

Abstract
The orphan nuclear receptor, retinoic acid receptor-related orphan nuclear receptor γt (RORγt), is required for the development and pathogenic function of interleukin-17A-secreting CD4+ T helper type 17 (Th17) cells. Whereas small molecule RORγt antagonists impair Th17 cell development and attenuate autoimmune inflammation in vivo, the broader effects of these inhibitors on RORγt-dependent gene expression in vivo has yet to be characterized. We show that the RORγt inverse agonist TMP778 acts potently and selectively to block mouse Th17 cell differentiation in vitro and to impair Th17 cell development in vivo upon immunization with the myelin antigen MOG35–55 plus complete Freund's adjuvant. Importantly, we show that TMP778 acts in vivo to repress the expression of more than 150 genes, most of which fall outside the canonical Th17 transcriptional signature and are linked to a variety of inflammatory pathologies in humans. Interestingly, more than 30 genes are related with SMAD3, a transcription factor involved in the Th17 cell differentiation. These results reveal novel disease-associated genes regulated by RORγt during inflammation in vivo, and provide an early read on potential disease indications and safety concerns associated with pharmacological targeting of RORγt.
Keywords: autoinflammatory disease, cytokines, T cells
Introduction
Naive CD4+ T cells differentiate into a variety of effector and regulatory subsets, including T helper type 1 (Th1), Th2, induced T regulatory (iTreg) cells, and Th17 cells.1–4 Th1 cells are characterized by transcription factor T-bet and the production of interferon-γ (IFN-γ), whereas Th2 cells are characterized by transcription factor GATA3 and the production of interleukin-4 (IL-4), Induced Treg cells, like their thymic-derived ‘nTreg’ cell counterparts, selectively express the transcription factor Foxp3 and suppress bystander T-cell activation to regulate inflammatory T-cell responses. In contrast, Th17 cells are defined by production of IL-17A, IL-17F, IL-21 and IL-22, and these cells play a central role in autoimmune pathogenesis.4–7 Human monoclonal antibodies against IL-17A or IL-17 receptor A (IL-17RA) have demonstrated clinical efficacy in both psoriasis and rheumatoid arthritis.7–9 Differentiation of Th17 cells requires the retinoic acid receptor-related orphan nuclear receptor RORγt,10,11 which is induced during human and mouse naive T-cell activation in the presence of cytokines such as IL-6, transforming growth factor-β (TGF-β) and IL-1β.4,6,12
The differentiation of Th17 cells in vivo is more complicated, and can lead to functionally distinct pro-inflammatory and anti-inflammatory Th17 cell subsets. Interleukin-23 is critical for both the maintenance of IL-17A expression as well as pathogenic Th17 cell function in experimental autoimmune encephalomyelitis (EAE).13–15 ‘Pathogenic’ Th17 cells express Th17 cytokines together with IFN-γ; they express both RORγt and T-bet, and they are distinguished from non-pathogenic Th17 cells by their selective expression of the multidrug transporter, MDR1.16 In addition to RORγt and T-bet, the Runx family transcription factors, namely Runx1 and Runx 3, have been recently shown to control IFN-γ production by pathogenic Th17 cells.17 In mouse and human CD4+ T cells, as well as in γδ Τ cells, IL-1β and IL-23 are important, both for high-level IL-17A production and development of autoimmune inflammation. Given that RORγt is expressed in both pathogenic and non-pathogenic Th17 cells in vivo, it is unlikely that the full repertoire of RORγt-dependent transcriptional activity in vivo has been captured by previous transcriptional profiling experiments using Th17 cells generated in vitro.
Several small molecule inhibitors targeting RORγt have been described previously.11,18–20 These studies provide a proof-of-principle that pharmacological inhibition of RORγt can regulate Th17 cell differentiation (and IL-17A production) in vitro. In vivo, administration of these compounds shows variable levels of efficacy in EAE. Previous work from our laboratory has described a novel, potent and selective RORγt inverse agonist TMP778.21,22 TMP778 inhibits human Th17 signature gene expression in vitro; and targeting of RORγt in mice via TMP778 administration reduces imiquimod-induced psoriasis-like cutaneous inflammation21 and severity of EAE progression.23 Here we demonstrate that TMP778 inhibits mouse Th17 cell development in vivo in two different mouse model systems. Further, we show that TMP778 regulates expression of more than 150 genes during inflammatory Th17 cell differentiation. Understanding the broader activity of RORγt inhibitors on gene expression during physiological inflammation is vital to advancing this class of compounds for clinical development.
Materials and methods
Mice
Female C57BL/6 mice (6–8 weeks) were purchased from The Jackson Laboratory and were housed in a pathogen-free barrier facility at Tempero Pharmaceuticals. Green fluorescent protein-tagged IL-17 (IL-17A-GFP) knock-in mice were licensed from Biocytogen, Inc. (Worcester, MA), bred at the Jackson Laboratory (Bar Harbor, ME), and shipped to Tempero for experiments. All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at GSK.
