

Abstract
Objective
We explore the role of DNA methylation in Alzheimer’s disease (AD). To elucidate where DNA methylation falls along the causal pathway linking risk factors to disease, we examine causal models to assess its role in the pathology of AD.
Methods
DNA methylation profiles were generated in 740 brain samples using the Illumina HumanMet450K beadset. We focused our analysis on CpG sites from 11 AD susceptibility gene regions. The primary outcome was a quantitative measure of neuritic amyloid plaque (NP), a key early element of AD pathology. We tested four causal models: (1) independent associations, (2) CpG mediating the association of a variant, (3) reverse causality, and (4) genetic variant by CpG interaction.
Results
Six genes regions (17 CpGs) showed evidence of CpG associations with NP, independent of genetic variation – BIN1 (5), CLU (5), MS4A6A (3), ABCA7 (2), CD2AP (1), and APOE (1). Together they explained 16.8% of the variability in NP. An interaction effect was seen in the CR1 region for two CpGs, cg10021878 (P = 0.01) and cg05922028 (P = 0.001), in relation to NP. In both cases, subjects with the risk allele rs6656401AT/AA display more methylation being associated with more NP burden, whereas subjects with the rs6656401TT protective genotype have an inverse association with more methylation being associated with less NP.
Interpretation
These observations suggest that, within known AD susceptibility loci, methylation is related to pathologic processes of AD and may play a largely independent role by influencing gene expression in AD susceptibility loci.
Introduction
Genome-wide association studies have recently identified nineteen genes in which genetic variation influence susceptibility to Alzheimer’s disease (AD [MIM104300]).1–8 The functional consequences of some of the genomic variants is being elaborated, such as the role of APOE (MIM 107741), ABCA7 (MIM 605414), CD2AP (MIM 604241), CR1 (MIM 120620), CD33 (MIM 159590), and PICALM (MIM 603025) in neuritic plaque (NP) pathology,9–11 a key early feature in the neuropathological cascade leading to AD. In addition, studies examining tissue from the cortex of AD and healthy aging brains have shown that some of the risk alleles are associated with the level of RNA expression of nearby genes, such as in the ABCA7, BIN1 (MIM 601248), CLU (MIM 185430), and MS4A2 (MIM 147138) loci,12–15 However, other genomic features that influence gene expression in these loci have not been characterized in detail for a possible role in AD susceptibility.
In previous literature we explored the relationship between DNA methylation and AD traits on a genome-wide level16 and found CpGs in two known AD loci, ABCA7 and BIN1, to be significantly associated with AD and NP burden on a methylome-wide level, nonetheless on that scale we were limited by the appropriate but stringent criteria of controlling for hundreds of thousands of comparisons. Here, by limiting our analyses to known AD loci we have the power to assess more complex models of association. Specifically, we examine the level of methylation in known AD susceptibility loci using a large cohort of prospectively collected, frozen brains from two cohort studies of aging: the Rush Memory and Aging Project (MAP) and the Religious Order Study (ROS). A sample of the dorsolateral prefrontal cortex (DLPFC), a hub in cognitive and mood circuits, was extracted from each subject, and we show that several of the loci implicated in AD susceptibility from studies of genetic variation contain CpGs whose level of methylation is associated with the deposition of NPs, a defining neuropathologic feature of AD. Then, we evaluated the role genetic variation on DNA methylation within these loci: we focused on the single-nucleotide polymorphism (SNP) in each locus that has previously been reported to best capture the association with AD susceptibility (index SNPs). In several loci, such index SNPs also influence methylation levels of nearby CpGs. Finally, we were able to test the hypothesis that DNA methylation levels mediate the effect of a genetic variant in two loci: APOE and CR1. Overall, we find that genetic variation and methylation levels have largely independent effects on AD pathology and susceptibility, with evidence for a haplotype-specific effect of DNA methylation in the CR1 gene region.
Materials and Methods
Subjects and materials
Subjects
Samples are from participants in two ongoing longitudinal studies of cognition, the ROS and the Rush MAP. The ROS, started in 1994, enrolls Catholic priests, nuns and brothers from more than 40 groups in 12 states. On enrollment, participants were older and free of dementia. The MAP, started in 1997, enrolls participants from retirement living communities, subsidized housing, church groups and social service agencies throughout the Chicagoland area. The studies have a large common core of data designed to facilitate combination of data, and are maintained by a single investigative team at the Rush Alzheimer’s Disease Center at Rush University Medical Center in Chicago, IL. At the time of generation of the DNA methylation data, 1 January 2010, 2475 participants completed baseline evaluations and 806 participants died and underwent autopsies. Participants in each study are followed annually with detailed clinical evaluations and have signed both an informed consent and an Anatomical Gift Act for donating their brains at time of death. More detailed descriptions of each study have been published previously.17,18 Both ROS and MAP were approved by the Rush University Medical Center institutional review board.
Genotyping
DNA was extracted from whole blood, lymphocytes, or frozen postmortem brain tissue. Genotyping was done in three subsets. The data for the first two subsets were generated in 2009 on the Affymetrix Genechip 6.0 platform (Affymetrix, Inc, Santa Clara, CA, USA) at the Broad Institute’s Center for Genotyping or the Translational Genomics Research Institute. The third subset was genotyped in 2012 on the Illumina HumanOmniExpress (Illumina, Inc, San Diego, CA, USA) at the Children’s Hospital of Philadelphia. All three data sets underwent the same quality control (QC) analysis, described in detail elsewhere19 and QCed genotypes were pooled.
