

Abstract
Neutrophil serine proteases (NSPs), especially elastase, are major agents of lung destruction in cystic fibrosis (CF) patients. This study investigated SerpinB1, a highly efficient inhibitor of NSPs, in CF lung disease.
Bronchoalveolar lavage fluid (BALF) from 31 children with CF and 24 control children was examined for amount and molecular species of SerpinB1, and its mechanism of action was studied.
CF BALF had more SerpinB1 than control BALF (geometric mean 3.9 (95% CI 2.60–5.62) versus 1.37 (1.20–1.55) μg·mL−1; p<0.001). BALF levels of SerpinB1 were higher for infected versus uninfected CF subjects (5.5 versus 2.7 μg·mL−1; p<0.004) and substantially higher for elastase-positive versus -negative CF subjects (8.41 versus 1.89 μg·mL−1; p<0.001). Most SerpinB1 in CF BALF had been cleaved. Adding recombinant SerpinB1 to CF BALF stoichiometrically inhibited endogenous elastase, indicating that the inhibitor functions in the CF microenvironment. In vitro simulations comparing SerpinB1 and α1-antitrypsin (SerpinA1) showed that both rapidly form irreversible inhibitory covalent complexes with elastase and that these differed in survival time. The SerpinB1–elastase complex survived only briefly due to fragmentation of bound elastase, releasing cleaved SerpinB1, the molecular form in CF BALF.
The findings define an innate role for SerpinB1 in CF airways.
Keywords: Cystic fibrosis, elastase, neutrophil serine proteases, protease inhibitors, Serpin
Neutrophils and neutrophil serine proteases (NSPs) play key roles in antimicrobial lung defense, but in situations of protease excess, they act as major agents of inflammation and injury. The NSPs, elastase, cathepsin G and proteinase-3, are high-turnover enzymes carried in circulating neutrophils [1]. Their major protective function is killing bacteria, and although NETs (neutrophil extracellular traps) play a role [2], killing of bacteria by NSPs primarily takes place intracellularly after the bacteria are taken up in phagosomes that fuse with NSP-containing granules [3]. NSPs are released when neutrophils degranulate or die by necrosis. Released NSPs can be rapidly inactivated by protease inhibitors, which is the usual outcome in plasma, but in the extravascular space, local conditions, such as inhibitor deficiency or excess of NSPs, can extend their functional life in time and space. Active NSPs, especially elastase, are frequently found in cystic fibrosis (CF) airway fluid, even in patients with mild disease [4, 5]. Extracellular actions of NSPs are largely pathologic. NSPs degrade structural proteins contributing to bronchiolectasis and bronchiectasis [6], exacerbate airway dysfunction by increasing mucin release [7, 8], propagate inflammation by inducing pro-inflammatory cytokines [91 and diminish antimicrobial defense by cleaving phagocyte receptors, antibodies, complement and surfactant proteins [10 14].
The frequent presence of active elastase in CF indicates that the naturally occurring protease inhibitors are collectively inadequate as an antiprotease shield. Nonetheless, it is relevant that we learn about endogenous inhibitors so that their levels can be therapeutically modulated and/or recombinant protein therapies developed. Several NSP inhibitors have been studied in CF, including SLPI (secretory leukocyte elastase inhibitor), elafin and, particularly, α1-antitrypsin (α1-AT; SerpinA1), and both SLPI and α1-AT, as well as synthetic NSP inhibitors, have been studied as potential therapeutics (reviewed in [15, 16]). Largely absent from these lists is SerpinB1, also known as MNEI (monocyte–neutrophil elastase inhibitor), a broadly expressed inhibitor found at especially high levels in myeloid cells. SerpinB1/MNEI is among the most efficient naturally occurring inhibitors of NSPs [17]. It was the goal of this study to determine the contribution of SerpinB1 to NSP regulation in CF.
MATERIALS AND METHODS
Bronchoalveolar lavage fluids
Bronchoalveolar lavage fluids (BALFs) were obtained with informed written consent and Institutional Review Board (The Children’s Hospital, Denver, CO, USA) approval as part of a cross-sectional study of airway inflammation and infection in CF. Bronchoalveolar lavage was performed at Pediatric Pulmonary Medicine (The Children’s Hospital) [14, 18] by instilling a total of 3 mL·kg−1 sterile saline for infants and 80 mL for older patients. Aliquots of the BALF were utilised for total and differential cell counts, and for quantitative bacterial and viral culture as described previously [18]. The remaining BALF was clarified by centrifugation and immediately frozen in aliquots at −80°C.
