

The data from this study contribute to the ongoing debate about the role of cancer stem cells in gliomagenesis. Results showed that CD15, a marker previously thought to be a cancer stem-like marker in glioblastoma, could not isolate a phenotypically or genetically distinct population. Moreover, isolated CD15-positive and -negative cells were able to generate mixed populations of glioblastoma cells in vitro.
Keywords: Glioblastoma, CD15, SSEA1, Hierarchy, Stem cell, Cancer
Abstract
Recent research has focused on the hypothesis that the growth and regeneration of glioblastoma (GB) is sustained by a subpopulation of self-renewing stem-like cells. This has led to the prediction that molecular markers for cancer stem cells in GB may provide a treatment target. One candidate marker is CD15: we wanted to determine if CD15 represented a credible stem cell marker in GB. We first demonstrated that CD15-positive (CD15+) cells were less proliferative than their CD15-negative (CD15−) counterparts in 10 patient GB tumors. Next we compared the proliferative activity of CD15+ and CD15− cells in vitro using tumor-initiating primary GB cell lines (TICs) and found no difference in proliferative behavior. Furthermore, TICs sorted for CD15+ and CD15− were not significantly different cytogenetically or in terms of gene expression profile. Sorted single CD15+ and CD15− cells were equally capable of reconstituting a heterogeneous population containing both CD15+ and CD15− cells over time, and both CD15+ and CD15− cells were able to generate tumors in vivo. No difference was found in the phenotypic or genomic behavior of CD15+ cells compared with CD15− cells from the same patient. Moreover, we found that in vitro, cells were able to interconvert between the CD15+ and CD15− states. Our data challenge the utility of CD15 as a cancer stem cell marker.
Significance
The data from this study contribute to the ongoing debate about the role of cancer stem cells in gliomagenesis. Results showed that CD15, a marker previously thought to be a cancer stem-like marker in glioblastoma, could not isolate a phenotypically or genetically distinct population. Moreover, isolated CD15-positive and -negative cells were able to generate mixed populations of glioblastoma cells in vitro.
Introduction
Glioblastoma (GB), the most common and aggressive neuroepithelial malignancy, is characterized by genetic instability and complex clonal evolutionary dynamics that result in histopathological diversity and clinical heterogeneity [1]. The evolution of disease complexity in cancer is generated through genetic and epigenetic mechanisms that form the basis of the clonal evolution model of oncogenesis [2]. More recently, the cancer stem cell hypothesis has challenged stochastic evolution by positing that cancers are hierarchically organized [3]. In this model, oncogenesis is driven by a subpopulation of cancer stem cells with progeny that undergo irreversible epigenetic changes, analogous to normal cell differentiation, resulting in nontumorigenic populations that contribute the bulk of the tumor. The hypothesis is attractive because elimination of the aggressive subpopulation could result in disease cure.
Although cancer stem cells (CSCs) and tumor-initiating cells (TICs) have been relatively established in some cancers, including leukemia [4, 5], germ cell tumors [6, 7], breast [8], and colon cancers [9, 10], their existence seems unlikely in other cancers including lymphoma and melanoma [11–13].
In neuroepithelial malignancies, the existence of CSCs and TICs remains controversial because no robust cell-surface marker has been identified to distinguish prospectively tumorigenic and nontumorigenic cell populations [14–19]. Initial data identifying CD133/prominin 1 as a marker that enriched for TICs in GB [20–22] were subsequently challenged [23–26]. More recently, the cell surface marker CD15/stage-specific embryonic antigen 1 has been identified as a possible TIC marker in GB and medulloblastoma [23, 27–30]. CD15 is a carbohydrate moiety expressed by a variety of cells including neural stem and progenitor cells and is developmentally regulated in the brain, where it is thought to play a role in cell-cell interaction during neuronal development [31–35].
Based on these data, we wanted to determine whether CD15-expressing cells in GB represented a TIC marker and a potential therapeutic target in clinically symptomatic patients with GB. We used a combined approach to characterize CD15-expressing cells in patient tumors, in vitro, and in xenograft animal models.