In addition to the APOE ε4 haplotype (MIM 107741), we limited out analyses to the list of SNPs that showed a genome-wide association with AD in previous literature and were replicated in additional publications. These included index SNPs from CR1 (MIM 120620; rs6656401), PICALM (MIM 603025; rs7110631), CLU (MIM 185430; rs11136000), ABCA7 (MIM 605414; rs3752246), BIN1 (MIM 601248; rs744373), CD2AP (MIM 604241; rs9349407), CD33 (MIM 159590; rs3865444), EPHA1 (MIM 179610; rs11767557), MS4A4A (MIM 606547; rs4938933) and MS4A6A (MIM 606548; rs610932).1,2,4,5,7–9,20 Data on APOE were generated by directly genotyping the two SNPs that comprise the APOE functional haplotype.
DNA methylation
DNA methylation profiles were created from samples of brain tissues from the DLPFC collected from 761 deceased ROS and MAP participants. Information on experimental and QC procedures and mapping of chromatin state is provided in detail in previous literature.16 In brief, frozen 100 mg sections of the DLPFC sent from the Rush Alzheimer’s Disease Center were processed at the Broad Institute in Cambridge, MA. White matter was carefully dissected from gray matter on the thawed sections and DNA extraction was performed using the Qiagen (cat: 51306, Qiagen, Inc, Valencia, CA, USA) QIAmp DNA mini-protocol. The DNA methylation data were generated by the Broad Institute’s Genomics Platform using the Illumina InfiniumHumanMethylation450 bead chip assay. The Illumina platform consists of 485,513 probes covering a total of 21,231 genes, with an average of 17.2 probes per gene region. Data generated from the Illumina 450K platform were processed using the GenomeStudio software Methylation Module v1.8 to produce beta-values and detection P-values for each CpG site. Probes were discarded based on the following criteria: (1) 14,664 probes with detection P-values >0.01 indicating poor quality, (2) 10,868 probes found on the sex chromosomes, (3) 29,233 probes that are predicted to cross-hybridize with sex chromosomes based on alignment,21 and (4) 14,964 probes that overlap with known SNP sites, limiting them to those with minor allele frequencies >1% in a European population based on 1000 Genomes database, leaving us with a total of 415,848 autosomal CpGs for analyses. Subjects were removed from analyses if they were deemed poor quality defined in two ways. First, we performed a principle component analysis (PCA) on a random selection of 50,000 probes and any subject not within ±3 SD of the mean of PC1, PC2, or PC3 was discarded (n = 13 subjects). Second, subjects were excluded based on poor bisulfite conversion (BC) efficiency, defined as having at least two of the 10 BC control probes fail to reach a value of 0.8 (n = 8) leaving us with 740 samples for analysis. A more detailed description of processing the DNA methylation data is described elsewhere.16
The map for the state of the chromatin was generated in collaboration with the Epigenomics Roadmap team (http://www.roadmapepigenomics.org/). It was derived from samples from the DLPFC of two individuals from the MAP study who were cognitively nonimpaired, with minimal AD-associated pathology at time of death. Using established methods22 the map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks.
Pathological outcomes
The autopsy rate in ROS and MAP is over 85% and the procedures are described in detail in previous manuscripts.17,18 Two pathological outcomes were used in these analyses: (1) a quantitative measure of AD pathology based on standardized counts of NPs, and (2) a pathological diagnosis of AD based on intermediate or high likelihood of AD by National Institute on Aging-Reagan criteria using Consortium to Establish a Registry for Alzheimer’s Disease estimates of NP density and Braak staging of neurofibrillary pathology, as previously described.23,24
Models
All analyses are focused on regions around known AD risk loci. Our primary outcome of interest is neuritic amyloid plaque (AD pathology), with secondary analyses on pathological diagnosis of AD.
First, we examine each side of the causal pathway, the CpGs association with outcomes, the index SNPs association with the outcomes and the CpGs association with the index SNP in each gene region. Second, we explore the four possible models of association: (1) Independent Associations, (2) CpG-mediated effect, (3) Reverse Causality, and (4) an interaction model. The steps for each model are described in Figure1. With our cross-sectional data, we are unable to distinguish between models (2) and (3) computationally, however, we will explore each hypothesized model individually, but limit our interpretation of (2) and (3) to the possibility of it being either CpG-mediated effect or reverse causality. The first three models require a significant association between the SNP of interest and the outcome, whereas (4) the interaction model does not; in fact, if a strong interaction effect is present it will suppress any association between SNP and outcome.
Figure 1.

Proposed models incorporating the role of DNA methylation in Alzheimer’s disease (AD) susceptibility. Four possible models are tested: (1) Independent associations of CpG and genetic variants with the outcome measure, (2) CpG-mediated effect in which DNA methylation mediates the effect of a genetic variant, (3) Reverse causality in which the outcome measure (such as AD) leads to DNA methylation changes, and (4) interaction model in which the association of a CpG varies based on a genetic variant. Note that models (2) and (3) can only be tested when all three components of the models are associated with one another: in our data, only the CR1 and APOE loci are tested. Dashed lines represent hypothesized associations tested in each model and solid lines are significant associations required to test the model.
Statistical analysis
Patient characteristics are summarized using means and standard deviations for continuous measures and frequencies for discrete variables. A linear regression model is used for the quantitative measure of NPs and a logistic regression model is used for the AD pathological diagnosis outcome. All models are adjusted for age at death, sex, batch, and a measure of technical variation, namely, BC ratio. Since the NP was skewed, the measure was transformed by taking the square root. Multiple comparisons are taken into account on a gene level, using the Benjamini-Hochberg procedure.25 All statistical analyses were performed using SAS version 9.3 (SAS Institute, Cary, NC).
