

Abstract
High levels of the flame retardant 2,2′,4,4′-tetrabromodiphenyl ether (BDE 47) have been detected in Pacific salmon sampled near urban areas, raising concern over the safety of salmon consumption. However, salmon fillets also contain the antioxidants eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), whose oxidation products induce cellular antioxidant responses. Because oxidative stress is a mechanism of BDE 47 toxicity, we hypothesized that oxidized EPA and DHA can ameliorate the cellular and mitochondrial toxicity of BDE 47. HepG2 cells were treated with a mixture of oxidized EPA and DHA (oxEPA/oxDHA) at a ratio relevant to salmon consumption (1.5/1 oxEPA/oxDHA) followed by exposure to 100 μM BDE 47. Pretreatment with oxEPA/oxDHA for 12 h prior to BDE 47 exposure prevented BDE 47-mediated depletion of glutathione, and increased expression of antioxidant response genes. oxEPA/oxDHA also reduced the level of reactive oxygen species production by BDE 47. The oxEPA/oxDHA antioxidant responses were associated with partial protection against BDE 47-induced loss of viability and also mitochondrial membrane potential. Mitochondrial electron transport system functional analysis revealed extensive inhibition of State 3 respiration and maximum respiratory capacity by BDE 47 were partially reversed by oxEPA/oxDHA. Our findings indicate that the antioxidant effects of oxEPA/oxDHA protect against short exposures to BDE 47, including a protective role of these compounds on maintaining cellular and mitochondrial function.
Keywords: Omega-3 polyunsaturated fatty acids, PBDE, BDE 47, mitochondria, salmon, oxidative stress
1.0 Introduction
Polybrominated diphenyl ether (PBDE) flame retardants are common lipophilic organic contaminants present in environmental media and human samples [1-3]. Despite governmental bans on the new manufacture of these compounds [4], most PBDE congeners are resistant to degradation and continue to persist in the environment [5]. The fact that PBDEs are often detected in human blood, adipose tissue, and breast milk underscores the risk of potential toxic effects [6-8]. Uptake of PBDEs from household dust appears to be the primary route of exposure for humans [9-11], but significant exposures can also occur through dietary consumption of contaminated meat, poultry, and especially seafood [12-16]. Elevated levels of PBDEs have been detected in the edible tissues of several food fish, including Pacific salmon [14, 15, 17, 18]. This may pose a particular problem for certain populations, including Tribal Nations, and Asian and Pacific Islander populations, whose dietary customs of high rates of fish consumption put them at particularly high risk of significant exposures to PBDEs and other persistent contaminants [19]. For this reason, a major focus for assessing the safety of human consumption of marine seafood species is to measure the toxicological effects of PBDEs found in fish.
The most prevalent and persistent PBDE congener detected in human and wildlife samples is 2,2′,4,4′-tetrabromodiphenyl ether (BDE 47) [6, 14], which may comprise up to 65% of the total PBDE body burden in salmon [18]. The dominance of BDE 47 among other congeners in salmon is likely a result of several factors, including its bioaccumulation potential, resistance to debromination, and that it can occur as a debromination product of other higher molecular weight PBDE congeners [20, 21]. BDE 47 is a developmental and neurological toxicant [8, 22, 23] and is reported to disrupt thyroid hormone status [24]. The mechanisms of BDE 47 toxicity are poorly understood, but substantial evidence indicates the induction of cellular oxidative stress, via generation of reactive oxygen species (ROS), may play a major role [25-29]. Importantly, BDE 47-induced oxidative stress is associated with a loss of cellular viability and proliferation, and a loss of mitochondrial membrane potential [28, 30-32].
Despite concerns about the presence of lipophilic toxicants, consumption of most marine fish is considered to be highly beneficial to human health. Salmon, in particular, are rich dietary sources of high quality protein, vitamins, and the beneficial antioxidant micronutrients known as omega-3 polyunsaturated fatty acids. The two major omega-3 fatty acids in marine fish oil are eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Salmon fillets rank among the highest EPA+DHA/weight ratios among food species of fish [33], and are estimated to contain 4.75 g of omega-3s in an average 180 g meal [34, 35]. Omega-3s are highly labile, and auto-oxidation of these compounds occurs readily under ambient environmental conditions both in vitro and in vivo [36]. Peroxidation of EPA and DHA by free radicals and ROS generates electrophilic cyclopentenone isoprostanes [36, 37]. Recent evidence suggests these products of oxidized EPA and oxidized DHA (oxEPA and oxDHA) may be critical mediators of the beneficial human health effects of fish oil omega-3s [38, 39], as oxEPA and oxDHA can activate nuclear factor erythroid 2-related factor 2 (Nrf2), leading to the upregulation of a suite of antioxidant genes that function to maintain cellular redox status [40] and also glutathione (GSH). In this regard, GSH has been shown to modulate the toxicity of PBDEs, including BDE 47 [23, 26, 31]. Hence, it has been proposed that the activation of Nrf2-regulated cellular antioxidant responses via oxEPA and oxDHA can be a protective mechanism against the progression of diseases with a cellular oxidative stress etiology [34, 41].
The fact that fish omega-3s are potent cellular antioxidants, whereas a major mechanism of BDE 47 cell injury is oxidative damage, provides a scenario in which omega-3s may chemoprotect against the toxicity of co-consumed PBDEs. This hypothesis is supported by studies showing that induction of intracellular GSH by the antioxidant N-acetylcysteine (NAC), a GSH precursor, protects human fetal hematopoietic stem cells [31], human hepatocytes [27], and T lymphocyte cell lines [28] against BDE 47 toxicity. Others have demonstrated that oxidative stress-mediated inflammation resulting from in vitro exposure to BDE 47 [32, 42] and similar persistent organic contaminants [39] can be reduced by treatment with dietary antioxidant compounds, including via activation of Nrf2 by free radical-oxidized EPA and DHA.
In the present study we investigated the hypothesis that the activation of cellular antioxidant responses by a mixture of oxidized omega-3s relevant to dietary exposures (1.5/1 oxEPA/oxDHA) can ameliorate the toxicity of BDE 47. Among other relevant target systems of PBDE toxicity, including cells of the developmental [43] and nervous systems [23, 44], the liver is a major target organ of BDE 47 toxicity and receives extensive PBDE exposures through dietary routes. Hence, chemoprotection against BDE 47-induced cellular toxicity and mitochondrial dysfunction were investigated in the human hepatocellular carcinoma cell line HepG2, a model in which activation of the Nrf2 antioxidant response via oxEPA and oxDHA has been previously characterized [37]. Our approach involved characterizing the modulatory effects of oxEPA/oxDHA on specific functional components of the mitochondrial electron transport system under conditions of BDE 47 exposure.
2.0 Methods and Materials
2.1. Chemicals and reagents
BDE 47 (2,2′,4,4′-tetrabromodiphenyl ether, >99% purity) was obtained from AccuStandard, Inc. (New Haven, CT, USA). 5-Sulfosalicylic acid dehydrate (SSA), Napthalinedicarboxyaldehyde (NDA), dimethyl sulfoxide (DMSO), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) were obtained from Sigma-Aldrich Corp (St. Louis, MO, USA). Sulforaphane (SFN) (R-Sulforaphane) was obtained from LKT Laboratories, Inc. (St. Paul, MN, USA). 2,2′-azobis-2-methyl-propanimidamide, dihydrochloride (AAPH) was obtained from Cayman Chemical (Ann Arbor, MI, USA). 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) was obtained from Life Technologies (Grand Island, NY, USA). All cell culture reagents were obtained from Invitrogen, GIBCO (Carlsbad, CA, USA).