To study SerpinB1, CF BALF (31 patients) and disease-control BALF (24 patients) were randomly drawn from the panel. The disease-control lavage had been performed for clinical indications in patients with chronic cough (n=5), asthma (n=4), interstitial lung disease n=(4), pulmonary haemosiderosis (n=2), immunodeficiency (n=2) and chronic wheezing, neuro-muscular disease, hypoxaemia, pneumonia, chronic aspiration, suspected aspiration and haemorrhage (n=1 for each). Information on patients and clinical specimens is summarised in table 1. Comparable urea levels indicate similar dilution factors in the two groups. Two additional BALF specimens (CF-32 and CF-33) were studied for inhibition of endogenous elastase by SerpinB1; these contained 15.8 and 34.3 μg·mL−1 elastase, respectively.
Table 1.
Subjects and cystic fibrosis (CF) bronchoalveolar lavage fluid (BALF) specimens
| Characteristic | Control patients | CF patients |
|---|---|---|
| Subjects n | 24 | 31 |
| Age yrs | 8.9 (5.0–11.9) | 9.3 (4.8–16.6) |
| White blood cells × 103 cells·mL−1 | 262 (185–390) | 524 (287–2015)¶ |
| Neutrophils × 103 cells·mL−1 | 8.8 (3.8–36) | 230 (37–1401)¶ |
| Lymphocytes × 103 cells·mL−1 | 33 (11–86) | 24 (1–100) |
| Macrophages × 103 cells·mL−1 | 145 (93–270) | 239 (115–525) |
| Eosinophils × 103 cells·mL−1 | 0 (0–0) | 0 (0–0) |
| Epithelial cells × 103 cells·mL−1 | 0 (0–5.9) | 0 (0–0) |
| Urea mg·dL−1 | 0.35 (0.20–0.60) | 0.30 (0.30–0.50) |
| Albumin μg·mL−1 | 66 (34–168) | 49 (37–92) |
| Pseudomonas-positive BALF | 0/24 | 8/31 |
| Positive for other organisms# | 2/24 | 9/31 |
Data are presented as median (interquartile range) or n/N, unless otherwise stated.
four CF patients were positive for Staphylococcus aureus, three for Alcaligenes xylosoxidans, two for Haemophilus influenzae (one of whom was also positive for S. aureus), and one for Mycobacterium avium; two control patients were positive for Streptococcus pneumoniae.
α<0.05.
Elastase assay
Elastase activity was quantified in BALF specimens by hydrolysis of methoxysuccinyl (MeO-Suc)-Ala-Ala-Pro-Ala-p-nitroanilide (Sigma Aldrich, St Louis, MO, USA), by spectro-photometry at 410 nm [18], with human neutrophil elastase as a standard and MeO-Suc-Ala-Ala-Pro-Val-CH2Cl to confirm specificity.
Serpin inhibitors, protease, and protease inhibition assays
Human recombinant SerpinB1 (rSerpinB1) was expressed in insect cells, purified and stored in aliquots (2 μg·mL−1) in PBS at −80°C [19]. Mercaptoethanol (2 mM) was added prior to use. Human neutrophil elastase from purulent sputum (Elastin Products, Owensville, MO, USA) and human neutrophil cathepsin G from blood leukocytes (Athens Research and Technology, Athens, GA, USA) were stored as aliquots (1 mg·mL−1) at −20°C. α1-AT purified from human plasma (Athens Research) was stored as 2-mg·mL−1 aliquots in PBS with 2 mM mercaptoethanol. For in vitro study of elastase inhibition, rSerpinB1 or purified α1-AT was incubated with elastase or elastase-positive BALF in PBS with 0.05% Tween-20 at 37°C for 5 min (or as indicated). Aliquots (30 μL) were assayed for elastase activity with 0.8 mM MeO-suc-Ala-Ala-Pro-Ala-p-nitroanilide in 1 mL of 20 mM tris-(hydroxymethyl)-aminomethane (Tris)–HCl (pH 7.4), 500 mM NaCl and 0.1% polyethylene glycol. To detect serpin complexes and other products, additional aliquots were treated with 2 mM diisopropylfluorophosphate for 2 min at room temperature and solubilised for electrophoresis with 5× sodium dodecylsulfate (SDS) solution at 100°C.