Results
A total of 740 subjects passed QC and were included in the analyses. Of these, 399 (54%) were from the ROS and 341 (46%) from the MAP. The mean (SD) age at death was 88.0 (6.7) years, 270 (36%) were male, and 447 (61%) had a pathological diagnosis of AD. We focus our study on the index SNPs that have been reported to best capture the association of a given locus with AD susceptibility. Associations of these index SNPs to our primary outcome, the burden of NP pathology and pathological diagnosis of AD (AD) in our data set are presented in Table1.
Table 1.
Associations between CpGs or index SNPs and NP burden and AD diagnosis
| SNP | Position | Gene | SNP to NP association | SNP to AD association |
|---|---|---|---|---|
| P-value | P-value | |||
| rs6656401 | 1:207692049 | CR1 | 0.0169 | 1.1E-04 |
| rs744373 | 2:127894615 | BIN1 | 0.1790 | 0.2243 |
| rs9349407 | 6:47453378 | CD2AP | 0.4393 | 0.6703 |
| rs11767557 | 7:143109139 | EPHA1 | 0.4565 | 0.6931 |
| rs11136000 | 8:27464519 | CLU | 0.4878 | 0.4364 |
| rs610932 | 11:59939307 | MS4A6A | 0.7040 | 0.8746 |
| rs4938933 | 11:60034429 | MS4A4A | 0.9301 | 0.9305 |
| rs7110631 | 11:85856187 | PICALM | 0.3863 | 0.5214 |
| rs3752246 | 19:1056492 | ABCA7 | 0.2102 | 0.6958 |
| anye4 | APOE | 3.8E-23 | 1.6E-13 | |
| rs3865444 | 19:51727962 | CD33 | 0.1233 | 0.0951 |
| BIN1 (53 tested) | Position | Chromatin state | CpG to NP association | CpG to AD association | ||
|---|---|---|---|---|---|---|
| P-value | Direction* | P-value | Direction* | |||
| cg22883290 | 127800646 | Weak transcribed/low signal proximal to active regions | 3.7E-08 | + | 1.5E-05 | + |
| cg19153828 | 127782651 | Strong promoter | 0.0002 | + | 0.0012 | + |
| cg04019522 | 127852450 | Active enhancer | 0.0002 | + | 1.1E-04 | + |
| cg09006514 | 127800616 | Weak transcribed/low signal proximal to active regions | 0.0013 | + | 0.0060 | + |
| cg18981277 | 127805910 | Strong transcription | 0.0022 | − | 0.0232 | − |
| Position | Chromatin state | CpG to NP | CpG to AD | |||
|---|---|---|---|---|---|---|
| CD2AP (20 tested) | ||||||
| cg20172563 | 47487173 | Low signal | 7.5E-06 | + | 0.0043 | + |
| CLU (40 tested) | ||||||
| cg16292768 | 27467783 | Active TSS flanking | 3.7E-05 | − | 0.0003 | − |
| cg08594681 | 27468684 | Active TSS flanking | 8.4E-04 | − | 0.0847 | − |
| cg11783834 | 27469331 | Active TSS flanking | 0.0029 | − | 0.0976 | − |
| cg13488078 | 27469338 | Active TSS flanking | 0.0029 | − | 0.0034 | − |
| cg20918219 | 27493854 | Weak enhancer | 0.0035 | + | 0.0025 | + |
| MS4A6A (12 tested) | ||||||
| cg03055440 | 59950405 | Low signal | 0.0015 | − | 0.0012 | − |
| cg07170641 | 59950068 | Low signal | 0.0016 | − | 0.0161 | − |
| cg18159934 | 59942032 | Low signal | 0.0030 | + | 0.0583 | + |
| ABCA7 (91 tested) | ||||||
| cg02308560 | 1071176 | PC repressed | 3.1E-08 | + | 5.0E-06 | + |
| cg04587220 | 1071208 | PC repressed | 5.0E-06 | + | 2.3E-04 | + |
| APOE (41 tested) | ||||||
| cg18799241 | 45412599 | Weak transcribed/low signal proximal to active regions | 6.2E-04 | + | 0.0241 | + |
Only those features meeting a Benjamini-Hochberg threshold of significance are presented. All models are adjusted for age at death, sex, study,batch effect, and bisulfide conversion QC probes. CR1 (31 CpGs), EPHA1 (60 CpGs), MS4A4A (11 CpGs), PICALM (25 CpGs) and CD33 (12 CpGs) showed no CpGs that met multiple comparisons cutoff.
“+” means more methylation is associated with more pathology or increased odds of AD, “−” means more methylation is assoicated with less pathology or decreased odds of AD.
Figure1 outlines the four different models that we considered in our analyses. For all 11 AD susceptibility loci, we explored whether DNA methylation is influenced by the index SNP and also whether DNA methylation is associated with our outcome measures, independent of genetic variation. Since, in our data, only the APOE and CR1 loci demonstrate a genetic association with the outcome measures – which is a prerequisite for testing mediation and reverse causality – all analyses testing for evidence of CpG-mediated effect of SNPs and reverse causality (Fig.1, models 2 and 3) were limited to these two loci. Finally, for all loci, we also tested for evidence of interaction between the index SNPs and methylation levels of the interrogated CpGs (Fig.1, model 4).