Mouse T-cell culture
Spleens were isolated from C57BL/6 mice and single-cell suspensions were prepared by passing the spleen through a 70-μm filter. Red blood cells were removed by lysis with ammonium chloride. Cells were stimulated for 3 days with anti-CD3 (1 µg/ml)/anti-CD28 (0·5 µg/ml) monoclonal antibodies (eBioscience, San Diego, CA) plus the indicated cytokines and antibodies (eBioscience) at the following concentrations: IL-6 (10 ng/ml), TGF-β (1 ng/ml), IL-12 (5 ng/ml), IL-4 (10 ng/ml), anti-IL-4 monoclonal antibody (2 µg/ml), anti-IL-12p40 monoclonal antibody (1 µg/ml). Cytokine titres and cell proliferation were assessed and values of IC50 were determined using graphpad prism (GraphPad, La Jolla, CA). For mouse Th17 differentiation experiments, naive mouse CD4+ T cells were stimulated with anti-CD3/anti-CD28 antibodies plus IL-6/TGF-β in the presence of TMP778 or Digoxin for 4 days. Cells were then re-stimulated with PMA (10 nm) and ionomycin (1 µm) in the presence of brefeldin A (5 µg/ml) (all from Sigma, St Louis, MO) for 3 hr before intracellular staining for IL-17 and IFN-γ as previously described.24
Ex vivo myelin oligodendrocyte glycoprotein recall studies
C57BL/6 mice were immunized with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35–55) in complete Freund's adjuvant (CFA), and 20 mg/kg TMP778 or Vehicle (3% dimethylacetamide, 10% solutol, and 87% saline) was administered twice daily via subcutaneous injection. On day 6, draining lymph node cells were isolated and stimulated with 50 µg/ml MOG35–55 in T-cell culture medium as described previously.21 Cells were harvested 20 hr later for RNA isolation and gene expression analysis, and cell supernatants were harvested 5 days after MOG stimulation for detection of IL-17 by Meso Scale Discovery (Rockville, MD). Interleukin-17A-GFP mice were immunized and treated with or without TMP778 as described above. Draining lymph node cells were isolated on day 9, and total mononuclear cells were stimulated with PMA/ionomycin for 3 hr in the presence of brefeldin A. Cells were then stained and analysed. The integrated mean fluorescence intensity MFI (iMFI) was calculated at the percentage of positive cells multiplied by the MFI. For each experiment, at least 10 mice were used for vehicle treatment at least 10 mice for each compound treatment.
Cell proliferation
Cell proliferation was measured using CellTiter-Glo as instructed by the manufacturer (Promega, Madison, WI), with luminescence read on a FLUOstar OPTIMA (BMG Labtech, Cary, NC).
Quantification of secreted cytokines
Interleukin-17A and Th1/Th2 cytokines were measured using electrochemiluminescent assays from Meso Scale Discovery as instructed by the manufacturer.
RNA extraction, quantitative RT-PCR
Total RNA was extracted using RNeasy mini kits including the optional DNaseI digestion (Qiagen, Valencia, CA). Complementary DNA synthesis and TaqMan Real Time PCR were performed as described previously.25,26 TaqMan quantitative PCR was performed on a 7900HT Real Time PCR System (Applied Biosystems, Foster City, CA). All TaqMan reagents were purchased from Applied Biosystems.
Microarray
After extraction of total RNA using an RNeasy mini kit, microarray assays were performed at the Boston University Microarray Resource Facility (Boston, MA). Briefly, the RNA samples were amplified and labelled following Ambion® WT Expression Kit Protocol (Life Technologies, Grand Island, NY) and GeneChip® Whole Transcript (WT) Sense Target Labeling Assay Manual (Affymetrix, Santa Clara, CA). The cRNA samples were then hybridized to Affymetrix mouse 1·0ST gene chips. Affymetrix data were extracted, normalized and analysed using both in-house-developed multiplex software based on genepattern software of Broad Institute (Cambridge, MA) and ingenuity IPA software (http://www.ingenuity.com). Transcriptional factor enrichment analysis was also conducted using IPA. Significance is defined as absolute value of Z-score ≥ 2 and P-value < 10−3. The network model was reconstructed using Advanced Network Analysis Tool, a tool for constructing and analysing functional networks using inference algorithms based on physical and regulatory associations.27
Calculation of percent of expected composition and statistics
Percentage of Expected Composition equals (Nobs / Nexp) × 100 in which Nobs represents the number of genes in the signature that belong to a given family; and Nobs is the number of genes expected to be present in the list if the list were taken at random. Different groups were evaluated for statistical significance using the two-tailed unpaired Student's t-test for Figs 2 and 3. The P-value in Fig. 4 is calculated using Fisher's Exact Test.
Figure 2.
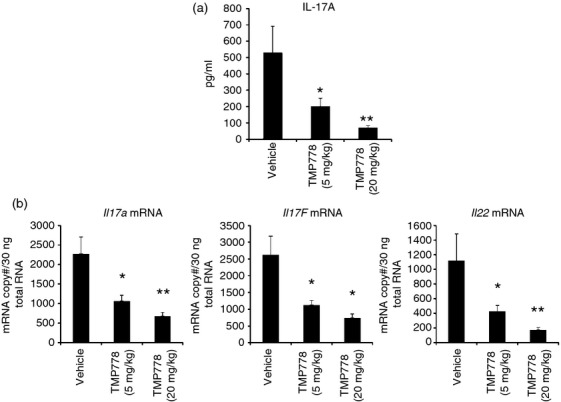
In vivo differentiation of T helper type 17 (Th17) cells was blocked by administration of TMP778. Mice were immunized with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35–55) in complete Freund's adjuvant (CFA). TMP778 (20 mg/kg or 5 mg/kg) or Vehicle was subcutaneously injected twice a day for 6 days. (a) The draining lymph node cells were isolated and stimulated with MOG35–55 for 5 days and the supernatants were harvested to detect interleukin-17 (IL-17) titres by Meso Scale Discovery. (b) Draining lymph node cells were isolated and stimulated with MOG35–55 for 20 hr. Cells were harvested for RNA extraction and real time RT-PCR analysis for mRNA expression of Il17a, Il17f and Il22. *P < 0·05 TMP778 versus Vehicle; **P < 0·01 TMP778 versus Vehicle. Data are representative of three separate experiments.