Immunoblots
BALF samples or rSerpinB1 (10, 33 or 100 ng) were solubilised with 2× SDS solution (2% SDS, 5% glycerol and 1% mercaptoethanol) at 100°C for 2 min. Samples (30 μL) were electrophoresed on Tris Gly 10% polyacrylamide gels (Novex, San Diego, CA, USA) with Tris HCl (pH 8.3) running buffer (Laemmli gels) [20] or with bis-(2-hydroxyethyl)-amino-Tris HCl buffered (pH 6.4) 10% polyacrylamide with 2-(N-morpholino)-ethanesulfonic acid (pH 7.7) running buffer (Bis-Tris gels; NuPAGE system, Novex). The latter system separates the 42-kDa from the 38-kDa SerpinB1 species (see Results section). The separated proteins were transferred to nitrocellulose, blocked with 20% milk solids in PBS with 0.05% Tween 20 (PBS Tween), incubated with rabbit anti-SerpinB1 (anti-MNEI) antiserum [21] (1:500) in 0.1% milk in PBS Tween for 2 h, and for 1 h with 0.3 μg·mL−1 125I-labelled goat antibodies to rabbit immunoglobulin G. SerpinB1 bands were quantified by densitometry using rSerpinB1 as standard and employing the Storm 860 Phosphorimager and Image Quant software (GE Healthcare, Little Chalfont, UK). Preliminary experiments established that the presence of endogenous SerpinB1 in BALF does not alter the incremental signal intensity due to rSerpinB1 standard (data not shown). For in vitro studies of pure proteins, the electrophoresis gels were stained with Coomassie blue and protein bands were quantified using a Bio-Rad Gel Documentation system (Bio-Rad Laboratories, Hercules, CA, USA).
Statistical evaluation
SerpinB1 data were transformed using a log10 transformation and are presented as geometric mean (95% CI). Cell count, albumin and urea data (nonparametric distribution) were analysed by Mann–Whitney rank-sum test and are presented as median (interquartile range (IQR)). Correlations between SerpinB1 and cell counts were assessed using Pearson correlation coefficients. An α-value of 0.05 was set to determine statistical significance. Analyses were performed in SigmaStat V3.5 (Systat Software Inc., Port Richmond, CA, USA) or SAS V9.2 (SAS Institute, Carey, NC, USA).
RESULTS
SerpinB1 is elevated in CF airways
The characteristics of the participants and BALF specimens are summarised in table 1. The median ages were 9.3 yrs for 31 CF subjects and 8.9 yrs for 24 disease-control subjects. The median white blood cell counts were higher in the CF group than the control group, largely reflecting 25-fold higher median neutrophil counts. Among the CF group, bacterial pathogens were isolated from 17 (54.8%) BALFs: eight BALFs positive for Pseudomonas aeruginosa and nine positive for other CF pathogens. BALF of two disease control subjects were culture-positive, in both cases for Streptococcus pneumoniae.
The content of SerpinB1 in patient BALF was determined by immunoblotting after SDS electrophoresis on Tris–Gly (Laemmli) gels. SerpinB1 migrates as a 42-kD band, which was quantified relative to the rSerpinB1 standard (fig. 1). Levels of SerpinB1 (“total SerpinB1”) were elevated in BALF of CF patients compared with BALF of disease controls (geometric mean 3.9 (95% CI 2.69–5.62) versus 1.37 (1.20–1.55) μg·mL−1; p<0.001). Among the CF group, SerpinB1 concentrations were higher in culture-positive (>300 organisms·mL−1) compared with culture-negative BALF (mean 5.5 (3.10–9.84) versus 2.7 (1.74–4.11) μg·mL−1; p<0.04) and were substantially higher in BALF that contained active elastase (elastase-positive BALF) compared with elastase-negative BALF (8.41 (5.4–13.22) versus 1.89 (1.46–2.44) μg·mL−1; p<0.001) (fig. 1). All disease-control BALF lacked active elastase. SerpinB1 concentration was not different for P. aeruginosa-positive CF BALF compared with CF BALF positive for other micro-organisms (3.38 (1.74–6.66) versus 8.95 (3.39–23.64) μg·mL−1; p=0.08).
FIGURE 1.