Assessing the relation of DNA methylation in AD loci to AD pathology (CpG → Y)
We first assessed whether methylation levels of CpGs found in AD susceptibility loci are associated with our primary outcome measure, an individual’s burden of NP pathology (Table1). The gene with one of the most CpGs associated with NP burden is BIN1 with five CpGs out of 53 tested meeting the Benjamini-Hochberg corrected threshold of significance, and four of the five showing a positive direction, indicating more methylation is associated with higher burden of NP pathology and increased odds of AD (Table1 and Fig.2). When accounting for the CpG with the strongest association (cg22883290, P = 3.7 × 10−8) two CpGs remain significant (cg04019522, P = 0.001 and cg18981277, P = 0.002), and when accounting for the effect of the top two, cg22883290 and cg04019522, cg18981277, is still significant (P = 0.003) suggesting that there are independent effects of CpGs within this locus (Table S1 and Fig.2). A similar relationship is seen in MS4A6A, where three CpGs out of 12 CpGs tested, are independently associated with pathology burden and all three CpGs remain significant when controlling for the other two, (cg03055440, P = 0.02, cg07170641, P = 0.04, and cg18159934, P = 0.004) (Table S1 and Fig.3). Another locus, CLU had five CpGs (out of 40 tested) significantly associated with NP pathology. In this case four out of the five CpGs showing a negative direction indicating that more methylation is associated with lower burden of NP pathology and lower odds of AD. When accounting for the top CpG (cg16292768, P = 3.7 × 10−5) one CpG remains significant (cg20918219, P = 0.0004) suggesting that this CpG appears to have an effect independent of the top CpG cg16292768 (Table S1 and Fig.4). The ABCA7, APOE, and CD2AP loci harbor only single CpGs with a significant association, and the remaining loci had no CpGs meeting the threshold of significance (Figs. S1–S3). Altogether, the 17 CpGs across the six gene regions that meet the threshold of significance explain 16.8% of the variability in NP. By reducing the analysis to the subset of subjects who were cognitively nonimpaired at the time of death, we see that the effect size of many of the associated CpGs is not altered (Table S2), indicating that the association of DNA methylation at certain sites with AD pathology is not a feature only of the demented state. These alterations in DNA methylation within known AD susceptibility loci therefore occur early in the pathologic process with BIN1, CD2AP, ABCA7, and APOE primarily in the positive direction and CLU and MS4A6A in the negative direction, but we cannot, at this time, distinguish whether they are a cause or an effect of NP pathology.
Figure 2.

AD susceptibility loci BIN1 displays multiple, independent associations of CpG methylation with neuritic plaque (NP) burden. The figure represents the results of analyses for the BIN1 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds report the results of the primary model relating CpG methylation and NP burden using a model adjusted for age at death, sex, batch, study, and bisulfite conversion fraction. The black diamonds report the results of the primary model with the additional adjustment for the top CpG, cg22883280, to identify secondary, independent associations such as cg04019522. The blue diamonds report the primary model adjusted for both cg22883290 and cg04019522 showing a third independently associated CpG, cg08663425. All P-values are reported in Table S1.
Figure 3.
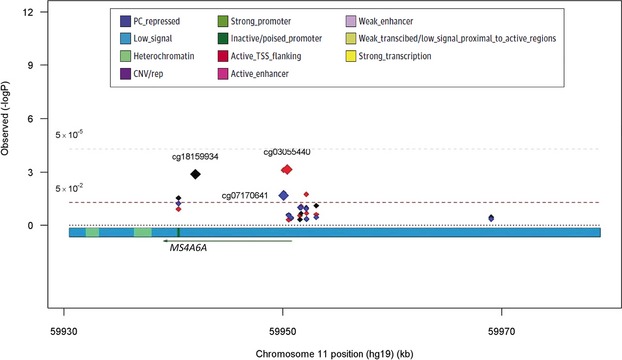
AD susceptibility loci MS4A6A displays multiple, independent associations of CpG methylation with neuritic plaque (NP) burden. The figure represents the results of analyses for the MS4A6A region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds are from the primary model testing for NP association using a model adjusted for age at death, sex, batch, study, and bisulfite conversion fraction. The black diamonds report the results of the primary model with the additional adjustment for the top CpG, cg03055440 and the blue diamonds show the results from the primary model adjusted for the top two independent CpGs, cg03055440 and cg1815993, showing a third independently associated CpG, cg07170641. All P-values are reported in Table S1.
Figure 4.

AD susceptibility loci CLU displays multiple, independent associations of CpG methylation with neuritic plaque (NP) burden. The figure represents the results of analyses for the CLU region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds are from the primary model testing for NP association. The black diamonds report the results of the primary model with the additional adjustment for the top CpG, cg16292768. All P-values are reported in Table S1.
AD susceptibility alleles influence DNA methylation in the aging brain (SNP → CpG)
Given that the selected loci all harbor genetic variation associated with AD susceptibility in published literature, we next assessed whether the index SNPs that have been identified and validated in the large genome-wide association studies influence the level of DNA methylation at the associated CpG (Table S3). With the exception of PICALM, all genes showed at least one CpG with an association to the index SNP meeting Benjamini-Hochberg correction (Table S3). For example, in the BIN1 locus, the index SNP (rs744373) influences the level of DNA methylation at 13 CpGs, 25% of the probes in the BIN1 region, suggesting that the SNP has a large effect on the methylation status of the region. However, the level of methylation of the affected CpGs is not strongly correlated with the level of NP pathology or AD susceptibility. To explore this question more explicitly, we investigated the possibility that the effects of SNPs and CpGs on pathology may be largely independent of one another.
Assessing the independent association model (1)
The first type of independent association model that we assess is one in which the level of CpG methylation is associated with both the index SNP and NP burden, but the SNP and NP burden are not associated (Fig.1, model 1). We see multiple examples of this in three genes. For example, in the BIN1 region, the level of methylation of one CpG (cg22883290) is associated with the index SNP, rs744373 (P = 1.2 × 10−4), and the NP trait (P = 3.7 × 10−8) (Table2). While we appreciate the fact that the lack of association between the BIN1 SNP and NP could be due to lack of power (pNP = 0.18, pAD = 0.22), the beta estimate for the association between the BIN1 index SNP rs744373 and NP is close to zero (β = 0.04) suggesting little to no association. Nevertheless, we tested whether controlling for the SNP modifies the association between the CpG and NP and found that the cg22883290 association with NP remains after adjusting for the index SNP, rs744373, (P = 5.4 × 10−7). This suggests that the association between cg22883290 and NP is independent of the BIN1 susceptibility SNP. This is a case where the index SNP does not appear to influence the associations between the CpG and NP.