Figure 3.
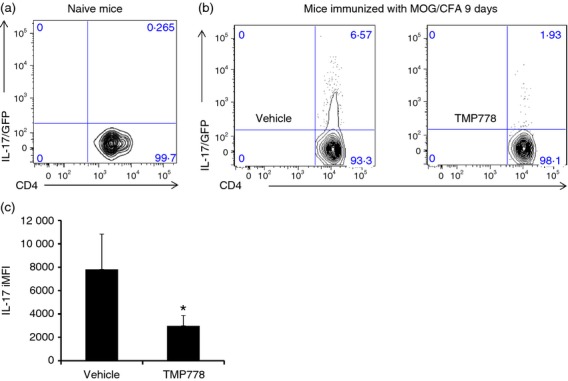
Retinoic acid receptor-related orphan nuclear receptor γt (RORγt) inverse agonist inhibits the generation of interleukin-17 (IL-17) -producing CD4+ T cells in an IL-17/GFP transgenic model. (a) IL-17/GFP expression from naive lymph nodes. (b) IL-17-IRES-GFP transgenic mice were immunized with myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35–55) in complete Freund's adjuvant (CFA). TMP778 (20 mg/kg) or Vehicle was subcutaneously injected twice a day for 9 days. The draining lymph node cells were purified and stimulated with PMA/ionomycin in the presence of brefeldin A for 3 hr before staining for mouse CD4. (c) Average of integrated MFI (iMFI) of individual animal CD4+ IL-17/GFP+ quadrant, as in Figure4(b). *P < 0·05 TMP778 20 mg/kg versus Vehicle. Data are representative of two separate experiments.
Figure 4.

The effect on genome-wide transcription by administration of retinoic acid receptor-related orphan nuclear receptor γt (RORγt) inverse agonist TMP778 in vivo. Total RNA from Fig.2(b) samples were used for microarray analysis using an affymetrix mouse gene 1.0 ST Array. Each RNA sample was the pool of four individual mouse RNA samples. (a) Data are shown as the log2-fold change of TMP778/vehicle versus the expression level, indicating genes increased or decreased by TMP778 treatment in vivo. Data shown are representative of microarray data of one RNA sample set (vehicle sample versus TMP778 sample) from two RNA sample sets. (b) Gene family signature enrichment. Genes up-regulated or down-regulated at least 1·5-fold with an expression level of 50 or greater were analysed. Each gene family enrichment is displayed as a percentage of the expected number of genes for a list of that size. When more genes than expected are found, the family is over-represented/enriched in the list (red bars). If the list contains fewer members of that family than expected, then that family is under-represented (blue bars). P-value is calculated using Fisher Exact Test and the value is used in colouring the bars (the more significant the P-value, the darker blue/red a bar is). All of the enriched genes in the figure happened to be down-regulated genes. (c) Canonical pathway analysis by ingenuity. Genes from (b) were analysed for their role in canonical pathways by IPA of ingenuity. Each canonical pathway has a different total number of genes as indicated. Pathways shown are the percentage of our down-regulated or up-regulated genes. Graphs were selected from data of which – log (P-value) is equal or higher than 4. P-value was also calculated using right-tailed Fisher Exact test. (d) Transcriptional factor enrichment analysis was conducted using IPA. The network model was reconstructed using the Advanced Network Analysis Tool (ANAT). SMAD3 was discovered by transcriptional factor enrichment analysis, the green nodes were genes down-regulated by TMP778 treatment, and the grey nodes were predicted genes connecting SMAD3. The arrow indicates direct regulation and the straight line indicates correlation between two nodes.
Results and Discussion
TMP778 inhibits mouse Th17 cell differentiation in vitro
Previously, we have reported that TMP778 blocks human Th17 cell differentiation in vitro.21 To study the effect of TMP778 in animal models, we first assessed whether TMP778 also inhibits mouse Th17 cell differentiation in vitro. Mouse CD4+ T cells in splenocytes differentiate to Th1, Th2, Treg or Th17 cells under different conditions. We stimulated mouse splenocytes under Th1 (IL-12/anti-IL-4), Th2 (IL-4/anti-IL-12), Treg (TGF-β) or Th17 (IL-23 or IL-6/TGF-β) polarizing conditions for 3 days and then examined IL-17 production. The IL-6/TGF-β stimulation induced the highest level of IL-17 (Fig.1a). Next, we asked whether TMP778 could inhibit IL-6/TGF-β induced IL-17 production. TMP778 blocked IL-17 production in a dose-dependent manner with an IC50 of 0·1 µm (Fig.1b,c). In addition, TMP536, the parental compound of TMP778, and TMP870, its diastereomer, also suppressed IL-17 production (Fig.1b). In contrast, the diastereomers TMP774 and TMP776 had no significant effect on mouse IL-17 production, consistent with a lack of activity displayed by these compounds in an IL-17F reporter system.21 The inhibition of IL-17 could be a result of the non-specific inhibition of T-cell proliferation. However, TMP778 treatment did not impair T-cell proliferation (Fig.1d). TMP778 regulation of Th17 cell development was also evident by intracellular IL-17A staining of purified naive mouse CD4+ T cells stimulated with plate-bound anti-CD3/anti-CD28 antibodies and cultured for 4 days with IL-6 and TGF-β (Fig.1e). Here, TMP778 had no effect on residual IFN-γ-producing Th1 cells in Th17 cultures (Fig.1e), nor did it affect production of Th1 or Th2 cytokines by Th1 or Th2 cells that were polarized in vitro (Fig.1f). The IC50 for inhibition of mouse Th17 cell differentiation by TMP778 was 0·1 µm (Fig.1g). In contrast to the single nanomolar potency of TMP778, another reported RORγt inhibitor, digoxin, showed much weaker activity against mouse Th17 cell differentiation (IC50 = 2·0 µm) (Fig.1g). There are a variety of small molecule RORγt inhibitors that have been recently discovered, such as digoxin, SR2211, SR1001 and ursolic acid.18–20 Although these compounds do inhibit Th17 cell differentiation, they all have far less potency than TMP778. In addition, they are not genome-wide profiled in vivo. Hence, these compounds can only be used as tool compounds but not possible clinical inhibitors.