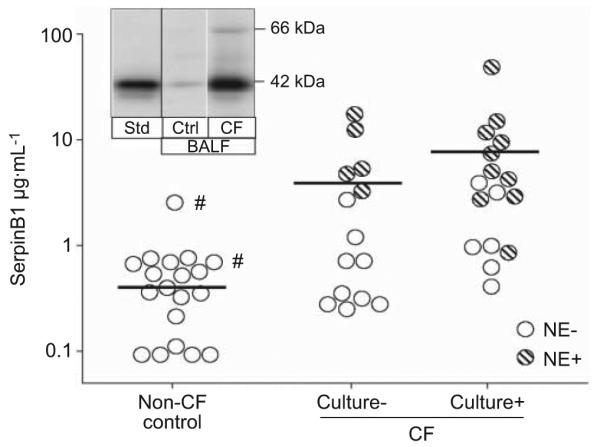
Quantitation of SerpinB1 in cystic fibrosis (CF) bronchoalveolar lavage fluid (BALF). SerpinB1 (42 kDa) was quantified by immunoblot (Laemmli gels; reducing conditions) and densitometry relative to a recombinant SerpinB1 standard (Std). Data for non CF control patients, culture negative (culture−) CF patients and culture positive (culture+; >300 organisms·mL−1) CF patients are shown. Each symbol represents the mean of triplicate assays for an individual patient. NE−: neutrophil elastase-negative specimens; NE+: neutrophil elastaste positive specimens. A subset of the findings were reported in abstract form previously [22]. #: culture+ non CF control patients.
SerpinB1 concentration correlated positively with neutrophil counts in the BALF (r=0.80; p<0.001; fig. 2a). Figure 2 also shows, as anticipated, that free elastase (fig. 2b) and α1–AT elastase (fig. 2c) correlated positively with neutrophil counts (r=0.73 (p<0.001) and r=0.76 (p<0.001), respectively). SerpinB1 levels correlated positively with free elastase (r=0.70; p<0.001) and α1-AT–elastase complex (r=0.82; p<0.001; data not shown). No correlation was detected for SerpinB1 with macrophage number (r=0.03; p=0.82).
FIGURE 2.

Correlation of bronchoalveolar lavage fluid concentration of a) SerpinB1, b) active elastase and c) α1-antitrypsin (α1-AT)–elastase complex with neutrophil counts. –––: lower limits of detection.
Molecular forms of SerpinB1 in CF BALF
The Laemmli SDS electrophoresis gels in figure 1 provided quantitation but, contrary to predictions [20], did not differentiate between active and inactive SerpinB1. To examine these different molecular forms, we used an alternative SDS electrophoresis system. When tested with pure SerpinB1 standards, Bis-Tris gels (fig. 3a, bottom panel) separated three major species: active SerpinB1 at 42 kDa, the 66-kDa covalent SerpinB1–elastase complex, the primary product of SerpinB1 inhibition of elastase, and cleaved post-complex SerpinB1 at 38 kDa. Based on earlier sequencing results [17], the 38-kDa band represents SerpinB1 cleaved after Cys344, the specificity-determining residue in the reactive center loop (termed “P1” in the nomenclature of Schechter and Berger [23]). An intermediate species representing partially degraded complex was also detected.
FIGURE 3.

Molecular forms of SerpinB1 in cystic fibrosis (CF) bronchoalveolar lavage fluid (BALF). a) Comparison of Tris-Gly (Laemmli; top) and Bis/Tris SDS gels (bottom). Standard SerpinB1 species were generated by reacting rSerpinB1 with increasing elastase. The molecular forms separated on bis-(2-hydroxyethyl)-amino-Tris (Bis-Tris) gels are the 42-kDa active SerpinB1 (bottom, left lane), 66-kDa SerpinB1–protease complex, partially degraded complex (#) and 38-kDa cleaved SerpinB1. b) Bis-Tris immunoblots of patient BALF. In the top panel, lanes 1–9 contain coded BALF specimens of CF patients. All but specimens 6 and 7 are elastase positive. Lane 10 contains the single disease-control specimen (out of 11 analysed) that had SerpinB1 detectable by this method. Std: mixture of sandards. The major species in CF BALF is the 38-kDa cleaved SerpinB1 with variable minor amounts of the 66-kDa complex. A degradation product is seen in BALFs 2, 3 and 9. The bottom panel shows a repeat analysis to verify size (38 kDa) of the species in CF patient BALF. Groups of recombinant SerpinB1 (Std), Std mixed with CF BALF and CF BALF sample alone were analysed. Lanes 2 and 3 contain a pool of four CF samples (two elastase-positive and two –negative sample), lanes 5 and 6 contain the CF BALF run in lane 4 of the top panel), lanes 8 and 9 contain an elastase-positive CF BALF, and lanes 11 and 12 contain the CF BALF sample run in lane 8 of the top panel.
To identify the molecular species in CF patients, the remaining CF BALF samples were evaluated on Bis-Tris gels. This analysis revealed 38-kDa cleaved SerpinB1 as the predominant molecular form in 16 out of 17 CF specimens analysed (one specimen was blank; fig. 3b). Depending on the specimen, minor amounts were found of the 66-kDa SerpinB1–elastase complex; active 42-kDa SerpinB1 was essentially nondetectable (fig. 3b). The single disease-control specimen that had SerpinB1 detectable by this method (lane 10) contained 42- and 38-kDa (active and inactive, respectively) SerpinB1.