Table 2.
Assessment of independent associations between CpGs and neuritic plaque burden
| BIN1 | Position | SNP to CG association P-value | CG to NP association | CG to NP association Adjusted for rs744373 | ||
|---|---|---|---|---|---|---|
| CG | β | P-value | β | P-value | ||
| cg22883290 | 127800646 | 1.2E-04 | 4.42 | 3.7E-08 | 4.25 | 5.4E-07 |
| CD2AP | Position | SNP to CG association | CG to NP association | CG to NP association Adjusted for rs9349407 | ||
|---|---|---|---|---|---|---|
| CG | P-value | β | P-value | β | P-value | |
| cg20172563 | 47487173 | 8.1E-25 | 3.48 | 7.5E-06 | 4.10 | 2.8E-06 |
| CLU | Position | SNP to CG association | CG to NP association | CG to NP association Adjusted for rs11136000 | ||
|---|---|---|---|---|---|---|
| CG | P-value | β | P-value | β | P-value | |
| cg16292768 | 27467783 | 1.2E-09 | −2.34 | 3.7E-05 | −2.32 | 1.0E-04 |
| cg08594681 | 27468684 | 1.1E-05 | −2.77 | 0.0008 | −2.68 | 0.002 |
| cg13488078 | 27469338 | 0.0020 | −2.63 | 0.0029 | −2.46 | 0.009 |
| MS4A6A | Position | SNP to CG association | CG to NP association | CG to NP association Adjusted for rs610932 | ||
|---|---|---|---|---|---|---|
| CG | P-value | β | P-value | β | P-value | |
| cg03055440 | 59950405 | 0.0099 | −2.16 | 0.0015 | −2.12 | 0.003 |
All models are adjusted for age at death, sex, study, batch, and bisulfite conversion fraction.
Similarly, in the CLU region, there are three CpGs associated with both the index SNP, rs11136000, (cg16292768, P = 1.2 × 10−9; cg08594681, P = 1.1 × 10−5; and cg13488078, P = 0.002) and NP (P = 3.7 × 10−5, 8.4 × 10−4, and 0.003, respectively) (Table2). Including the CLU SNP in the model does not significantly alter the relation of CpG and NP. As with BIN1, the index CLU SNP is not significantly associated with NP (β = −0.02, P = 0.48), again suggesting that the effects of CpG methylation on NP appear to be independent of AD susceptibility alleles. The same is true in two other AD loci, CD2AP and MS4A6A in which index SNPs influence methylation levels (Table2). Thus, in these AD loci, the role of susceptibility alleles and DNA methylation appear to be largely independent in the CpGs sampled by the array. Similar results are found in CLU, BIN1, and MS4A6A when we consider an AD diagnosis as the outcome measure (Table S4).
A second type of independent association occurs when the SNP is associated with the outcome, and methylation level is associated with the outcome, but the SNP and the methylation level are not associated (Fig.1, model 1). We can only test this hypothesis within APOE and CR1 because it is only in these two loci that genetic variation are associated with NP burden in our data set of moderate size. In the CR1 region, there are no CpGs that reach significance with the Benjamini-Hochberg correction; however, in the APOE region there is one CpG that reaches significance for NP, cg18799241 (P = 6.2 × 10−4) (Table1). That CpG is also modestly associated with APOE ε4 (P = 0.04), suggesting that cg18799241 may have some effect independent of APOE ε4. To assess this effect, we calculated that the APOE ε4 haplotype (adjusting for age at death, sex, study, and batch) explains 15.4% of the variance in NP in our data. cg18799241 explains 4.7% of the variance in this trait, and the model that includes both the APOE ε4 haplotype and cg18799241 explains 16.3% of the variance. Thus, in the case of the APOE locus, it is unclear if the variation in methylation contributes to an individual’s risk of developing neuritic pathology independently of the effect of genetic variation.
Assessing the CpG-mediated effect model (2)
Since a significant association between the SNP and outcome is required to test the CpG-mediated effect model, we can only address the interesting question of whether methylation of certain CpGs mediates the effect a susceptibility allele on AD pathology in the APOE (P = 3.8 × 10−23) and CR1 index SNP, rs6656401 (P = 0.017) (Table1). The analysis is outlined in our proposed models for the role of DNA methylation (Fig.1, model 2); here, we assess whether adding the CpG to the model relating SNP to NP attenuates the effect of the SNP on NP.
Of the 41 CpGs interrogated in the region of the APOE gene by the Illumina beadset, the level of methylation of eight CpGs is significantly associated with the APOE ε4 haplotype (P-values between 0.006 and 5.1 × 10−11); however, none of those CpGs met the multiple comparison threshold for association with NP burden, although four displayed suggestive evidence of association (P < 0.05) (Table3). When the CpGs are added to the model associating APOE ε4 to NP, they do not diminish this association, suggesting that the effect of the APOE ε4 haplotype on NP is not mediated by CpGs in which the level of methylation is influenced by the APOE ε4 haplotype.
Table 3.