Figure 1.

Retinoic acid receptor-related orphan nuclear receptor γt (RORγt) inverse agonist inhibits mouse interleukin-17 (IL-17) expression and T helper type 17 (Th17) cell differentiation in vitro. (a) Mouse splenocytes were stimulated with anti-CD3/anti-CD28 monoclonal antibody (mAb) plus the indicated cytokine and antibody for 3 days. The IL-17 titres in the supernatants were then determined by Meso Scale Discovery (MSD). (b) Mouse splenocytes were stimulated with anti-CD3/anti-CD28 mAb plus IL-6 and transforming growth factor-β (TGFβ) in the presence of 1 µm of indicated RORγt inverse agonists for 3 days. The IL-17 titres in the supernatants were then determined by MSD. (c) Different doses of TMP778 were added into the culture as described in Fig.2(b) for 3 days. The IL-17 titres in the supernatants were then determined and the IC50 was calculated. (d) Cell proliferation from (c) was determined using CellTiter-Glo. (e) Naive mouse CD4+ T cells were stimulated with anti-CD3/anti-CD28 plus IL-6/TGF-β in the presence of small molecule compound for 4 days. Cells were then re-stimulated with PMA/ionomycin for 3 hr before intracellular staining for IL-17 and interferon-γ (IFN-γ). (f) Mouse splenocytes were stimulated under ThN (anti-CD3/28 mAb), Th17 (anti-CD3/28 mAb, IL-6, TGFβ), Th2 (anti-CD3/28 mAb, IL-4, anti-IL12 mAb), or Th1 (anti-CD3/28 mAb, IL-12, anti-IL4) skewing conditions with or without 1 μmTMP778 for 3 days. The Th1/Th2/Th17 cytokines were then determined by Meso Scale Discovery. Data shown are the log2-fold change between TMP778 and DMSO control. (g) The percentage of IL-17-positive T cells after treatment with TMP778 or digoxin as in (e) was used to calculate their IC50. Data are representative of two to five separate experiments.
RORγt inverse agonist TMP778 inhibits mouse Th17 cell differentiation in vivo
Given its potent and selective activity in blocking mouse Th17 cell differentiation in vitro, we next asked whether TMP778 also regulates Th17 cell development during inflammatory T-cell activation in vivo. EAE models have recently been shown to be largely caused by the induction of pathogenic Th17 cells.6,13,28 Mice develop EAE within 10–14 days after immunization with MOG35–55 in CFA with pertussis toxin. In vivo administration of TMP778 for 3 weeks significantly delayed the onset of EAE and substantially reduced the severity of disease progression.23 Here, we modified the model where C57BL/6 mice were immunized with MOG in CFA without pertussis toxin and the draining lymph nodes were harvested to assess their function ex vivo. Lymph node cells from naive mice did not produce IL-17A upon stimulation with MOG, whereas lymph node cells from mice immunized with MOG for 10 days produced detectable levels of IL-17A upon ex vivo re-stimulation with MOG (data not shown). Further, we performed a time–course study to determine the length of immunization required for the development of naive T cells into Th17 cells as measured by IL-17A production from draining lymph node cells upon stimulation with MOG35–55 specific antigen. Lymph node cells produced a high level of IL-17A after immunization for only 6 days (data not shown), indicating differentiation of Th17 cells. After determining the pharmacokinetics and exposure levels of TMP778, we asked whether TMP778 regulates the development of Th17 cells in response to immunization with MOG35–55 in CFA. MOG/CFA-immunized mice were dosed with TMP778 (20 mg/kg or 5 mg/kg) or vehicle for 6 days. Whereas TMP778 did not influence the overall numbers or proportions of CD4+, CD8+, B220+, NK1.1+ or CD11c+ cells in draining lymph nodes (see Supporting information, Fig. S1), re-stimulation of ex vivo-isolated draining lymph node cells with MOG showed that cells from immunized mice treated with TMP778 produced substantially less IL-17A than cells dosed with vehicle alone (Fig.2a). The effects of TMP778 on MOG recall-induced IL-17A production was dose-dependent (Fig.2a), and TMP778 treatment also reduced Il17a, Il17f and Il22 mRNA expression in ex vivo-stimulated draining lymph node cells, as measured by RT-PCR (Fig.2b).
Similar to TMP778, the parental compound TMP53621 inhibited mouse Th17 cell differentiation in vitro (IC50 = 0·3 µm) (see Supporting information, Fig. S2a); dosing of MOG/CFA-immunized mice with TMP536 also inhibited MOG recall-induced IL-17A production (Fig. S2b). A third RORγt inverse agonist, TMP934, which is structurally distinct from TMP778 and TMP536 (Fig. S2c), also inhibited mouse Th17 cell differentiation in vitro (IC50 = 1·1 µm) (Fig. S2d). TMP934 also reduced ex vivo IL-17A production upon dosing in MOG/CFA-immunized mice (Fig. S2e). These results suggest that RORγt inverse agonists block the in vivo development of effector cells capable of expressing IL-17A upon recall stimulation.