2.2. Preparation of oxidized omega-3s
The ratios of omega-3s reported in salmon fillets (1.5/1 EPA/DHA) are relatively consistent and are often reflected in the ratio of EPA/DHA in many commercially-available omega-3 dietary supplements. A ratio of 1.5/1 EPA/DHA is often used in clinical trials investigating the effects of omega-3 dietary supplementation [45-47], and this ratio was also used in our experiments. EPA, DHA, and SFN were dissolved in DMSO, and frozen stocks were held at −20 °C. Methods for preparation and dosing with oxEPA/oxDHA were conducted as described by Majkova et al., 2011 [39], with minor modifications. For each experiment, EPA and DHA were oxidized separately by diluting the fatty acid to 1 mM in PBS containing 2 mM of the free radical generator AAPH, and incubated at 37 °C for 16 h. After oxidation, oxEPA and oxDHA were combined as a 1.5/1 mixture, sterile filtered, and diluted in Minimum Essential Media α (MEMα) containing 5% FBS such that the final concentrations in the pretreatment media were 60 and 40 μM of oxEPA and oxDHA, respectively.
2.3. Cell culture, and in vitro exposures
HepG2 human hepatocellular carcinoma cells were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). HepG2 cells were cultured in MEMα media supplemented with 5% fetal bovine serum (FBS), 1% HEPES buffer, and 1% penicillin-streptomycin. Cells were maintained in 75 cm2 flasks at 37 °C and 95% CO2 and split at 90-100% confluency every five days. For all experiments, cells were seeded into wells at a density such that confluency was ≥80% at time of dosing (48 h after seeding). MEMα pretreatment media contained 5% FBS and the oxEPA/oxDHA mixture, SFN, or an equal volume of DMSO (vehicle control). For all experiments, the final volume of DMSO in media was <0.4%. Cells were treated with oxEPA/oxDHA for a time period to induce a maximal antioxidant response. Pretreatment media was then aspirated and cells were washed with PBS. Cells were then exposed to 50 or 100 μM BDE 47 (or DMSO) in serum-free MEMα media for a period of 24 h, unless specified otherwise. These toxicant concentrations were selected based on our previous studies in human cells [31], as well as range-finding experiments in HepG2 cells, to elicit a moderate 50-75% loss in cell viability. The effect of oxEPA/oxDHA on BDE 47 cell toxicity was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay [48] in three separate experiments, each containing three replicate treatments.
2.4. Glutathione status
Intracellular GSH was measured after experimental treatment with DMSO or antioxidants to determine an exposure time to elicit a robust antioxidant response in HepG2 cells. Following these optimization experiments, GSH was then measured in cells following pretreatment with DMSO or oxEPA/oxDHA for the determined exposure period, and subsequent toxicant challenge with BDE 47 (100 μM) for 24 h. Briefly, cells were washed with PBS, detached with 0.25% trypsin-EDTA (Invitrogen), and pelleted by centrifugation at 800 rpm for 5 min at 4 °C. Cells were resuspended in ice-cold PBS and lysed on ice by sonication. Redox status of reduced glutathione (GSH) was ensured by acidification of cell lysates in 5% SSA as described [49], and lysates were immediately stored at −80 °C. GSH concentrations in acidified lysates were determined by conjugation with NDA, which generates a stable fluorescent product detected at excitation 472 nm/emission 528 nm [49]. GSH concentrations were normalized to total protein by the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA).
2.5. Quantitative PCR analysis of oxidative stress marker genes
Two inducible antioxidant genes under Nrf2 regulation were selected as markers of oxidative stress elicited by BDE 47: glutamate-cysteine ligase catalytic subunit (GCLC) and NAD(P)H-quinone oxidoreductase 1 (NQO1). After pretreatment with or without oxEPA/oxDHA (12 h), followed by exposure to BDE 47 or DMSO (24 h), mRNA transcript levels of gclc and nqo1 were measured in cells. Cells were harvested in TRIzol® reagent and immediately stored at −80°C. Procedures for isolation of total RNA, cDNA synthesis, PCR primer product validation, and subsequent quantitative PCR analyses with SYBR Green were conducted as described [50]. Primer sequences and information are presented in Table 1. Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) did not differ between treatment groups and was used for normalization purposes. Measurements were carried out in three separate experiments, each containing three replicate treatments.
Table 1.
Sequences of gene-specific primers used in quantitative PCR experiments
| Gene | Primers (5′—3′) | Accession #/reference |
|---|---|---|
| GCLC | Forward: AGGCATTGATCATCTCCTGG Reverse: AGGAGGGGGCTTAAATCTCA |
NM_001498.3 |
| NQO1 | Forward: ACTGCCCTCTTGTGGTGCAT Reverse: GCTCGGTCCAATCCCTTCAT |
NM_001025434 |
| GAPDH | Forward: TCCTGCACCACCAACTGCTT Reverse: GAGGGGCCATCCACAGTCTT |
BC08351/Shao et al., (2007) |
2.6. Mitochondrial membrane potential assay
Mitochondrial membrane potential (ΔΨm) was assessed using the fluorescent probe JC-1 (Cayman Chemical, Ann Arbor, MI, USA). Following pretreatment with or without oxEPA/oxDHA (12 h), followed by exposure to BDE 47 or DMSO (24 h), cells were then incubated for 1 h with JC-1 probe (diluted 1:10 in the media of each well). Quantification of ΔΨm was based on the ratio of red fluorescent J-aggregates to green fluorescent J-monomers, detected at 560 nm/595 nm and 485 nm/535 nm excitation/emission, respectively. Measurements were carried out in three separate experiments each containing three replicate treatments using a 96-well fluorescent plate reader.
2.7. Assessment of electron transport system function
Electron transport system (ETS) functional parameters were measured in saponin-permeabilized HepG2 cells by high-resolution respirometry using the Oxygraph-2k (Oroboros Instruments, Innsbruck, Austria). After BDE 47 exposure, with or without pretreatment with oxEPA/oxDHA, cells were harvested and resuspended in 1 mL of ice-cold respiration buffer: distilled H2O, 0.25 M mannitol, 10 mM MgCl2, 10 mM KHPO4 buffer, pH=7.2. Solubility of oxygen in respiration buffer was assumed to be 0.920 μmol/L, and the oxygraph chambers were set to 30 °C and calibrated by determining dissolved oxygen in 2.2 mL respiration buffer under ambient atmospheric conditions as described by Gnaiger et al. [51]. Cells were permeabilized on ice with 12 μL of permeabilization buffer (50 μg/mL saponin in respiration buffer, 20 min, 4 °C) prepared prior to each experiment.