The paucity of active inhibitor in CF BALF and the presence of the 38-kDa proteolytically cleaved species indicate that the elevated levels of total SerpinB1 were insufficient or barely sufficient to inhibit the high levels of NSPs in patient airways. This finding is consistent with the presence of active elastase in many of the specimens. The paucity of the 66-kDa SerpinB1–elastase complex was surprising, because the classical serpin mechanism of protease inhibition [24] predicted that the SerpinB1–elastase complex, like the α1-AT–elastase complex, would be a readily detectable product.
rSerpinB1 inhibits endogenous elastase in CF BALF
Since endogenous SerpinB1 in CF BALF was inactive, we next questioned whether exogenously added SerpinB1 would be functional. Pure neutrophil elastase with activity matched to CF BALF was examined as standard (fig. 4a). Endogenous elastase in each of two CF BALFs was completely inhibited by rSerpinB1 (fig. 4b and c). The amount of rSerpinB1 required for 50% inhibition was indistinguishable from predicted stoichiometric values, and the amount for complete inhibition was not different from that required to inhibit pure protease. These findings indicate that SerpinB1 efficiently inhibits neutrophil elastase in the presence of components of CF BALF.
FIGURE 4.

Recombinant SerpinB1 (rSerpinB1) inhibits endogenous elastase in cystic fibrosis (CF) bronchoalveolar lavage fluid (BALF). a) Neutrophil elastase, or elastase-positive BALF b) CF 32 or c) CF 33 (all containing 500 ng of elastase activity), were allowed to react with varying amounts of rSerpinB1 in a 50-μL reaction for 5 min, and remaining elastase activity was assayed. Results are the average of duplicate assays. CF-32 and CF-33 BALF contain 15.8 and 34.3 μg·mL1 active elastase, respectively.
Mechanism of action of SerpinB1
To explain the paucity of the SerpinB1–elastase complex in CF BALF, we modelled the in vivo situation by reacting SerpinB1 with varying amounts of elastase for ≤18 h. Previous studies established that SerpinB1 inhibition of elastase is extremely rapid, with an association rate constant >107 M−1·s−1, and inhibition is thus complete within minutes at the concentrations studied [17]. For comparison, we examined the similarly rapid reaction of α1-AT (SerpinA1) with elastase. A fixed amount of inhibitor was combined with an increasing molar ratio of elastase, including excess elastase (protease/inhibitor ratio 1.25) to simulate conditions of the CF airway. As anticipated, maximal amounts of the respective covalent complexes were observed at the earliest time point (5 min) for both α1-AT (fig. 5a, top panel) and SerpinB1 (fig. 5a, bottom panel). The generation of these complexes reflects the classical serpin mechanism: attempted proteolysis by elastase at the exposed serpin P1 residue (Met358 in α1-AT and Cys344 in SerpinB1), resulting in 1) cleavage at the P1 bond with 2) generation of an ester bond from P1 to the elastase active site serine and 3) large-scale conformational rearrangement of the serpin, the driving force for the reaction. Elastase, covalently bound to the cleaved serpin, is inactivated due to conformational distortion of its active site [25]. On further incubation, the α1-AT–elastase complex remained largely intact at 30 min, 3 h and, for the lower elastase ratios, even at 18 h. In contrast, the SerpinB1–elastase complex was short-lived, being partially (30 min) or completely (3 h) degraded, coincident with generation of 38-kDa SerpinB1 at the two higher elastase ratios and completely lost at 18 h, even at low elastase. Elastase activity did not change over the time period 5 min–18 h for any serpin:protease combination (data not shown), indicating that inhibition was irreversible (i.e. that active elastase was not released from either complex under these conditions), consistent with previous findings for α1-AT [25]. Densitometry to quantify the α1-AT and SerpinB1 complexes for the reactions with excess elastase also show that the SerpinB1–elastase complex is short-lived (fig. 5b). Thus, the anticipated (dominant) accumulated product of elastase inhibition by SerpinB1 is the 38-kDa cleaved form.
FIGURE 5.