To gain further insight into the effects of RORγt inhibition on Th17 cell differentiation in vivo, we next tracked the development of Th17 cells upon MOG/CFA immunization using a transgenic IL-17A-GFP reporter system and flow cytometry.29 We immunized IL-17A-GFP mice with MOG/CFA, treated these animals +/− TMP778, and then analysed the development of IL-17A-GFP+ effector (CD4+ CD25− CD44hi CD62Llo) T cells from draining lymph nodes after 9 days by ex vivo FACS analysis. Detectable GFP expression in effector T cells from IL-17A-GFP mice required immunization with MOG/CFA (Fig.3a,b), and treatment of these immunized mice with TMP778 almost completely abolished GFP expression (Fig.3b). The effects of TMP778 on in vivo Th17 cell development in this model were evident, both in terms of the number of GFP+ effector T cells (Fig.3b) and the level of GFP expression iMFI observed in residual GFP+ effector cells (Fig.3c). Collectively, these data indicate that pharmacological inhibition of RORγt potently blocks Th17 cell development in vivo.
A number of groups have discovered small molecule inhibitors targeting RORγt.18–20 These molecules inhibited Th17 cell differentiation and demonstrated efficacy in EAE. The efficacy of these molecules in vivo could be due to either inhibition of Th17 cell differentiation or acute regulation of Th17 signature gene expression by mature Th17 cells, or both. Here, we demonstrated that RORγt inverse agonist TMP778 inhibited in vivo Th17 cell differentiation in two different models, indicating the important role of RORγt inverse agonists in treating Th17-related autoimmune inflammatory diseases.
Whole genome transcriptional profile of in vivo RORγt inhibition by TMP778
We next sought to analyse the broader effects of RORγt inhibition on gene expression during Th17 cell differentiation in vivo. For these experiments, we analysed the RNA of lymph node cells from vehicle- or TMP778-treated mice, prepared as in Fig.2(b) above, by microarray. As expected, expression of Th17-signature genes, including Il17a, Il17f, Il22, Il23r, Rorc and Ccr6 were reduced in cells from TMP778-treated mice (Fig.4a). In addition, a large number of genes outside the established Th17 transcriptional signature were significantly different in cells from TMP778-treated versus vehicle-treated mice, with the vast majority of these differentially regulated genes being reduced by TMP778 treatment. For example, expression of Il1a and Il1b was reduced by TMP778 treatment in vivo. Expression of other, non-canonical Th17-related genes, namely Ifng, Il2, Tnf, Il9 and Ahr, which can be expressed in pathogenic and non-pathogenic Th17 cells at varying levels, were modestly decreased by TMP778 treatment. The vast majority of other T helper cell signature genes were not influenced by in vivo inhibition of RORγt (Fig.4a).
Outside the conventional T helper cell-related genes, more than 150 genes were found to be reduced at least 1·5-fold in TMP778-treated mice compared with control animals, whereas 13 genes were up-regulated. Among them, Dcpp, Muc19 and Abpb were markedly repressed by in vivo TMP778 treatment. In defining gene families among the non-Th17 signature genes significantly influenced by in vivo RORγt inhibition, we found that chemokines, cytokines and receptors were highly enriched, and all were down-regulated by in vivo TMP778 treatment (Fig.4b). We next analysed TMP778-modulated genes for functional pathways using IPA. TMP778-modulated genes were classified into at least 51 distinct pathways, where the −log (P-value) is equal to or is higher than 2·0 or P ≤ 0·01 (data not shown). Twenty-seven of these pathways showed −log (P-value) ≥ 4·0 or P ≤ 0·0001 (Fig.4c). Only one gene, Glycam1, was up-regulated in these 27 pathways. Interestingly, the top two pathways modulated by in vivo TMP778 treatment are involved in granulocyte/agranulocyte adhesion and diapedesis (Fig.4c and Table1). Other pathways regulated by TMP778 treatment are those involved in cell activation, cytokine signalling and regulation, and are also associated with autoimmune inflammatory diseases such as rheumatoid arthritis, atherosclerosis, psoriasis, chronic obstructive pulmonary disease and airway allergic inflammation. Other classes of TMP778-modulated pathways were those associated with bacterial/viral infection, liver function, cancer signalling and graft-versus-host disease signalling. The individual genes of these pathways are summarized in Table1.
Table 1.