Respiratory substrates and inhibitors were added sequentially in the following order: 5 mM pyruvate, 2 mM malate, 10 mM glutamate, and 2.5 mM ADP (to induce State 3 respiration with complex I substrates only); 10 mM succinate (to induce maximum State 3 respiration with substrates of complexes I and II); 2.5 μM oligomycin (to induce proton leak, or State 4 respiration); 2.5 μM CCCP (to induce maximum respiratory capacity, or uncoupled respiration); 0.5 μM rotenone (to measure uncoupled respiration with complex I inhibition, i.e., uncoupled flux through complex II); 2.5 μM antimycin A (to determine non-mitochondrial respiration); 0.5 mM N,N,N′,N′-tetramethyl-ρ-phenylenediamine (TMPD), 2 mM ascorbate (to determine flux through complex IV); 1 mM potassium cyanide (normalization by inhibition of complex IV) [52]. The non-mitochondrial rate of oxygen consumption was subtracted from all measured functional parameters before reporting. Similarly, the rate of oxygen consumption after addition of potassium cyanide was subtracted from the rate of flux through complex IV for normalization purposes. Respiratory control ratios (RCRs) were determined for all treatment groups as the ratio of maximally-induced State 3 respiration/State 4 respiration.
2.8. Reactive oxygen species (ROS) production
Generation of ROS was evaluated using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) (Life Technologies, Carlsbad, CA, USA). Cells were plated in black 96-well plates, pretreated with or without oxEPA/oxDHA, and subsequently exposed to BDE 47 (or DMSO) for: 0.5 min, 1.5 min, 3, 6, 12, and 24 h. Cells pretreated with DMSO (12 h) and subsequently exposed to hydrogen peroxide (250 μM) were used as a positive control for ROS production at each time point. After each exposure period, cells were directly incubated with 10 μM H2DCF-DA for 30 min at 37 °C. Intracellular ROS production is correlated to cellular oxidation of H2DCF-DA to the highly fluorescent 2,7-dichlorofluorescein (DCF). Oxidation of H2DCF-DA to DCF was measured in a fluorescence plate reader at 485 excitation/530 emission. Fluorescence was normalized to total protein by the Bio-Rad Protein Assay. Measurements were carried out in three separate experiments, each containing three replicate treatments.
2.9. Statistical analyses
All statistical analyses were conducted in GraphPad Prism Ver. 5.0 (Graph Pad Software Inc., San Diego, CA, USA). The effect of treatments on cellular GSH status, gene expression, cell viability, mitochondrial membrane potential, mitochondrial ETS functional parameters, and ROS production were assessed by One-way ANOVA with a Bonferonni’s Multiple Comparisons post-test. Differences between means of control and treatment groups were considered significant at p≤0.05, and homogeneity of variances was confirmed by F test. All data sets were examined for potential outliers using the Grubb’s test [53], and outlier values were excluded at significance level p<0.05.
3.0 Results
3.1. Glutathione status
Treatment of HepG2 cells with oxEPA/oxDHA for 12 h significantly increased intracellular GSH to two-fold control levels (Fig. 1A). The level of GSH induction at 12 h by oxEPA/oxDHA exceeded that observed for SFN, a positive control of GSH induction, although the potency of GSH induction by oxEPA/oxDHA relative to SFN was not statistically different (p>0.05). Because the elevation in intracellular GSH by oxEPA/oxDHA was consistent over 12-18 h of incubation, we selected a 12 h oxEPA/oxDHA pretreatment for all subsequent chemoprotection experiments. GSH was measured in cells after pretreatment with or without oxEPA/oxDHA for 12 h, and subsequent exposure to BDE 47 (or DMSO) for 24 h. After oxEPA/oxDHA treatment, and in the absence of toxicant, GSH levels were 1.8-fold above those of control cells (Fig. 1B). Exposure to BDE 47 in the absence of antioxidants elicited a 39% increase in GSH relative to controls, although the level of induction was not statistically significant. By contrast, GSH levels were 2.2-fold above control levels in cells pretreated with oxEPA/oxDHA before BDE 47 exposure (Fig. 1B).
Figure 1. Effect of oxEPA/oxDHA and BDE 47 on GSH status.

(A) Induction of GSH was measured in HepG2 cells exposed to a 1.5/1 mixture of oxEPA/oxDHA (60 μM oxEPA/40 μM oxDHA), sulforaphane (SFN, 40 μM), or equal volume of DMSO (vehicle control). Alphabetical letters are used to indicate statistically significant differences between means at p≤0.05. Letters shared in common between or among the groups indicate no significant difference at p>0.05. (B) GSH status in cells pretreated with oxEPA/oxDHA (or DMSO, denoted as “-oxEPA/oxDHA in figure legend) for 12 h, prior to exposure to 100 μM BDE 47 (or DMSO, denoted as “-BDE 47” in figure legend) for 24 h. ***p≤0.001 relative to corresponding controls. Data are expressed as fold over control values and are means ± SEM of n=3 experiments.
3.2. Effect of treatments on antioxidant gene expression
As observed in Figure 2, exposure to BDE 47 caused a significant modulation of gclc and nqo1 mRNA expression. Specifically, BDE 47 increased gclc expression to 3.3-fold control levels (Fig. 2A), and also reduced nqo1 expression by 25% relative to controls (Fig. 2B). By contrast, both gclc and nqo1 were significantly induced in cells pretreated with oxEPA/oxDHA prior to BDE 47 exposure. Specifically, a 4-fold induction of gclc relative to controls (Fig. 2A), and a minor 24% increase in nqo1 (Fig. 2B) was observed in cells pretreated with oxEPA/oxDHA and challenged with BDE 47. Expression of gclc and nqo1 in cells treated with oxEPA/oxDHA before incubation in the absence of toxicant for 24 h did not differ from controls.
Figure 2. Effect of oxEPA/oxDHA and BDE 47 on oxidative stress marker genes.
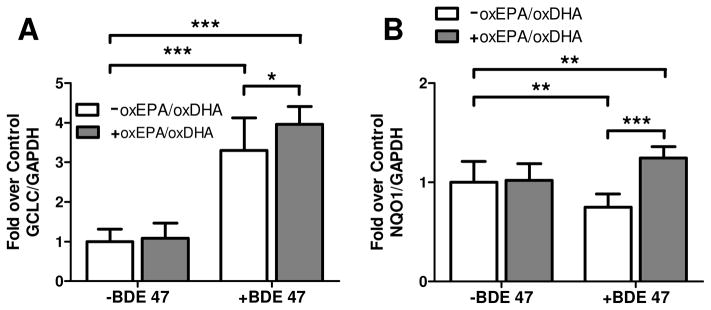
Fold induction relative to controls of (A) gclc, or (B) nqo1 mRNA transcripts was measured in cells pretreated with or without oxEPA/oxDHA for 12 h, followed by exposure to 100 μM BDE 47 for 24 h. *p≤0.05, **p≤0.01, ***p≤0.001 relative to corresponding controls. Data are expressed as fold over control values and are means ± SEM of n=3 experiments.
3.3. Effect of treatments on HepG2 cell viability
Treatment of cells with oxEPA/oxDHA under conditions that elevated intracellular GSH concentrations (i.e. 12 h pretreatment) was associated with a minor, albeit significant 13% increase in cell viability in the absence of toxicant (Fig. 3). As observed, pretreatment with oxEPA/oxDHA prior to exposure to BDE 47 provided partial protection against BDE 47-induced loss of cell viability (Fig. 3). Specifically, the 50 μM dose of BDE 47 caused a 55% loss of viability in HepG2 cells, and oxEPA/oxDHA pretreatment reduced the observed loss in viability to 48%, which was not statistically significant (p=0.052). At the 100 μM dose, BDE 47 caused a 67% loss of viability, and oxEPA/oxDHA pretreatment provided a minor, albeit significant 6% protection of viability (Fig. 3).