a) Reaction of α1-antitrypsin (α1-AT) and SerpinB1 with elastase and stability over time of the resulting covalent complexes. α1 AT or SerpinB1 (1.3 mM) were allowed to react with varying molar ratios of elastase (indicated by protease:inhibitor (P/I) ratio ranging from subthreshold to excess elastase. The reactions were stopped after 5 min, 30 min, 3 h or 18 h, and the products analysed on Coomassie blue stained Bis Tris gels. α1-AT (top panel, left lane) migrates at 52 kDa and its complex (cpx) with elastase at 76 kDa; pure SerpinB1 (bottom panel, left lane) migrates at 42 kDa and its cpx with elastase at 66 kDa. b) Quantitation of α1-AT–elastase and SerpinB1–elastase complexes remaining over time for the reactions with excess elastase (P/I 1.25). The 5-min values were considered 100%. Data are presented as mean±SEM of three experiments.
DISCUSSION
A key finding of the study is that total SerpinB1 concentration is elevated in BALF of CF subjects compared with disease controls. Within the CF group, levels of SerpinB1 were higher for infected versus uninfected subjects and substantially higher for elastase-positive versus -negative subjects. SerpinB1 concentration correlated positively with neutrophil counts, free elastase and α1-AT–elastase levels. Since neutrophil counts and free elastase are among the best established biomarkers of lung inflammation in CF, the findings suggest that SerpinB1 could serve as biomarker of inflammation in CF.
The major molecular form of SerpinB1 in BALF of CF patients was 38-kDa SerpinB1, identified in previous studies as SerpinB1 cleaved at the reactive center loop. Although other scenarios, including direct proteolysis by a different protease, cannot be eliminated, a straightforward interpretation is that the 38-kDa species in CF BALF is post-complex SerpinB1, the major product of the in vitro inhibitory reaction under conditions of excess elastase and extended time, conditions that apply in the CF lung. The absence of active (42-kDa) SerpinB1 in CF BALF is consistent with the release of high levels of elastase in patient airways, as documented by the presence of free elastase in half of the CF specimens. The further finding of elastase-negative CF BALF containing 38-kDa SerpinB1 suggests (although it is not proof) that excess elastase has been released in patient lungs and successfully inhibited by SerpinB1. The presence of α1-AT–elastase complex in elastase-negative BALF also indicates that excess elastase has been released and successfully inhibited.
Free elastase is an indicator of current lung inflammation and a target for therapy in CF. It has been recently identified as the best inflammatory correlate of pulmonary function in induced sputum [26]. This finding is no surprise, given the plethora of elastase’s deleterious actions: matrix destruction, induction of inflammatory cytokines, and degradation of anti-inflammatory and antimicrobial proteins. Moreover, the concept was recently advanced that, among airway inflammatory proteases, neutrophil elastase occupies a position at the apex of a regulatory hierarchy, as assessed by the capacity of elastase inhibitor to decrease the induction of macrophage metalloprotease-2 and the cysteinyl protease cathepsin B [27]. The findings suggest that therapy to inhibit elastase may abrogate more than its own deleterious proteolytic effects.
Endogenous elastase in CF BALF was stoichiometrically inhibited by rSerpinB1, suggesting that the inhibitor functions in the CF airway microenvironment. A caveat of this is that BALF specimens are dilute relative to airway fluids. However, concentrations of rSerpinB1 only slightly higher than stoichiometric completely inhibited endogenous elastase in CF sputum sol, which is only minimally diluted relative to sputum [21]. Altogether, the ability of (near-)stoichiometric amounts to inhibit endogenous elastase suggests that SerpinB1 is not prevented from its elastase inhibitory function by components of the CF airway.
The functional capacity and efficacy of delivering rSerpinB1 have been shown also in models of lung injury and infection in rats [21, 28]. Also, in mice, deletion of serpinB1 decreased, and delivery of rSerpinB1 rescued, antibacterial and anti-inflammatory lung protection [29].
The high levels of SerpinB1 in CF airway fluid introduce several questions. First, it is not clear where the SerpinB1 originates. The gene is broadly expressed, and levels are highest in myeloid cells, especially neutrophils [30], and so necrotic neutrophils probably contribute to this. SerpinB1, like all clade-B serpins, lacks a leader sequence [31, 32], and thus may be actively produced by viable neutrophils, monocytes or airway epithelial cells. An increase of expression in inflammation is suggested by the presence of a nuclear factor-κB regulatory element in the promoter of the human gene [33].
The most fundamental finding of this study is the demonstration that SerpinB1 is a naturally occurring contributor to the anti-elastase shield of normal and CF lungs and, thus, joins previously characterised inhibitors, such as SLPI, elafin and α1-AT. Although SLPI and α1-AT are present in normal concentrations in CF BALF, the majority of α1-AT and SLPI were inactive molecular forms, either complexed or degraded [5], consistent with the increase of free elastase in CF. The concentration of SerpinB1 reported here for CF BALF approaches that reported for α1-AT.