Down-regulated molecules in the top canonic pathways
| Canonical pathways | Down-regulated molecules |
|---|---|
| Granulocyte adhesion and diapedesis | IL1A, IL1RL1, MMP14, CXCL 1, CCL17, CCL22, SDC3, CLDN7, CXCL 10, IL1R2, CXCL3, IL36G, CLDN4, MMP8, Ccl2, PPBP, MMP12, Ccl6, C5AR1, PF4, IL1 R1, FPR1, Ccl9, ITGAM, CCL7, IL1 RN, CLDN1, IL1 B, MMP9 |
| Agranulocyte adhesion and diapedesis LXR/RXR activation | IL1A, C5AR1, PF4, MMP14, CXCL1, CCL 17, CCL22, IL1R1, CLDN7, Ccl9, CXCL10, CXCL3, IL36G, CCL7, CLDN4, CLDN1, IL1RN, MMP8, Ccl2, PPBP, IL1 B, MMP12, Ccl6, MMP9 |
| IL-10 signalling PPAR signalling | IL1A, MSR1, IL1RL1, CD36, ARG2, IL1R1, IL1R2, IL36G, CCL7, IL1RN, IL 1B, S100A8, PTGS2, MMP9, IL1R2, HMOX1, FOS, IL36G, IL1A, IL 1RN, IL1RL1, IL 1B, ARG2, IL1 R1 |
| Hepatic fibrosis I Hepatic stellate cell activation osteoblasts, osteoclasts and chondrocytes in RA regulation of cytokine in intestinal cells by IL-17 atherosclerosis signalling | IL1R2, FOS, IL36G, IL1A, PDGFA, IL 1RN, IL1RL1, IL 1B, IL1R1, PTGS2, IL1R2, MET, CXCL3, IL1A, EDNRB, EDN1, PDGFA, IL1RL1, IL 1B, IL1R1, MMP9, IL1R2, FOS, IL36G, IL1A, IL 1RN, IL1RL1, MMP8, MMP14, IL1B, IL1R1, CSF1R, LRP1, IL17A, IL1A, LCN2, CXCL 1, IL1B, IL17F, IL17A |
| Role of PRR in recognition of bacteria and viruses role of cytokines in communication in immune cells altered T-cell and B-cell signalling in RA communication in innate and adaptive immune cells | IL36G, IL1A, MSR1, PDGFA, IL1RN, CD36, IL1B, S1OOA8, PLA2G7, MMP9, CLEC7A, OAS1, IRF7, C5AR1, OAS2, IL1B, CLEC6A, OAS3, C3AR1, IL36G, IL1A, IL1RN, IL1B, IL22, IL17F, IL17A, IL36G, IL1A, IL1RN, FCER1G, Tlr13, IL 1B, IL22, IL17A |
| Macrophages, fibroblasts and endothelial cells in RA hypercytokinaemia/hyperchemokinaemia in influenza graft-versus-host disease signalling | CXCL1O, IL36G, IL1A, IL1RN, FCER1G, Tlr13, IL1B, Ccl9 |
| Role of IL-17A in psoriasis IL-6 signalling | IL1R2, FOS, IL36G, IL1A, C5AR1, IL1RN, IL1RL1, PDGFA, IL1B, Tlr13, IL1R1, LRP1, IL17A, CXCL10, IL36G, IL1A, IL1RN, IL1B, IL17A |
| Bladder cancer signalling | IL36G, IL1A, IL1RN, FCER1G, IL1B, GZMB, CXCL3, CXCL1, S100A8, IL17A, IL1R2, FOS, IL36G, IL1A, IL1RN, IL1RL1, IL1B, IL1R1 |
| Regulation of cytokine in immune cells by IL-17 inhibition of matrix metalloproteases | CDH1, THBS1, MMP14, MMP8, CDKN1A, MMP12, MMP9, CXCL1, IL1B, IL17F, IL17A |
| Airway pathology in COPD | MMP14, MMP8, MMP12, MMP9, LRP1, CXCL3, MMP8, MMP9 |
| IL-17F in allergic inflammatory airway diseases p38 MAPK signalling | CXCL1O, CCL7, CXCL1, IL1B, IL17F, IL1R2, IL36G, IL1A, IL1RN, IL1RL1, IL1B, IL1R1, CXCL10, FOS, CXCL1, IL17A |
| IL-17A signalling in gastric cells role of IL-17A in arthritis hepatic cholestasis | CXCL3, CCL7, CXCL1, PTGS2, IL17A, IL1R2, IL36G, IL1A, IL1RN, IL1RL1, IL1B, IL1R1 |
The table lists the specific down-regulated gene names in Fig. 4(c) except Glycaml, which is up-regulated in agranulocyte adhesion and diapedesis pathway.
Using IPA's transcriptional factor enrichment analysis, we found that several transcription factors were correlated with the 150 genes regulated by our RORγt inverse agonist (data not shown). These data were further analysed to generate a network model using the Advanced Network Analysis Tool.27 Interestingly, more than 30 genes were downstream of or related with transcription factor SMAD3 (Fig.4d). As we know, TGF-β is critical for the differentiation of Th17 cells and Th17-mediated pathology.30 SMAD3 is required for TGF-β-induced gene expression. After phosphorylation by TGF-β signalling, SMAD3 binds SMAD4, which then cooperates with c-Jun/c-Fos to medicate TGF-β-induced transcription.31–33 Recently, Dong and colleagues discovered that SMAD3 interacts with RORγt and decreases its transcriptional activity. Furthermore, Smad3-deficiency led to enhanced Th17 differentiation in vitro and in vivo.34 Here, we discovered that RORγt inverse agonist inhibited more than 30 SMAD3 downstream genes (Fig.4d), indicating that RORγt could in turn regulate the function of SMAD3. The precise mechanism remains to be determined.
We have previously reported that TMP778 does not affect the gene expression of Il1a and Il1b in human CD4+ T cells in vitro.21 Here, Il1a and Il1b gene expression was significantly inhibited by TMP778 treatment in vivo. As CD4+ T cells express a low level of Il1, the Il1a and Il1b we observed may be mainly expressed in macrophages within the draining lymph nodes after the MOG immunization. The inhibition of Il1a and Il1b gene expression by TMP778 treatment may be an indirect effect such as through the change of SMAD3 activity (Fig.4d); the exact mechanism remains to be determined. Importantly, as IL-1 signalling is critical for the differentiation of Th17 cells and IL-17 production by γδ T cells, inhibition of mRNA expression of Il1a and Il1b could result in a synergistic effect on the inhibition of Th17 cell differentiation and IL-17 production.