Figure 3. Effect of oxEPA/oxDHA on loss of cellular viability from BDE 47 exposure.

HepG2 cells were treated with oxEPA/oxDHA for 12 h to induce intracellular GSH prior to 24 h exposure to BDE 47 (50 or 100 μM). Cellular viability was determined by MTT assay. *p≤0.05, **p≤0.01 relative to corresponding controls. Data are expressed as percent of control values and are means ± SEM of n=3 experiments.
3.4. Effect of treatments on mitochondrial membrane potential and electron transport system function
Exposure to BDE 47 (100 μM) for 24 h resulted in a significant 22% loss of mitochondrial membrane potential (ΔΨm) (Fig. 4). By contrast, pretreatment of HepG2 cells with oxEPA/oxDHA fully protected against the loss of ΔΨm by the toxicant. Pretreatment of HepG2 cells with oxEPA/oxDHA in the absence of BDE 47 had no effect on ΔΨm. The loss of ΔΨm elicited by BDE 47 was accompanied by a decline in mitochondrial respiratory capacity (Fig. 5). As observed in the ΔΨm experiments, the decline in mitochondrial respiratory capacity was partially mitigated by pretreatment with oxEPA/oxDHA. Mitochondrial State 3 respiration using complex I substrates was reduced by 54% following BDE 47 exposure, and oxEPA/oxDHA pretreatment reduced the magnitude of loss by 33% (Fig. 5A). Maximally-induced State 3 respiration using complex I+II substrates was also reduced by 59% following BDE 47 exposure (Fig. 5B), with oxEPA/oxDHA attenuating this reduction by 20% (Fig. 5B). Maximum respiratory capacity as measured under conditions of uncoupled mitochondrial respiration was decreased by 66% following BDE 47 exposure, and oxEPA/oxDHA attenuated the BDE 47-associated decrease in maximum uncoupled respiration by 14% (Fig. 5C). State 4 respiration was reduced by 25% following BDE 47 exposure, but this effect was not statistically significant (Fig. 5D). No modulation of State 4 respiration was observed in the group pretreated with oxEPA/oxDHA prior to BDE 47 exposure. Exposure to BDE 47 reduced oxygen flux capacity through complexes II and IV by 32% (Fig. 5E), and 28% (Fig. 5F), respectively. Pretreatment with oxEPA/oxDHA partially prevented the loss of flux through complexes II and IV by 9%, and 20%, respectively. BDE 47 exposure reduced the respiratory control ratios (RCR) of HepG2 cells by 41%, and pretreatment with oxEPA/oxDHA prior to BDE 47 exposure did not prevent loss of RCR (Fig. 5G).
Figure 4. Effect of oxEPA/oxDHA on loss of mitochondrial membrane potential from BDE 47 exposure.

Mitochondrial membrane potential (ΔΨm) was quantified in cells by measuring JC-fluorescence in cells pretreated with, or in the absence of oxEPA/oxDHA for 12 h, followed by 24 h exposure to 100 μM BDE 47 (or DMSO). *p≤0.05, ***p≤0.001 relative to the corresponding control group. Data are expressed as percent of control values and are means ± SEM of n=3 experiments.
Figure 5. Effect of oxEPA/oxDHA on loss of mitochondrial electron transport system function from BDE 47 exposure.

Oxygen consumption (flux) was determined in permeabilized HepG2 cells exposed to: 1) vehicle control (DMSO, 36 h), 2) DMSO for 12 h followed by BDE 47 (100 μM) for 24 h, or 3) pretreated with oxEPA/oxDHA for 12 h before BDE 47 exposure for 24 h. Experimental conditions included: (A) State 3 respiration with complex I substrates only; (B) maximally-induced State 3 respiration with substrates of complex I and II; (C) maximum uncoupled respiration using the uncoupling agent CCCP; (D) State 4 respiration induced by oligomycin. Oxygen flux capacity through (E) complex II, and (F) complex IV were also determined. (G) The respiratory control ratios (RCRs) were calculated as the ratio of maximally-induced State 3 respiration/State 4 respiration. *p≤0.05, ***p≤0.001 relative to control group. All data are means ± SEM of n=4 experiments.
3.5. ROS production
Treatment with oxEPA/oxDHA in the absence of toxicant did not modulate ROS production relative to control cells. By contrast, exposure of cells to BDE 47 in the absence of antioxidants caused a significant 60% increase in ROS production relative to controls by 24 h exposure. As observed in Figure 6, the greatest increase in BDE 47-stimulated ROS generation (31% increase) occurred rapidly after 1.5 h exposure to BDE 47 (Fig. 6). By contrast, oxEPA/oxDHA pretreatment prior to BDE 47 exposure prevented the BDE 47 stimulated ROS production observed at 1.5 h, and significantly limited BDE 47-induced ROS generation for 12 h such that levels were not statistically different from controls (Fig. 6). By 24 h, however, the protective effect of the antioxidants diminished and there were no differences in ROS production among antioxidant pretreated and control cells in the presence of BDE 47.
Figure 6. Effect of oxEPA/oxDHA on BDE 47-induced ROS production.

Production of ROS was measured in HepG2 cells pretreated with (or without) oxEPA/oxDHA prior to exposure to 100 μM BDE 47 (or DMSO). Alphabetical letters are used to indicate statistically significant differences between means at p≤0.05. Letters shared in common between or among the groups indicate no significant difference at p>0.05. Data are means ± SEM of n=3 experiments.
4.0. Discussion
In the present study we have demonstrated the protective interaction among the major antioxidants and a major contaminant present in Pacific salmon, relevant to a scenario involving salmon consumption. Our results are consistent with reports that oxidized omega-3s play a critical role in activating beneficial cellular antioxidant responses [36, 37, 39]. The fact that oxEPA/oxDHA induced intracellular GSH in HepG2 cells more effectively than treatment with sulforaphane, a model dietary isothiocyanate and a positive control for Nrf2 activation and GSH induction [39, 54], and that oxEPA/oxDHA increased overall cell viability in the absence of toxicant, are noteworthy. Treatment with oxEPA/oxDHA stimulated a significant increase of GSH that remained stably elevated in cells for 24 h after removal of the treatment media, whereas expression of gclc and nqo1 were similar to control levels 24 h after initial treatment. This finding suggests oxEPA/oxDHA stimulates a biphasic effect on expression of Nrf2-regulated genes, inducing an initial increase in expression that eventually returns to near control levels. Indeed, similar studies using HepG2 cells treated with dietary antioxidants are consistent with the observed effects of oxEPA/oxDHA, reporting a biphasic increase and subsequent decrease toward control levels of Nrf2-mediated antioxidant response gene expression, including gclc [55-57], by 8-12 h after initial treatment. Taken together, the lasting stimulatory effect on GSH status, and protection against BDE 47-induced depletion of GSH, indicates the activation of cellular antioxidant responses by oxEPA/oxDHA is associated with protective functional impacts in cells.