Assessment of CpG mediation model with neuritic plaque burden and CpGs and AD diagnosis
| CR1 | Position | CR1 to CpG association | CR1 to NP association Adjusting for CpG1 | CR1 to AD association Adjusting for CpG | ||
|---|---|---|---|---|---|---|
| CG | P-value | β | P-value | β | P-value | |
| rs6656401 without adjusting for CpG | 0.09 | 0.0169 | 0.65 | 1.0E-04 | ||
| cg19373649 | 207817630 | 5.2E-09 | 0.09 | 0.0227 | 0.67 | 9.3E-05 |
| cg26119563 | 207817838 | 1.8E-07 | 0.09 | 0.0208 | 0.66 | 1.0E-04 |
| cg23026906 | 207819196 | 7.8E-04 | 0.08 | 0.0359 | 0.65 | 1.0E-04 |
| APOE | Position | APOE e4 to CpG association | APOE e4 to NP association | APOE e4 to AD association | ||
|---|---|---|---|---|---|---|
| Adjusting for CpG1 | Adjusting for CpG | |||||
| CG | P-value | β | P-value | β | P-value | |
| any e4 without adjusting for CpG | 0.41 | 3.7E-23 | 1.57 | 1.7E-13 | ||
| cg09379229 | 45417668 | 5.1E-11 | 0.41 | 3.8E-22 | 1.63 | 1.7E-13 |
| cg02613937 | 45395297 | 5.5E-11 | 0.44 | 2.9E-24 | 1.69 | 2.4E-14 |
| cg14123992 | 45407868 | 2.5E-10 | 0.43 | 8.7E-24 | 1.63 | 1.1E-13 |
| cg04406254 | 45407945 | 2.7E-10 | 0.42 | 3.4E-23 | 1.64 | 7.8E-14 |
| cg07773593 | 45417793 | 4.0E-05 | 0.41 | 2.2E-22 | 1.57 | 2.6E-13 |
| cg00397545 | 45417526 | 5.9E-04 | 0.43 | 7.2E-25 | 1.63 | 4.3E-14 |
| cg08121984 | 45429870 | 0.004 | 0.41 | 7.0E-23 | 1.57 | 2.1E-13 |
| cg01032398 | 45408121 | 0.006 | 0.41 | 2.2E-22 | 1.56 | 2.8E-13 |
All models are adjusted for age at death, sex, study, batch effect, and bisulfide conversion QC probes.
Weak transcribed/low signal proximal to active regions.
In the region of the CR1 locus, there are 31 CpGs targeted by the Illumina chip. We see a significant association between the CR1 index SNP rs6656401 and NP (P = 0.017) (Table1). We also see associations between rs6656401 and three CpGs, cg19393649 (P = 5.2 × 10−9), cg26119563 (P = 1.8 × 10−7) and cg23026906 (P = 7.8 × 10−4) (Table S3). When controlling for each of these CpGs, the association between rs6656401 and NP burden did not change (Table3) indicating that methylation at these CpGs does not mediate the effect of CR1 on NP accumulation.
Assessing reverse causality model (3)
Using methods similar to those used in testing model 2, we see no evidence of reverse causality in either the APOE or the CR1 loci. Neither NP burden nor AD diagnosis mediate the association between the genetic variants (APOE e4 haplotype or CR1 risk allele) and the CpGs (data not shown).
Assessing interaction model (4)
The final set of analyses explored the possibility of interactions between the effect of CpGs and genetic variation (Fig.1, Model 4). We see a significant interaction between the index SNP rs6656401 and two CpGs in the CR1 region, cg10021878 and cg05922028 and suggestive for cg00175709 for both AD diagnosis (P’s for interactions, cg10021878 P = 0.001, cg05922028 P = 0.01, cg00175709 P = 0.05) and NP burden (P’s for interactions cg10021878 P = 0.01, cg05922028 P = 0.009, and cg00175709 P = nonsignificant) (Table4). In all three CpGs, we see that the level of methylation is negatively associated with NP burden and odds of AD diagnosis in those subjects homozygous for the protective allele (rs6656401TT) and positively associated in those subjects bearing at least one copy of the risk allele (rs6656401AT/AA) (Table4). This interaction is also seen in the variance explained by each CpG and a combination of all three. In all three CpGs and for both NP and AD diagnosis, the CpGs explain more of the variance in the outcome for those with the risk allele (rs6656401AT/AA) than those without (rs6656401TT). For example, cg10021878 explains 19.2% of the variance in AD diagnosis for those with rs6656401AT/AA but only 10.4% for those with rs6656401TT, suggesting that DNA methylation within the CR1 locus plays a larger role in AD diagnosis for those with the CR1 risk allele than those without (Table4). We did not find evidence of significant interaction for any other SNP.
Table 4.
Interaction effects between CR1 and DNA methylation
| TT (n = 453) | AT/AA (n = 239) | P-interaction | ||||||
|---|---|---|---|---|---|---|---|---|
| β (SE) | P-value | R 2 | β (SE) | P-value | R 2 | |||
| Neuritic plaque burden | ||||||||
| cg10021878 | 207669922 | −0.84 (0.35) | 0.02 | 3.7% | 0.76 (0.57) | 0.18 | 4.7% | 0.014 |
| cg05922028 | 207669334 | −0.61 (0.89) | 0.49 | 2.6% | 3.57 (1.33) | 0.008 | 6.9% | 0.009 |
| cg00175709 | 207670014 | −2.73 (1.06) | 0.01 | 3.9% | 1.26 (1.86) | 0.48 | 4.2% | 0.5 |
| All three together | 3.5% | 6.5% | ||||||
| AD diagnosis | ||||||||
| cg10021878 | 207669922 | −4.4 (1.5) | 0.003 | 10.4% | 4.9 (3.0) | 0.1 | 19.2% | 0.001 |
| cg05922028 | 207669334 | −3.9 (3.6) | 0.27 | 8.2% | 12.1 (6.8) | 0.07 | 19.4% | 0.01 |
| cg00175709 | 207670014 | −11.8 (4.4) | 0.007 | 9.9% | 5.5 (8.7) | 0.53 | 17.9% | 0.05 |
| All three together | 10.4% | 21.0% | ||||||
All models are adjusted for age at death, sex, study, batch effect, and bisulfide conversion QC probes.