Here, we discovered that Dcpp, Abpb and Muc19 were expressed in lymph node cells from mice immunized with MOG in CFA and their expression was down-regulated by TMP778 treatment. However, their roles in the immune system have not yet been determined. Dcpp gene is expressed in embryo-containing oviduct.35 Hence, RORγt might have some role in embryo development. Abpb is the β-subunit of the androgen-binding protein and is expressed in salivary gland and lacrimal glands;36 its function in the immune system needs to be explored. Mucus covers the epithelial surface of various tissues, providing lubrication and protection. The gel-like properties of mucus are generally attributed to the physical properties and structural features of gel-forming mucins, including Muc19.37 Mucus has a protective function for the intestine,38 and genome-wide association studies have shown an association between Muc19 and Crohn's disease.39 As Muc19 is down-regulated by TMP778 administration in vivo, RORγt inverse agonist should be evaluated in animal models of inflammatory bowel diseases such as Crohn's disease. In addition, digestive system symptoms should be monitored when treating other diseases by RORγt small molecule inverse agonists in clinic.
Most of the works in this study were carried out in mixed cell population (spleen cells or lymph node cells). We purposely designed the study to use mixed cell populations so that the compound data we obtained could be one step closer to the clinic because a RORγt inverse agonist would eventually be administered to human patients. The RORγt inverse agonist affect could be due to its inhibition of RORγt function in different cell types not just CD4+ Th17 cells. RORγt is expressed not only in CD4+ T cells, but also CD8+ Tc17 cells,21,40 innate lymphocytes,41,42 γδ T cells,41,42 and macrophage.43 Hence, RORγt inverse agonists should also affect the function of these cells.
This whole genomic approach has led us to discover that in vivo administration of TMP778 inhibited a variety of gene expression in addition to the Th17 signature gene, which is different from in vitro studies. These transcriptomic results provide new and important insight into the broader impact of targeting RORγt in vivo. Further, these studies provide an unbiased view of potential disease indications and side-effects for RORγt inhibitors, and so may facilitate clinical development.
In summary, we have demonstrated that the RORγt inverse agonist TMP778 blocks Th17 cell differentiation in vivo. This transcriptomics study provided critical information for understanding the possible mechanisms of RORγt inhibitors in vivo. This study should enable disease indication identification for RORγt inhibitors, and may also help to predict adverse effects in clinic and hence improve clinical trials for the RORγt small molecule inverse agonists.
Acknowledgments
We thank Vijay Kuchroo, Christophe Benoist, Diane Mathis and Alexander Rudensky for their advice and suggestions for our RORγt project. We thank Spiros Jamas for his support of the project. We thank Gregory Wands, Tetsuya Yamagata, Jing Hua and Quentin Wright for their technical support and/or discussions for the RORγt project. We thank GSK Drug Discovery and HTS groups for their support of the RORγt project. We thank ChemPartner, Indigo Biosciences and Lotus Separations for their technical support. Tempero Pharmaceuticals is fully owned and funded by GlaxoSmithKline (GSK).
Disclosures
All authors are present or former employees of Tempero Pharmaceuticals, Inc. ML is an employee of GlaxoSmithKline.
Supporting Information
Figure S1. No change of cell surface markers by administration of TMP778. C57BL/6 mice were immunized with MOG35–55 in complete Freund's adjuvant. TMP778 (20 mg/kg or 5 mg/kg) or Vehicle was subcutaneously injected twice a day for 6 days as in Fig. . Draining lymph node cell were harvested, stained for cell surface markers, and analysed by flow cytometry.
Figure S2. In vivo differentiation of T helper type 17 (Th17) cells was blocked by administration of TMP536 or TMP934. (a and d) C57BL/6 mouse splenocytes were stimulated with anti-CD3/anti-CD28 monoclonal antibody plus interleukin-6 (IL-6) and transforming growth factor-β (TGF-β) in the presence of the indicated concentration of RORγt inverse agonist TMP536 (a) or TMP934 (d) for 3 days. The IL-17 titres in the supernatants were then determined and the IC50 were calculated. (b and e) C57BL/6 mice were immunized with MOG35–55 in CFA. TMP536 (20 or 50 mg/kg) (b) or TMP934 (50 mg/kg) (e) were subcutaneously injected twice a day for 6 days. The draining lymph node cells were isolated and stimulated with MOG35–55 for 5 days, after which the supernatants were harvested to detect IL-17 titers by Meso Scale Discovery. (c) Chemical structure of TMP934.