SerpinB1 and SerpinA1 (α1-AT), the two serpin elastase inhibitors in lung, share several features. Both rapidly inhibit each of the NSPs (elastase, cathepsin G and proteinase-3; summarised in [17]), but despite this shared capacity, the two serpins are only distantly related within the superfamily, and their reactive centre sequences are different, indicating that they represent independent convergent evolutionary pathways. Interestingly, both SerpinB1 and α1-AT are susceptible to oxidative inactivation, suggesting that temporal and/or spatial restriction may be requisite for pulmonary NSP inhibitors. Again, the mechanisms differ. α1-AT is subject to oxidative inactivation of its reactive-centre methionine by hydrogen peroxide and peroxidases produced by inflammatory neutrophils. SerpinB1, due to its reactive-centre cysteine, can be reversibly inactivated by dimerisation in an environment devoid of free sulfhydryls (unpublished observations). However, SerpinB1, unlike α1-AT, is highly resistant to inactivation by hydrogen peroxide (S. Van Patten, Genzyme Corp., Cambridge, MA, USA; personal communication).
Collectively, these findings characterise the innate role of SerpinB1 in the CF airway and provide baseline information for potential therapies involving SerpinB1.
ACKNOWLEDGEMENTS
We thank P. Emmett and I. Osberg (both Children’s Hospital, Denver, CO, USA), and F. Rubio and K. Ronaszeki (both Immune Disease Institute, Boston, MA, USA) for assistance with sample and data collection. We thank C. Benarafa (Theodor Kocher Institute, Berne, Switzerland) for insightful discussion of data.
SUPPORT STATEMENT
The study was supported by National Institutes of Health (grants HL066548, HL41579, HL081335 and RR0069) and the Cystic Fibrosis Foundation.
Footnotes
STATEMENT OF INTEREST
None declared
REFERENCES
- 1.Fouret P, du Bois RM, Bernaudin JF, et al. Expression of the neutrophil elastase gene during human bone marrow cell differentiation. J Exp Med. 1989;169:833–845. doi: 10.1084/jem.169.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 3.Reeves EP, Lu H, Jacobs HL, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 4.McElvaney NG, Hubbard RC, Birrer P, et al. Aerosol a1 antitrypsin treatment for cystic fibrosis. Lancet. 1991;337:392–394. doi: 10.1016/0140-6736(91)91167-s. [DOI] [PubMed] [Google Scholar]
- 5.Birrer P, McElvaney NG, Rudeberg A, et al. Protease antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994;150:207–213. doi: 10.1164/ajrccm.150.1.7912987. [DOI] [PubMed] [Google Scholar]
- 6.Janoff A. Elastase in tissue injury. Annu Rev Med. 1985;36:207–216. doi: 10.1146/annurev.me.36.020185.001231. [DOI] [PubMed] [Google Scholar]
- 7.Schuster A, Fahy JV, Ueki I, et al. Cystic fibrosis sputum induces a secretory response from airway gland serous cells that can be prevented by neutrophil protease inhibitors. Eur Respir J. 1995;8:10–14. doi: 10.1183/09031936.95.08010010. [DOI] [PubMed] [Google Scholar]
- 8.Witko-Sarsat V, Halbwachs-Mecarelli L, Schuster A, et al. Proteinase 3, a potent secretagogue in airways, is present in cystic fibrosis sputum. Am J Respir Cell Mol Biol. 1999;20:729–736. doi: 10.1165/ajrcmb.20.4.3371. [DOI] [PubMed] [Google Scholar]
- 9.Bedard M, McClure CD, Schiller NL, et al. Release of interleukin 8, interleukin 6, and colony stimulating factors by upper airway epithelial cells: implications for cystic fibrosis. Am J Respir Cell Mol Biol. 1993;9:455–462. doi: 10.1165/ajrcmb/9.4.455. [DOI] [PubMed] [Google Scholar]
- 10.Tosi MF, Zakem H, Berger M. Neutrophil elastase cleaves C3bi on opsonized Pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J Clin Invest. 1990;86:300–308. doi: 10.1172/JCI114699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartl D, Latzin P, Hordijk P, et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13:1423–1430. doi: 10.1038/nm1690. [DOI] [PubMed] [Google Scholar]