Discussion
In our focused analysis of regions previously associated with genetic susceptibility to AD, we find a number of alterations in DNA methylation that relate to NP burden. In the ABCA7, CD2AP, CLU, and MS4A6A loci, we find evidence for greater methylation of certain CpGs in relation to AD pathology and find that these changes occur early, prior to the appearance of cognitive impairment, as evidenced by the fact that the association remains after subjects with mild cognitive impairment or AD proximate to death are removed from the analysis. In these loci, the associated methylation changes are not driven by the AD-associated genetic variants, and there appears to be a single effect in each locus. In the BIN1 locus, which has strong evidence of association at multiple different sites (Fig.2), we see that the CpG that best captures the association, cg22883290, is influenced in part by rs744373, the index BIN1 SNP, however, its association with NP remains when we account for the effect of this SNP. This suggests that other, unknown factors influence its methylation and perhaps AD susceptibility through alterations of DNA methylation. Similarly, we see associations of methylation levels to AD pathology that are unrelated to susceptibility variants in the ABCA7, CD2AP, CLU, and MS4A6A loci. Overall, it appears that methylation levels in many AD susceptibility loci relate to NP pathology and are relatively independent of genetic variation. While these alterations are not dependent on the presence of cognitive impairment, it is not yet clear whether methylation changes are a cause or an effect of AD pathology. However, the presence of multiple, independent associations of CpG with NP in loci such as BIN1, CLU, and MS4A6A further refines our perspective on epigenomic changes in AD: the independence of these associations suggests that they are not driven by a single mechanism such as a simple increase in NP burden. They are either driven by (1) different risk factors whose effects on AD pathology converge on different genomic segments within BIN1 and the other genes or (2) different molecular processes that are responding to the presence of AD pathophysiology. Both models are plausible and although our subjects are from a longitudinal study, the brain samples are cross sectional which limits our ability to assess pathways and causality. Parsing out pathways and models will require experimental investigation.
In two of the AD loci, we had the opportunity to evaluate the possibility that DNA methylation mediates the effect of the associated variant since both the CR1 index SNP and the APOE e4 haplotype as well as some of the CpGs in each locus are associated with NP burden and AD, which is a prerequisite to formally test mediation. In the APOE locus, we see that there are large DNA methylation changes that are related to the APOE e4 haplotype, but these do not appear to mediate the effect of the haplotype on the outcome measures. On the other hand, we find evidence of a different CpG, cg18799241, whose increased level of methylation is associated with greater NP burden, independent of APOE e4. This suggests, as in the other loci above, that nongenetic factors may be influencing AD pathology through alterations in DNA methylation which can manifest as alterations in transcriptional patterns.
In the CR1 locus, we find something unique among the AD susceptibility loci: evidence for interaction between the AD susceptibility allele and the level of methylation of certain CpGs (Table4). It is intriguing that the level of methylation within a small segment of the CR1 gene may have a differential effect on the accumulation of AD pathology based on the presence of an intronic variant 22 kb away. The index SNP rs6656401 tags the haplotype that contains a chromosomal segment duplication that includes a duplicated LHR domain, LHR-S26 (Fig.5), and the resulting protein isoform (CR1 slow form, CR1-S) encoded by this haplotype is structurally and perhaps functionally26 different from the reference CR1 protein isoform, CR1 fast (CR1-F). Thus, mechanistically, altering the transcriptional state of CR1 may have different biologic effects when the expression of the reference CR1-F or CR1-S form of CR1 is expressed. Previous studies suggested that the haplotype containing CR1-S is also associated with greater expression of this LHR-S form, so influencing expression using a nongenetic mechanism could also affect AD pathology. The associated CpGs are found in a segment of DNA which is assigned to an “inactive, poised promoter” conformation in our reference map derived from cognitively nonimpaired subjects displaying minimal brain pathology on autopsy.27 This suggests that alterations in DNA methylation take place in a region of the CR1 gene that can respond to dynamic changes.
Figure 5.
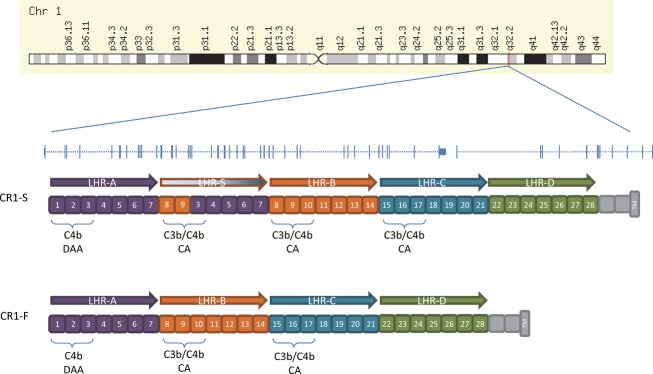
Genomic architecture of the CR1 locus in relation to the major CR1 isoforms.
Overall, our study targeted genomic regions implicated in AD susceptibility because they harbor AD risk alleles, indicating that other forms of genomic variation – in this case DNA methylation – in the target organ are independently associated with the risk of developing AD. One limitation of our analysis is that our interrogation of these loci is not comprehensive since we are limited to the CpGs tested by the Illumina beadset; however, so far, we do not see evidence that the effect of disease-associated genetic variation is mediated by alterations in chromatin conformation, as marked by changes in DNA methylation. A picture of integration among different sources of variation (genetic and epigenomic) is therefore emerging in the brain of aging individuals, with these different factors contributing to the accumulation of AD pathology and, ultimately, a diagnosis of AD. Further, with the effects of loci such as BIN1 and CR1 that are detailed in this manuscript, there are now precise hypotheses that we can pursue in model systems to test the challenging question of the direction of causality for the associated methylation changes.