We hypothesized a more dramatic chemoprotective effect of oxidized omega-3s on cell viability than the partial protection observed in the current study. However, the partial protection of the oxidized omega-3s against cell toxicity is consistent with the extent of antioxidant-mediated amelioration reported in previous studies of human cells exposed to BDE 47. For example, pretreatment of human fetal hematopoietic stem cells with 8 mM NAC induced a significant, but partial 20% protection of viability against 50 μM BDE 47 [31]. Similarly, co-treatment of Jurkat cells with 5 mM NAC has been shown to reduce the rate of apoptosis induced by 50 μM BDE 47 from 14% to 3% [28]. The partial chemoprotective effect of oxEPA/oxDHA against 50 and 100 μM BDE 47-induced loss of viability observed in HepG2 cells may reflect the fact that BDE 47 cell toxicity involves multiple mechanisms, including those other than oxidative stress, and are thus not mitigated by antioxidant responses.
As discussed, a major goal of this study was to better understand the mechanisms of BDE 47-mediated mitochondrial injury. Previous studies in our laboratory using both human and fish cells indicated BDE 47-induced generation of ROS is associated with a loss of ΔΨm [30, 31]. Significant loss of ΔΨm can activate mitochondrial-regulated apoptosis pathways or cellular necrosis via opening of the mitochondrial permeability transition pore (MPTP) [58]. Interestingly, the onset of MPTP opening can be delayed by dietary supplementation with DHA [59, 60], supporting a role of DHA in stabilizing ΔΨm. Dietary supplementation with omega-3s, particularly DHA, is reported to have significant beneficial effects on mitochondrial membranes and function [61]. Our results suggest the oxidation products of omega-3s may mediate some of the beneficial effects on mitochondrial membranes by significantly limiting loss of ΔΨm from BDE 47 exposure.
An interesting finding from our study was that exposure of HepG2 cells to BDE 47 reduced all electron transport system functional parameters with the possible exception of State 4 respiration, although we observed a similar trend toward reduction of State 4 respiration in the presence of 100 μM BDE 47 as well. The fact that extensive reduction of State 3 and maximally-induced State 3 respiration was observed following BDE 47 exposure, suggests inhibition of the ETS within complexes I-V, or a loss of electron transfer capacity of the ETS. Although we did not determine the effect of BDE 47 on complex V, we observed significant loss of maximum uncoupled respiration, which indicates impairment within complexes I-IV. In this regard, a reduction of oxygen flux capacity through complexes I, II, and IV by BDE 47 was verified in our experiments. Additionally, the significant reduction of uncoupled respiration supports the hypothesis that BDE 47 causes a loss of respiratory capacity of the ETS.
A previous report in isolated zebrafish mitochondria exposed to the hydroxylated BDE 47 metabolite 6-hydroxy-BDE 47 (6-OH-BDE47) showed a significant inhibition of complex II function and uncoupling of mitochondrial respiration [62]. By contrast, our data did not support a specific antagonistic effect of BDE 47 against a particular ETS complex, as we observed loss of respiratory capacity occurring at several complexes. Rather, BDE 47 caused a general inhibition of the ETS in HepG2 cells, significantly reducing State 3 respiration and maximum uncoupled respiratory capacity. A previous report in isolated rat mitochondria exposed to 25 and 50 μM BDE 47 similarly demonstrated a reduction of State 3 respiration using complex I and complex II substrates [63], consistent with our findings. Further experiments are needed to elucidate the mechanisms underlying the observed effects on complexes III and V in BDE 47-induced loss of ETS function. The observed protection of State 3 and maximally-induced State 3 respiration suggests the antioxidant effects of oxEPA/oxDHA play a beneficial role of preserving coupling of oxidative phosphorylation in mitochondria. Also, the protection of maximum uncoupled respiration indicates oxEPA/oxDHA can prevent against loss of function within complexes I-IV. Indeed, partial protection of flux capacity through complexes II and IV was observed, and the most extensive protective effect of oxEPA/oxDHA was observed for complex I (i.e. rescue of State 3 respiration using complex I substrates). Rescue of respiratory coupling by oxEPA/oxDHA may also involve beneficial protective effects on complexes III and V, but this was not determined in the current study.
The significant reduction of maximally-induced State 3 respiration in BDE 47-exposed HepG2 cells resulted in significant reduction of the RCR, a measure of coupling of oxidative phosphorylation [64]. Compared to this treatment group, nearly identical RCRs were calculated in our study in cells pretreated with oxEPA/oxDHA before BDE 47 treatment. This suggests that oxEPA/oxDHA did not prevent loss of overall mitochondrial respiratory function caused by BDE 47, despite significant protection of critical respiratory parameters in mitochondria. The lack of a more marked protective effect on mitochondrial function by oxEPA/oxDHA supports the hypothesis that other mechanisms unrelated to oxidative stress may contribute to BDE 47 mitochondrial toxicity in HepG2 cells. However, the fact that oxEPA/oxDHA antioxidant induction was associated with some preservation of respiratory function in HepG2 cells strongly suggests a role of oxidative stress in the toxicity of BDE 47 to the mitochondrial ETS.
A limitation of our study is that we did not conduct the relevant co-treatment of cells with oxEPA/oxDHA and BDE 47, as this exposure scenario can be similarly representative of dietary intake of contaminated seafood. However, our approach was to better understand if an antioxidant response elicited by the dietary antioxidants would be sufficient to ameliorate BDE 47 toxicity. Thus, we preferred to pretreat cells with oxEPA/oxDHA to elicit the antioxidant response and subsequently challenge with BDE 47 as in the context of a chemoprevention study. We also wanted to minimize the potential for artifacts from co-administration, specifically with regards to the presence of serum in the treatment media, which can decrease the bioavailability of lipophilic toxicants including BDE 47 in vitro [65, 66]. Supplementation of the pretreatment exposure media with 5-10% FBS was required for administration of oxEPA/oxDHA to HepG2 cells [37], whereas BDE 47 exposure media utilized in similar in vitro studies are supplemented with very low concentrations of serum (1%) [32, 42], or is serum-free [25, 67]. Thus, co-treatment of cells with oxEPA/oxDHA and BDE 47 should be addressed in future experiments, but was somewhat out of the scope of the current manuscript due to reasons including the aforementioned differences in experimental exposure methods that are required for administering these compounds to cells.
It is important to note that the doses of BDE 47 utilized in the current study were selected to induce significant cellular and mitochondrial toxicity in HepG2 cells based on previous studies in human cells [27, 28, 31]. In human [32, 42], fish [29, 30], and rodent cells [26] exposed to BDE 47, oxidative injury is not typically observed below concentrations in the 20-50 μM range. Hence, it is plausible that the antioxidant effects of oxEPA/oxDHA may provide greater protection against, or ameliorate, the toxicity of lower doses of BDE 47 representative of environmental exposures. Additionally, the fact that oxEPA/oxDHA significantly attenuated BDE 47-induced ROS production for 12 h suggests that cellular toxicity resulting from a dose of 100 μM BDE 47 for an exposure period of 24 h selected in our experiments may have overwhelmed the protective antioxidant effects of oxEPA/oxDHA. Therefore, we hypothesize that oxEPA/oxDHA pretreatment can ameliorate or have a greater protective effect against shorter subsequent exposure periods to BDE 47 (i.e. 0.5-12 h) than assessed in the current study. While experimental PBDE doses in the micromolar range are relevant to toxicological studies, these doses likely far exceed PBDE levels reaching target human tissues from fish consumption [14, 68-71]. The fact that an average meal size of 227 grams of salmon contains approximately 1x106-fold more omega-3s on a wet weight basis than persistent organic such as PBDEs [35] suggests that in vitro results from studies such as ours likely underestimate the chemoprotection elicited by omega-3s in vivo. In this regard, it is of importance that risk assessment paradigms addressing the adverse effects of persistent compounds in seafood that elicit oxidative stress as a mechanism of toxicity account for the protective antioxidant effects from omega-3s and other micronutrients.