- 12.Fick RB, Jr, Naegel GP, Squier SU, et al. Proteins of the cystic fibrosis respiratory tract. Fragmented immunoglobulin G opsonic antibody causing defective opsonophagocytosis. J Clin Invest. 1984;74:236–248. doi: 10.1172/JCI111407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Bredow C, Wiesener A, Griese M. Proteolysis of surfactant protein D by cystic fibrosis relevant proteases. Lung. 2003;181:79–88. doi: 10.1007/s00408-003-1008-z. [DOI] [PubMed] [Google Scholar]
- 14.Rubio F, Cooley J, Accurso FJ, et al. Linkage of neutrophil serine proteases and decreased surfactant protein-A (SP-A) levels in inflammatory lung disease. Thorax. 2004;59:318–323. doi: 10.1136/thx.2003.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly E, Greene CM, McElvaney NG. Targeting neutrophil elastase in cystic fibrosis. Expert Opin Ther Targets. 2008;12:145–157. doi: 10.1517/14728222.12.2.145. [DOI] [PubMed] [Google Scholar]
- 16.Griese M, Kappler M, Gaggar A, et al. Inhibition of airway proteases in cystic fibrosis lung disease. Eur Respir J. 2008;32:783–795. doi: 10.1183/09031936.00146807. [DOI] [PubMed] [Google Scholar]
- 17.Cooley J, Takayama TK, Shapiro SD, et al. The serpin MNEI inhibits elastase like and chymotrypsin like serine proteases through efficient reactions at two active sites. Biochemistry. 2001;40:15762–15770. doi: 10.1021/bi0113925. [DOI] [PubMed] [Google Scholar]
- 18.Khan TZ, Wagener JS, Bost T, et al. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- 19.Cooley J, Mathieu B, Remold-O’Donnell E, et al. Production of recombinant human monocyte/neutrophil elastase inhibitor (rM/ NEI) Protein expression and purification. 1998;14:38–44. doi: 10.1006/prep.1998.0951. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Rees DD, Rogers RA, Cooley J, et al. Recombinant human monocyte/neutrophil elastase inhibitor protects rat lungs against injury from cystic fibrosis airway secretions. Am J Respir Cell Mol Biol. 1999;20:69–78. doi: 10.1165/ajrcmb.20.1.3306. [DOI] [PubMed] [Google Scholar]
- 22.Cooley J, Rubio F, Sontag MK, et al. MNEI (monocyte/neutrophil elastase inhibitor) is found at increased levels in cystic fibrosis lavage fluid. Pediatric Pulmonology. 2001;22:271a. [Google Scholar]
- 23.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 24.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 25.Dementiev A, Dobo J, Gettins PG. Active site distortion is sufficient for proteinase inhibition by serpins: structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J Biol Chem. 2006;281:3452–3457. doi: 10.1074/jbc.M510564200. [DOI] [PubMed] [Google Scholar]
- 26.Mayer-Hamblett N, Aitken ML, Accurso FJ, et al. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:822–828. doi: 10.1164/rccm.200609-1354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geraghty P, Rogan MP, Greene CM, et al. Alpha 1 antitrypsin aerosolised augmentation abrogates neutrophil elastase induced expression of cathepsin B and matrix metalloprotease 2 in vivo and in vitro. Thorax. 2008;63:621–626. doi: 10.1136/thx.2007.088559. [DOI] [PubMed] [Google Scholar]
- 28.Woods DE, Cantin A, Cooley J, et al. Aerosol treatment with MNEI suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Pediatr Pulmonol. 2005;39:141–149. doi: 10.1002/ppul.20167. [DOI] [PubMed] [Google Scholar]
- 29.Benarafa C, Priebe GP, Remold-O’Donnell E. The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J Exp Med. 2007;204:1901–1909. doi: 10.1084/jem.20070494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benarafa C, Cooley J, Zeng W, et al. Characterization of four murine homologs of the human ov serpin monocyte neutrophil elastase inhibitor MNEI (SERPINB1) J Biol Chem. 2002;277:42028–42033. doi: 10.1074/jbc.M207080200. [DOI] [PubMed] [Google Scholar]
- 31.Remold-O’Donnell E. The ovalbumin family of serpin proteins. FEBS Lett. 1993;315:105–108. doi: 10.1016/0014-5793(93)81143-n. [DOI] [PubMed] [Google Scholar]
- 32.Silverman GA, Bird PI, Carrell RW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- 33.Zeng W, Remold-O’Donnell E. Human monocyte/neutrophil elastase inhibitor (MNEI) is regulated by PU.1/Spi-1, Sp1, and NF-κkB. J Cell Biochem. 2000;78:519–532. doi: 10.1002/1097-4644(20000915)78:4<519::aid-jcb1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]