Acknowledgments
We thank all the participants of the Religious Orders Study and the Rush Memory and Aging Project, as well as the staff at the Rush Alzheimer’s Disease Center for this work. This work was supported by the National Institutes of Health (grant K25AG041906 P30AG10161, R01AG15819, R01AG17917, R01AG36042, R01AG36836, and U01AG46152).
Conflict of Interest
Dr. Bennett and Dr. Schneider reports grants from NIH, grants from Illinois Dept Public Health, during the conduct of the study.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. AD susceptibility loci ABCA7 display single associations of CpG methylation with NP burden. The figure represents the results of analyses for the ABCA7 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds report the results of the primary analysis relating CpG methylation in the ABCA7 region and NP burden using a model adjusted for age at death, sex, batch, study, and bisulfite conversion fraction.
Figure S2. AD susceptibility loci APOE display single associations of CpG methylation with NP burden. The figure represents the results of analyses for the ABCA7 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds report the results of the primary analysis relating CpG methylation in the APOE region and NP burden using a model adjusted for age at death, sex, batch, study, and bisulfite conversion fraction.
Figure S3. AD susceptibility loci CD2AP display single associations of CpG methylation with NP burden. The figure represents the results of analyses for the ABCA7 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16
Table S1. Associations between CpG and NP burden and CpGs and AD diagnosis, comparing models adjusting for one, two or three associated CpGs.
Table S2. Associations between CpG and neuritic plaque burden only among those with no cognitive impairment at death.
Table S3. Associations between index SNPs and CpGs, with only those meeting multiple comparison criteria.
Table S4. Assessment of independent associations between CpGs and AD diagnosis.
References
- Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo MM, Zou F, Pankratz VS, et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer’s disease. Nat Genet. 2009;41:192–198. doi: 10.1038/ng.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Genome-wide association studies in Alzheimer’s disease. Hum Mol Genet. 2009;18(R2):R137–R145. doi: 10.1093/hmg/ddp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibnik LB, Shulman JM, Leurgans SE, et al. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann Neurol. 2011;69:560–569. doi: 10.1002/ana.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan BT, Shulman JM, Chibnik LB, et al. A coding variant in CR1 interacts with APOE-epsilon4 to influence cognitive decline. Hum Mol Genet. 2012;21:2377–2388. doi: 10.1093/hmg/dds054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, Chen K, Keenan BT, et al. Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol. 2013;70:1150–1157. doi: 10.1001/jamaneurol.2013.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Zou F, Chai HS, et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology. 2012;79:221–228. doi: 10.1212/WNL.0b013e3182605801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis J, Hansmannel F, Gistelinck M, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013;18:1225–1234. doi: 10.1038/mp.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj T, Shulman JM, Keenan BT, et al. Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am J Hum Genet. 2012;90:720–726. doi: 10.1016/j.ajhg.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez JB, Fardo DW, Estus S. ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci Lett. 2013;556:58–62. doi: 10.1016/j.neulet.2013.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Buchman AS, et al. Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res. 2012;9:646–663. doi: 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the religious orders study. Curr Alzheimer Res. 2012;9:628–645. doi: 10.2174/156720512801322573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Shulman JM, Chibnik LB, et al. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol Aging. 2012;33:1017.e1–1017.e15. doi: 10.1016/j.neurobiolaging.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Beecham GW, Martin ER, et al. Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genet. 2010;6:e1001130. doi: 10.1371/journal.pgen.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Tang Y, et al. The effect of social networks on the relation between Alzheimer’s disease pathology and level of cognitive function in old people: a longitudinal cohort study. Lancet Neurol. 2006;5:406–412. doi: 10.1016/S1474-4422(06)70417-3. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, et al. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer’s disease. Neurology. 2003;60:246–252. doi: 10.1212/01.wnl.0000042478.08543.f7. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol. 1995;57:289–300. [Google Scholar]
- Brouwers N, Van Cauwenberghe C, Engelborghs S, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Muller F, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. AD susceptibility loci ABCA7 display single associations of CpG methylation with NP burden. The figure represents the results of analyses for the ABCA7 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds report the results of the primary analysis relating CpG methylation in the ABCA7 region and NP burden using a model adjusted for age at death, sex, batch, study, and bisulfite conversion fraction.
Figure S2. AD susceptibility loci APOE display single associations of CpG methylation with NP burden. The figure represents the results of analyses for the ABCA7 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16 The red diamonds report the results of the primary analysis relating CpG methylation in the APOE region and NP burden using a model adjusted for age at death, sex, batch, study, and bisulfite conversion fraction.
Figure S3. AD susceptibility loci CD2AP display single associations of CpG methylation with NP burden. The figure represents the results of analyses for the ABCA7 region, with the physical position presented on the x axis and −log10 P-values presented in the y axis. Diamonds represent the individual CpGs across the gene region that were tested in the analysis. The state of the chromatin in the dorsolateral prefrontal cortex of a cognitively nonimpaired individual is presented below the x axis; this map is based on the derivation of chromatin state in 200 bp segments using data generated from the immunoprecipitation of chromatin marks. The chromatin states are defined in a box at the top right of the figure.16
Table S1. Associations between CpG and NP burden and CpGs and AD diagnosis, comparing models adjusting for one, two or three associated CpGs.
Table S2. Associations between CpG and neuritic plaque burden only among those with no cognitive impairment at death.
Table S3. Associations between index SNPs and CpGs, with only those meeting multiple comparison criteria.
Table S4. Assessment of independent associations between CpGs and AD diagnosis.