Supplementary Material
Supplementary Table 1. Summary of effects of oxEPA/oxDHA and BDE 47 on mitochondrial ETS complexes
Highlights.
BDE 47 is a toxic PBDE flame retardant and major contaminant in Pacific salmon.
Oxidized omega-3 fatty acids (oxEPA/oxDHA) partially prevented BDE 47 toxicity.
oxEPA/oxDHA partially protected viability and mitochondrial membrane potential.
oxEPA/oxDHA partially protected mitochondrial electron transport system function.
Our results indicate oxEPA/oxDHA protects against shorter exposures to BDE 47.
Acknowledgments
Funding was provided by the Washington Sea Grant (R/OCEH-5), the UW NIEHS Superfund Research Program (NIEHS P42ES004696), the UW NIEHS training grant in Environmental Pathology and Toxicology (T32 ES007032), and NIH grant RC2 AG036606.
Footnotes
Conflict of Interest.
The authors declare that there are no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gomara B, Herrero L, Pacepavicius G, Ohta S, Alaee M, Gonzalez MJ. Occurrence of co-planar polybrominated/chlorinated biphenyls (PXBs), polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) in breast milk of women from Spain. Chemosphere. 2011;83:799–805. doi: 10.1016/j.chemosphere.2011.02.080. [DOI] [PubMed] [Google Scholar]
- 2.Hale RC, Alaee M, Manchester-Neesvig JB, Stapleton HM, Ikonomou MG. Polybrominated diphenyl ether flame retardants in the North American environment. Environ Int. 2003;29:771–779. doi: 10.1016/S0160-4120(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 3.Miller MF, Chernyak SM, Domino SE, Batterman SA, Loch-Caruso R. Concentrations and speciation of polybrominated diphenyl ethers in human amniotic fluid. Science of the Total Environment. 2012;417:294–298. doi: 10.1016/j.scitotenv.2011.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.State Regulation of Flame Retardants in Consumer Products. http://www.ncsl.org/issues-research/env-res/flame-retardants-in-consumer-products.aspx.
- 5.Darnerud PO. Toxic effects of brominated flame retardants in man and in wildlife. Environ Int. 2003;29:841–853. doi: 10.1016/S0160-4120(03)00107-7. [DOI] [PubMed] [Google Scholar]
- 6.Joyce M, Donohue HG-G, Manibusan Mary. Agency UEP, editor. Toxicological review Of 2,2′,4,4′ tetrabromodiphenyl ether (BDE-47) 2008. [Google Scholar]
- 7.Jakobsson K, Fang J, Athanasiadou M, Rignell-Hydbom A, Bergman A. Polybrominated diphenyl ethers in maternal serum, umbilical cord serum, colostrum and mature breast milk. Insights from a pilot study and the literature. Environ Int. 2012;47:121–130. doi: 10.1016/j.envint.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Herbstman JB, Sjodin A, Kurzon M, Lederman SA, Jones RS, Rauh V, Needham LL, Tang D, Niedzwiecki M, Wang RY, Perera F. Prenatal exposure to PBDEs and neurodevelopment. Environ Health Perspect. 2010;118:712–719. doi: 10.1289/ehp.0901340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meeker JD, Johnson PI, Camann D, Hauser R. Polybrominated diphenyl ether (PBDE) concentrations in house dust are related to hormone levels in men. Sci Total Environ. 2009;407:3425–3429. doi: 10.1016/j.scitotenv.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson PI, Stapleton HM, Sjodin A, Meeker JD. Relationships between polybrominated diphenyl ether concentrations in house dust and serum. Environ Sci Technol. 2010;44:5627–5632. doi: 10.1021/es100697q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stapleton HM, Eagle S, Sjodin A, Webster TF. Serum PBDEs in a North Carolina Toddler Cohort: Associations with Handwipes, House Dust, and Socioeconomic Variables. Environmental Health Perspectives. 2012;120:1049–1054. doi: 10.1289/ehp.1104802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohta S, Ishizuka D, Nishimura H, Nakao T, Aozasa O, Shimidzu Y, Ochiai F, Kida T, Nishi M, Miyata H. Comparison of polybrominated diphenyl ethers in fish, vegetables, and meats and levels in human milk of nursing women in Japan. Chemosphere. 2002;46:689–696. doi: 10.1016/s0045-6535(01)00233-8. [DOI] [PubMed] [Google Scholar]
- 13.Tittlemier SA, Forsyth D, Breakell K, Verigin V, Ryan JJ, Hayward S. Polybrominated diphenyl ethers in retail fish and shellfish samples purchased from Canadian markets. J Agric Food Chem. 2004;52:7740–7745. doi: 10.1021/jf048665y. [DOI] [PubMed] [Google Scholar]
- 14.Hites RA, Foran JA, Schwager SJ, Knuth BA, Hamilton MC, Carpenter DO. Global assessment of polybrominated diphenyl ethers in farmed and wild salmon. Environ Sci Technol. 2004;38:4945–4949. doi: 10.1021/es049548m. [DOI] [PubMed] [Google Scholar]
- 15.Schecter A, Pavuk M, Papke O, Ryan JJ, Birnbaum L, Rosen R. Polybrominated diphenyl ethers (PBDEs) in U.S. mothers’ milk. Environ Health Perspect. 2003;111:1723–1729. doi: 10.1289/ehp.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S, Kannan K, Moon HB. Assessment of exposure to polybrominated diphenyl ethers (PBDEs) via seafood consumption and dust ingestion in Korea. Science of the Total Environment. 2013;443:24–30. doi: 10.1016/j.scitotenv.2012.10.099. [DOI] [PubMed] [Google Scholar]
- 17.O’Neill SM, JEW, Ylitalo GM, Sloan CA, Krahn MM, Collier TK. Concentrations of polybrominated diphenyl ethers (PBDEs) in fish from Puget Sound, WA, USA. ETAC World Congress and 25th Annual Meeting in North America Society of Environmental Toxicology and Chemistry; Portland, OR, USA. 2004. [Google Scholar]
- 18.Sloan CA, Anulacion BF, Bolton JL, Boyd D, Olson OP, Sol SY, Ylitalo GM, Johnson LL. Polybrominated diphenyl ethers in outmigrant juvenile Chinook salmon from the lower Columbia River and Estuary and Puget Sound, Washington. Arch Environ Contam Toxicol. 2010;58:403–414. doi: 10.1007/s00244-009-9391-y. [DOI] [PubMed] [Google Scholar]
- 19.Ecology WSDo; Ecology Do, editor. Fish Consumption Rates Technical Support Document: A Review of Data and Information about Fish Consumption in Washington. 2013. Version 2.0 edition. [Google Scholar]
- 20.Browne EP, Stapleton HM, Kelly SM, Tilton SC, Gallagher EP. In vitro hepatic metabolism of 2,2′,4,4′,5-pentabromodiphenyl ether (BDE 99) in Chinook salmon (Onchorhynchus tshawytscha) Aquat Toxicol. 2009;92:281–287. doi: 10.1016/j.aquatox.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stapleton HM, Letcher RJ, Baker JE. Debromination of polybrominated diphenyl ether congeners BDE 99 and BDE 183 in the intestinal tract of the common carp (Cyprinus carpio) Environ Sci Technol. 2004;38:1054–1061. doi: 10.1021/es0348804. [DOI] [PubMed] [Google Scholar]