References
- Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–44. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–88. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28:445–53. doi: 10.1016/j.immuni.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–3. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–98. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2:52ra72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190–9. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366:1181–9. doi: 10.1056/NEJMoa1109017. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. 2014;35:493–500. doi: 10.1016/j.tips.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–24. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–9. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh R, Kozhaya L, McKevitt K, et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. 2014;211:89–104. doi: 10.1084/jem.20130301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Godec J, Ben-Aissa K, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity. 2014;40:355–66. doi: 10.1016/j.immuni.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Leung MW, Huang P, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature. 2011;472:486–90. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt LA, Kumar N, Nuhant P, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–4. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Wang X, Zhong B, et al. Ursolic acid suppresses interleukin-17 (IL-17) production by selectively antagonizing the function of RORβγt protein. J Biol Chem. 2011;286:22707–10. doi: 10.1074/jbc.C111.250407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skepner J, Ramesh R, Trocha M, et al. Pharmacologic inhibition of RORγt regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol. 2014;192:2564–75. doi: 10.4049/jimmunol.1302190. [DOI] [PubMed] [Google Scholar]
- Yang J, Skepner J, Mark T, Ghosh S. Small molecule inhibitors targeting the Th17 cell transcription factor RORγt for the treatment of autoimmune diseases. J Clin Cell Immunol. 2013;4:e111. [Google Scholar]
- Xiao S, Yosef N, Yang J, et al. Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity. 2014;40:477–89. doi: 10.1016/j.immuni.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson TJ, Pellerin A, Djuretic IM, et al. Halofuginone-induced amino acid starvation regulates STAT3-dependent Th17 effector function and reduces established autoimmune inflammation. J Immunol. 2014;192:2167–76. doi: 10.4049/jimmunol.1302316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Castle BE, Hanidu A, et al. Sphingosine kinase 1 is a negative regulator of CD4+ Th1 cells. J Immunol. 2005;175:6580–8. doi: 10.4049/jimmunol.175.10.6580. [DOI] [PubMed] [Google Scholar]
- Yang J, Yang M, Htut TM, et al. Epstein–Barr virus-induced gene 3 negatively regulates IL-17, IL-22 and RORγt. Eur J Immunol. 2008;38:1204–14. doi: 10.1002/eji.200838145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Zalckvar E, Rubinstein AD, et al. ANAT: a tool for constructing and analyzing functional protein networks. Sci Signal. 2011;4:l1. doi: 10.1126/scisignal.2001935. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of TH17 cells. Nature. 2008;453:1051–7. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Okuyama Y, Ogura H, et al. Local microbleeding facilitates IL-6- and IL-17-dependent arthritis in the absence of tissue antigen recognition by activated T cells. J Exp Med. 2011;208:103–14. doi: 10.1084/jem.20100900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature. 1998;394:909–13. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-β responses. Cell. 1998;95:737–40. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Massague J, Xi Q. TGF-β control of stem cell differentiation genes. FEBS Lett. 2012;586:1953–8. doi: 10.1016/j.febslet.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez GJ, Zhang Z, Chung Y, et al. Smad3 differentially regulates the induction of regulatory and inflammatory T cell differentiation. J Biol Chem. 2009;284:35283–6. doi: 10.1074/jbc.C109.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Xu JS, Lee YL, Yeung WS. Demilune cell and parotid protein from murine oviductal epithelium stimulates preimplantation embryo development. Endocrinology. 2006;147:79–87. doi: 10.1210/en.2005-0596. [DOI] [PubMed] [Google Scholar]
- Remington SG, Nelson JD. Secretoglobins: sexually dimorphic expression of androgen-binding protein mRNA in mouse lacrimal glands. Invest Ophthalmol Vis Sci. 2005;46:31–8. doi: 10.1167/iovs.04-0216. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhao YH, Kalaslavadi TB, et al. Genome-wide search and identification of a novel gel-forming mucin MUC19/Muc19 in glandular tissues. Am J Respir Cell Mol Biol. 2004;30:155–65. doi: 10.1165/rcmb.2003-0103OC. [DOI] [PubMed] [Google Scholar]
- Johansson ME, Ambort D, Pelaseyed T, et al. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci. 2011;68:3635–41. doi: 10.1007/s00018-011-0822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Mack DR, Marcil V, et al. Genome-wide association study signal at the 12q12 locus for Crohn's disease may represent associations with the MUC19 gene. Inflamm Bowel Dis. 2013;19:1254–9. doi: 10.1097/MIB.0b013e318281f454. [DOI] [PubMed] [Google Scholar]
- Curtis MM, Way SS, Wilson CB. IL-23 promotes the production of IL-17 by antigen-specific CD8 T cells in the absence of IL-12 and type-I interferons. J Immunol. 2009;183:381–7. doi: 10.4049/jimmunol.0900939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelyushin S, Haak S, Ingold B, et al. Rorγt+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. J Clin Invest. 2012;122:2252–6. doi: 10.1172/JCI61862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Pantelyushin S. Hiding under the skin: interleukin-17-producing γδ T cells go under the skin? Nat Med. 2012;18:1748–50. doi: 10.1038/nm.3016. [DOI] [PubMed] [Google Scholar]
- Gu Y, Yang J, Ouyang X, et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38:1807–13. doi: 10.1002/eji.200838331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. No change of cell surface markers by administration of TMP778. C57BL/6 mice were immunized with MOG35–55 in complete Freund's adjuvant. TMP778 (20 mg/kg or 5 mg/kg) or Vehicle was subcutaneously injected twice a day for 6 days as in Fig. . Draining lymph node cell were harvested, stained for cell surface markers, and analysed by flow cytometry.
Figure S2. In vivo differentiation of T helper type 17 (Th17) cells was blocked by administration of TMP536 or TMP934. (a and d) C57BL/6 mouse splenocytes were stimulated with anti-CD3/anti-CD28 monoclonal antibody plus interleukin-6 (IL-6) and transforming growth factor-β (TGF-β) in the presence of the indicated concentration of RORγt inverse agonist TMP536 (a) or TMP934 (d) for 3 days. The IL-17 titres in the supernatants were then determined and the IC50 were calculated. (b and e) C57BL/6 mice were immunized with MOG35–55 in CFA. TMP536 (20 or 50 mg/kg) (b) or TMP934 (50 mg/kg) (e) were subcutaneously injected twice a day for 6 days. The draining lymph node cells were isolated and stimulated with MOG35–55 for 5 days, after which the supernatants were harvested to detect IL-17 titers by Meso Scale Discovery. (c) Chemical structure of TMP934.