. 1974;54:1344–1352. doi: 10.1172/JCI107881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regoeczi E, Bolyos M, Chindemi PA. Archives of Biochemistry and Biophysics. 1989;268:637–642. doi: 10.1016/0003-9861(89)90331-7. [DOI] [PubMed] [Google Scholar]
- Retzer MD, Kabani A, Button LL, Yu RH, Schryvers AB. The Journal of Biological Chemistry. 1996;271:1166–1173. doi: 10.1074/jbc.271.2.1166. [DOI] [PubMed] [Google Scholar]
- Sakajiri T, Yamamura T, Kikuchi T, Yajima H. Protein Journal. 2009;28:407–414. doi: 10.1007/s10930-009-9208-x. [DOI] [PubMed] [Google Scholar]
- Schade AL, Reinhart RW, Levy H. Archives of Biochemistry. 1949;20:170–172. [PubMed] [Google Scholar]
- Schlabach MR, Bates GW. The Journal of Biological Chemistry. 1975;250:2182–2188. [PubMed] [Google Scholar]
- Schneider C, Owen MJ, Banville D, Williams JG. Nature. 1984;311:675–678. doi: 10.1038/311675b0. [DOI] [PubMed] [Google Scholar]
- Seligman PA, Schleicher RB, Allen RH. The Journal of Biological Chemistry. 1979;254:9943–9946. [PubMed] [Google Scholar]
- Sharp P, Srai SK. World Journal of Gastroenterology. 2007;13:4716–4724. doi: 10.3748/wjg.v13.i35.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PA. International Journal for Vitamin and Nutrition Research. 2010;80:231–242. doi: 10.1024/0300-9831/a000029. [DOI] [PubMed] [Google Scholar]
- Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, et al. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- Sheftel AD, Mason AB, Ponka P. Biochimica et Biophysica Acta. 2012;1820:161–187. doi: 10.1016/j.bbagen.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AN, Byrne SL, Chasteen ND, Mason AB. Biochimica et Biophysica Acta. 2012;1820:326–333. doi: 10.1016/j.bbagen.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AN, Byrne SL, Chasteen ND, Smith VC, MacGillivray RT, Mason AB. Journal of Biological Inorganic Chemistry. 2010;15:1341–1352. doi: 10.1007/s00775-010-0694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AN, Chasteen ND, Miller BF, Smith VC, Macgillivray RT, Mason AB. Biochemistry. 2012;51:2113–2121. doi: 10.1021/bi3001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AN, Roberts SE, Byrne SL, Chasteen ND, Bobst CE, Kaltashov IA, et al. Protein Expression and Purification. 2010;72:32–41. doi: 10.1016/j.pep.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein LM, Graf TN, Ikeda RA. Protein Expression and Purification. 1995;6:619–624. doi: 10.1006/prep.1995.1081. [DOI] [PubMed] [Google Scholar]
- Steinlein LM, Ikeda RA. Enzyme and Microbial Technology. 1993;15:193–199. doi: 10.1016/0141-0229(93)90137-q. [DOI] [PubMed] [Google Scholar]
- Subramaniam VN, Summerville L, Wallace DF. Cell Biochemistry and Biophysics. 2002;36:235–239. doi: 10.1385/CBB:36:2-3:235. [DOI] [PubMed] [Google Scholar]
- Sun H, Li H, Sadler PJ. Chemical Review. 1999;99:2817–2842. doi: 10.1021/cr980430w. [DOI] [PubMed] [Google Scholar]
- Theil EC, Goss DJ. Chemical Review. 2009;109:4568–4579. doi: 10.1021/cr900052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CP, McCarty BM, Chasteen ND. Biochimica et Biophysica Acta. 1986;870:530–537. doi: 10.1016/0167-4838(86)90262-1. [DOI] [PubMed] [Google Scholar]
- Turkewitz AP, Amatruda JF, Borhani D, Harrison SC, Schwartz AL. The Journal of Biological Chemistry. 1988;263:8318–8325. [PubMed] [Google Scholar]
- Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, et al. Nature Genetics. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- Wally J, Halbrooks PJ, Vonrhein C, Rould MA, Everse SJ, Mason AB, et al. The Journal of Biological Chemistry. 2006;281:24934–24944. doi: 10.1074/jbc.M604592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver J, Pollack S. The Biochemical Journal. 1989;261:787–792. doi: 10.1042/bj2610787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Jr, Giannetti AM, Herr AB, Bennett MJ, Nangiana JS, Pierce JR, et al. Journal of Molecular Biology. 2001;313:385–397. doi: 10.1006/jmbi.2001.5048. [DOI] [PubMed] [Google Scholar]
- Williams J, Chasteen ND, Moreton K. The Biochemical Journal. 1982;201:527–532. doi: 10.1042/bj2010527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J, Moreton K. The Biochemical Journal. 1980;185:483–488. doi: 10.1042/bj1850483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lum JB, McGill JR, Moore CM, Naylor SL, van Bragt PH, et al. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:2752–2756. doi: 10.1073/pnas.81.9.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak O, Aisen P. Biochemistry. 2002;41:1647–1653. doi: 10.1021/bi015868w. [DOI] [PubMed] [Google Scholar]
- Zak O, Aisen P. Biochemistry. 2003;42:12330–12334. doi: 10.1021/bi034991f. [DOI] [PubMed] [Google Scholar]
- Zak O, Tam B, MacGillivray RT, Aisen P. Biochemistry. 1997;36:11036–11043. doi: 10.1021/bi970628v. [DOI] [PubMed] [Google Scholar]
- Zhang D, Nandi S, Bryan P, Pettit S, Nguyen D, Santos MA, et al. Protein Expression and Purification. 2010;74:69–79. doi: 10.1016/j.pep.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccola . The crystal structure of monoferric human serum transferrin. Atlanta, GA: Georgia Institute of Technology; 1993. [Google Scholar]