- 22.Costa LG, Giordano G. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology. 2007;28:1047–1067. doi: 10.1016/j.neuro.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giordano G, Kavanagh TJ, Costa LG. Neurotoxicity of a polybrominated diphenyl ether mixture (DE-71) in mouse neurons and astrocytes is modulated by intracellular glutathione levels. Toxicol Appl Pharmacol. 2008;232:161–168. doi: 10.1016/j.taap.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Branchi I, Capone F, Alleva E, Costa LG. Polybrominated diphenyl ethers: Neurobehavioral effects following developmental exposure. Neurotoxicology. 2003;24:449–462. doi: 10.1016/S0161-813X(03)00020-2. [DOI] [PubMed] [Google Scholar]
- 25.He WH, He P, Wang AG, Xia T, Xu BY, Chen XM. Effects of PBDE-47 on cytotoxicity and genotoxicity in human neuroblastoma cells in vitro. Mutation Research-Genetic Toxicology and Environmental Mutagenesis. 2008;649:62–70. doi: 10.1016/j.mrgentox.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 26.He P, He WH, Wang AG, Xia T, Xu BY, Zhang M, Chen XM. PBDE-47-induced oxidative stress, DNA damage and apoptosis in primary cultured rat hippocampal neurons. Neurotoxicology. 2008;29:124–129. doi: 10.1016/j.neuro.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 27.An J, Yin LL, Shang Y, Zhong YF, Zhang XY, Wu MH, Yu ZQ, Sheng GY, Fu JM, Huang YC. The combined effects of BDE47 and BaP on oxidatively generated DNA damage in L02 cells and the possible molecular mechanism. Mutation Research-Genetic Toxicology and Environmental Mutagenesis. 2011;721:192–198. doi: 10.1016/j.mrgentox.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Yan C, Huang DJ, Zhang YM. The involvement of ROS overproduction and mitochondrial dysfunction in PBDE-47-induced apoptosis on Jurkat cells. Experimental and Toxicologic Pathology. 2011;63:413–417. doi: 10.1016/j.etp.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 29.Shao J, Dabrowski MJ, White CC, Kavanagh TJ, Gallagher EP. Flow cytometric analysis of BDE 47 mediated injury to rainbow trout gill epithelial cells. Aquatic Toxicology. 2010;97:42–50. doi: 10.1016/j.aquatox.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao J, Eckert ML, Lee LE, Gallagher EP. Comparative oxygen radical formation and toxicity of BDE 47 in rainbow trout cell lines. Mar Environ Res. 2008;66:7–8. doi: 10.1016/j.marenvres.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao J, White CC, Dabrowski MJ, Kavanagh TJ, Eckert ML, Gallagher EP. The role of mitochondrial and oxidative injury in BDE 47 toxicity to human fetal liver hematopoietic stem cells. Toxicol Sci. 2008;101:81–90. doi: 10.1093/toxsci/kfm256. [DOI] [PubMed] [Google Scholar]
- 32.Park HR, Kamau PW, Loch-Caruso R. Involvement of reactive oxygen species in brominated diphenyl ether-47-induced inflammatory cytokine release from human extravillous trophoblasts in vitro. Toxicology and Applied Pharmacology. 2014;274:283–292. doi: 10.1016/j.taap.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kris-Etherton PM, Taylor DS, Yu-Poth S, Huth P, Moriarty K, Fishell V, Hargrove RL, Zhao G, Etherton TD. Polyunsaturated fatty acids in the food chain in the United States. Am J Clin Nutr. 2000;71:179S–188S. doi: 10.1093/ajcn/71.1.179S. [DOI] [PubMed] [Google Scholar]
- 34.Palacios-Pelaez R, Lukiw WJ, Bazan NG. Omega-3 essential fatty acids modulate initiation and progression of neurodegenerative disease. Mol Neurobiol. 2010;41:367–374. doi: 10.1007/s12035-010-8139-z. [DOI] [PubMed] [Google Scholar]
- 35.Domingo JL, Bocio A, Falco G, Llobet JM. Benefits and risks of fish consumption Part I. A quantitative analysis of the intake of omega-3 fatty acids and chemical contaminants. Toxicology. 2007;230:219–226. doi: 10.1016/j.tox.2006.11.054. [DOI] [PubMed] [Google Scholar]
- 36.Musiek ES, Brooks JD, Joo M, Brunoldi E, Porta A, Zanoni G, Vidari G, Blackwell TS, Montine TJ, Milne GL, et al. Electrophilic cyclopentenone neuroprostanes are anti-inflammatory mediators formed from the peroxidation of the omega-3 polyunsaturated fatty acid docosahexaenoic acid. J Biol Chem. 2008;283:19927–19935. doi: 10.1074/jbc.M803625200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 38.Anderson EJ, Taylor DA. Stressing the heart of the matter: re-thinking the mechanisms underlying therapeutic effects of n-3 polyunsaturated fatty acids. F1000 Med Rep. 2012;4:13. doi: 10.3410/M4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majkova Z, Layne J, Sunkara M, Morris AJ, Toborek M, Hennig B. Omega-3 fatty acid oxidation products prevent vascular endothelial cell activation by coplanar polychlorinated biphenyls. Toxicol Appl Pharmacol. 2011;251:41–49. doi: 10.1016/j.taap.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 41.Bousquet M, Gibrat C, Saint-Pierre M, Julien C, Calon F, Cicchetti F. Modulation of brain-derived neurotrophic factor as a potential neuroprotective mechanism of action of omega-3 fatty acids in a parkinsonian animal model. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1401–1408. doi: 10.1016/j.pnpbp.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 42.Park HR, Loch-Caruso R. Protective effect of nuclear factor E2-related factor 2 on inflammatory cytokine response to brominated diphenyl ether-47 in the HTR-8/SVneo human first trimester extravillous trophoblast cell line. Toxicol Appl Pharmacol. 2014;281:67–77. doi: 10.1016/j.taap.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schreiber T, Gassmann K, Gotz C, Hubenthal U, Moors M, Krause G, Merk HF, Nguyen NH, Scanlan TS, Abel J, et al. Polybrominated Diphenyl Ethers Induce Developmental Neurotoxicity in a Human in Vitro Model: Evidence for Endocrine Disruption. Environmental Health Perspectives. 2010;118:572–578. doi: 10.1289/ehp.0901435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giordano G, Kavanagh J, Costa LG. Mouse cerebellar astrocytes protect cerebellar granule neurons against toxicity of the polybrominated diphenyl ether (PBDE) mixture DE-71. Neurotoxicology. 2009;30:326–329. doi: 10.1016/j.neuro.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viral Brahmbhatta MO, Brianda Muriel, Perrisseaua Geneviève, Schmida Viktoria Bastic, Destaillatsa Frédéric, Pace-Asciakb Cecil, Benyacouba Jalil, Bosco Nabil. Protective effects of dietary EPA and DHA on ischemia–reperfusion-induced intestinal stress. The Journal of Nutritional Biochemistry. 2013;24:104–111. doi: 10.1016/j.jnutbio.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 46.Jackson PA, Deary ME, Reay JL, Scholey AB, Kennedy DO. No effect of 12 weeks’ supplementation with 1 g DHA-rich or EPA-rich fish oil on cognitive function or mood in healthy young adults aged 18–35 years. Br J Nutr. 2012;107:1232–1243. doi: 10.1017/S000711451100403X. [DOI] [PubMed] [Google Scholar]
- 47.Wojtowicz JC, Butovich I, Uchiyama E, Aronowicz J, Agee S, McCulley JP. Pilot, prospective, randomized, double-masked, placebo-controlled clinical trial of an omega-3 supplement for dry eye. Cornea. 2011;30:308–314. doi: 10.1097/ICO.0b013e3181f22e03. [DOI] [PubMed] [Google Scholar]
- 48.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 49.Giordano G, White CC, Costa LG. Assessment of glutathione homeostasis. Methods Mol Biol. 2011;758:205–214. doi: 10.1007/978-1-61779-170-3_14. [DOI] [PubMed] [Google Scholar]
- 50.Espinoza HM, Williams CR, Gallagher EP. Effect of cadmium on glutathione S-transferase and metallothionein gene expression in coho salmon liver, gill and olfactory tissues. Aquatic Toxicology. 2012;110:37–44. doi: 10.1016/j.aquatox.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 52.Siegel MP, Kruse SE, Knowels G, Salmon A, Beyer R, Xie H, Van Remmen H, Smith SR, Marcinek DJ. Reduced coupling of oxidative phosphorylation in vivo precedes electron transport chain defects due to mild oxidative stress in mice. PLoS One. 2011;6:e26963. doi: 10.1371/journal.pone.0026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grubbs FE. Procedures for Detecting Outlying Observations in Samples. Technometrics. 1969;11:1. [Google Scholar]
- 54.Ahn YH, Hwang Y, Liu H, Wang XJ, Zhang Y, Stephenson KK, Boronina TN, Cole RN, Dinkova-Kostova AT, Talalay P, Cole PA. Electrophilic tuning of the chemoprotective natural product sulforaphane. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9590–9595. doi: 10.1073/pnas.1004104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kay HY, Won Yang J, Kim TH, Lee da Y, Kang B, Ryu JH, Jeon R, Kim SG. Ajoene, a stable garlic by-product, has an antioxidant effect through Nrf2-mediated glutamate-cysteine ligase induction in HepG2 cells and primary hepatocytes. J Nutr. 2010;140:1211–1219. doi: 10.3945/jn.110.121277. [DOI] [PubMed] [Google Scholar]
- 56.Huerta-Olvera SG, Macias-Barragan J, Ramos-Marquez ME, Armendariz-Borunda J, Diaz-Barriga F, Siller-Lopez F. Alpha-lipoic acid regulates heme oxygenase gene expression and nuclear Nrf2 activation as a mechanism of protection against arsenic exposure in HepG2 cells. Environ Toxicol Pharmacol. 2010;29:144–149. doi: 10.1016/j.etap.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Gong P, Cederbaum AI. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology. 2006;43:144–153. doi: 10.1002/hep.21004. [DOI] [PubMed] [Google Scholar]
- 58.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Shea KM, Khairallah RJ, Sparagna GC, Xu W, Hecker PA, Robillard-Frayne I, Des Rosiers C, Kristian T, Murphy RC, Fiskum G, Stanley WC. Dietary omega-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J Mol Cell Cardiol. 2009;47:819–827. doi: 10.1016/j.yjmcc.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khairallah RJ, Sparagna GC, Khanna N, O’Shea KM, Hecker PA, Kristian T, Fiskum G, Des Rosiers C, Polster BM, Stanley WC. Dietary supplementation with docosahexaenoic acid, but not eicosapentaenoic acid, dramatically alters cardiac mitochondrial phospholipid fatty acid composition and prevents permeability transition. Biochim Biophys Acta. 2010;1797:1555–1562. doi: 10.1016/j.bbabio.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khairallah RJ, Kim J, O’Shea KM, O’Connell KA, Brown BH, Galvao T, Daneault C, Des Rosiers C, Polster BM, Hoppel CL, Stanley WC. Improved Mitochondrial Function with Diet-Induced Increase in Either Docosahexaenoic Acid or Arachidonic Acid in Membrane Phospholipids. PLoS One. 2012;7 doi: 10.1371/journal.pone.0034402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Boxtel AL, Kamstra JH, Cenijn PH, Pieterse B, Wagner JM, Antink M, Krab K, van der Burg B, Marsh G, Brouwer A, Legler J. Microarray analysis reveals a mechanism of phenolic polybrominated diphenylether toxicity in zebrafish. Environ Sci Technol. 2008;42:1773–1779. doi: 10.1021/es0720863. [DOI] [PubMed] [Google Scholar]
- 63.Pazin M, Pereira LC, Dorta DJ. Toxicity of brominated flame retardants, BDE-47 and BDE-99 stems from impaired mitochondrial bioenergetics. Toxicol Mech Methods. 2014:1–8. doi: 10.3109/15376516.2014.974233. [DOI] [PubMed] [Google Scholar]
- 64.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hestermann EV, Stegeman JJ, Hahn ME. Serum alters the uptake and relative potencies of halogenated aromatic hydrocarbons in cell culture bioassays. Toxicological Sciences. 2000;53:316–325. doi: 10.1093/toxsci/53.2.316. [DOI] [PubMed] [Google Scholar]
- 66.Mundy WR, Freudenrich TM, Crofton KM, DeVito MJ. Accumulation of PBDE-47 in primary cultures of rat neocortical cells. Toxicological Sciences. 2004;82:164–169. doi: 10.1093/toxsci/kfh239. [DOI] [PubMed] [Google Scholar]
- 67.Tagliaferri S, Caglieri A, Goldoni M, Pinelli S, Alinnovi R, Poli D, Pellacani C, Giordano G, Mutti A, Costa LG. Low concentrations of the brominated flame retardants BDE-47 and BDE-99 induce synergistic oxidative stress-mediated neurotoxicity in human neuroblastoma cells. Toxicology in Vitro. 2010;24:116–122. doi: 10.1016/j.tiv.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 68.Stone D. Polybrominated diphenyl ethers and polychlorinated biphenyls in different tissue types from chinook salmon (Oncorhynchus tshawytscha) Bulletin of Environmental Contamination and Toxicology. 2006;76:148–154. doi: 10.1007/s00128-005-0901-y. [DOI] [PubMed] [Google Scholar]
- 69.Hayward D, Wong J, Krynitsky AJ. Polybrominated diphenyl ethers and polychlorinated biphenyls in commercially wild caught and farm-raised fish fillets in the United States. Environmental Research. 2007;103:46–54. doi: 10.1016/j.envres.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 70.Jenssen BM, Sormo EG, Baek K, Bytingsvik J, Gaustad H, Ruus A, Skaare JU. Brominated Flame Retardants in North-East Atlantic Marine Ecosystems. Environmental Health Perspectives. 2007;115:35–41. doi: 10.1289/ehp.9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pulkrabova J, Hajslova J, Poustka J, Kazda R. Fish as Biomonitors of Polybrominated Diphenyl Ethers and Hexabromocyclododecane in Czech Aquatic Ecosystems: Pollution of the Elbe River Basin. Environmental Health Perspectives. 2007;115:28–34. doi: 10.1289/ehp.9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Summary of effects of oxEPA/oxDHA and BDE 47 on mitochondrial ETS complexes