Materials and Methods
Cell Line Derivation
Tissue collection protocols were compliant with the U.K. Human Tissue Act 2004 (HTA License ref 12315) and approved by the local regional ethics committee (LREC ref 04/Q0108/60). Informed consent was obtained from each patient through our research clinic [36]. Cell derivation and the tumor formation assay have been described previously [19]. Briefly, anonymized tissue was mechanically minced, and cells were seeded in defined serum-free (SF) medium and allowed to form primary aggregates. These spheroid aggregates were collected and plated, without dissociation, onto flasks coated with extracellular matrix (ECM; 1:10 dilution; Sigma-Aldrich, St. Louis, MO, https://www.sigmaaldrich.com) and allowed to form a primary monolayer. As the primary monolayer approached confluence, cells were passaged to generate the subsequent monolayers. Cells were cultured in 10 ml SF medium (phenol red-free Neurobasal A, Invitrogen; Thermo Fisher Scientific, Waltham, MA, http://www.thermofisher.com) with 2 mM l-glutamine and 1% volume/volume (vol/vol) penicillin/streptomycin/Fungizone solution with 20 ng/ml human epidermal growth factor (Sigma-Aldrich), 20 ng/ml basic fibroblast growth factor (R&D Systems, Minneapolis, MN, http://www.rndsystems.com), 20 ng/ml heparin (Sigma-Aldrich), 2% vol/vol B27 SF supplement (Invitrogen; Thermo Fisher Scientific), and 1% N2 SF supplement (Invitrogen; Thermo Fisher Scientific).
For the clonal analysis experiments, single CD15-positive (CD15+) and CD15-negative (CD15−) cells were seeded into 96-well plates and allowed to grow for 8 weeks, at which point the number of colonies was quantified. All experiments were conducted in triplicate.
For the 12-day clonal analysis time course, single cells were seeded into 96-well plates, but prior to seeding, 96-well plates were coated in 1:10 ECM. This allowed cells to proliferate as a monolayer.
Staining and Imaging
Table 1 summarizes the primary and secondary antibodies used for immunohistochemistry and immunocytochemistry in this study.
Table 1.
Primary antibodies
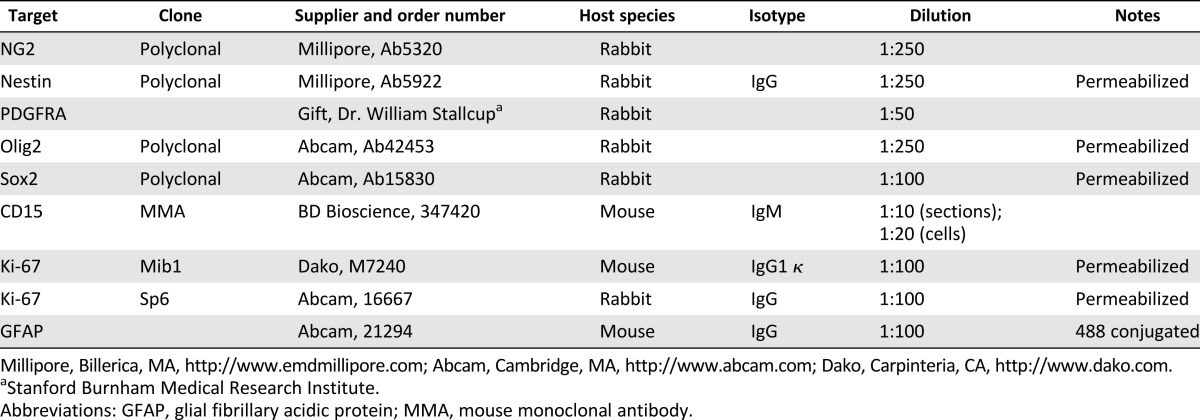
The immunocytochemistry protocol was conducted at room temperature with three phosphate-buffered saline (PBS; pH 7.4) washes between each incubation; samples were gently shaken during incubation. Cells were fixed for 10 minutes with 4% paraformaldehyde (PFA; Sigma-Aldrich) and then washed with PBS and permeabilized with 1% Triton X for 5 minutes (if indicated in Table 1) and incubated with blocking buffer (5% bovine serum albumin [BSA; Sigma-Aldrich] in PBS) for 45 minutes. Block was removed, and cells were then incubated with primary antibodies with 1% BSA for 2 hours. Appropriate secondary antibodies (Alexa Fluor conjugates from Invitrogen Molecular Probes; Thermo Fisher Scientific) were used at a dilution of 1:500 in PBS for 1 hour. Cells were then incubated with a 1:10,000 dilution of Hoechst 33342 (Enzo Life Sciences, Farmingdale, NY, http://www.enzolifesciences.com) for 5 minutes before a final washing step. Samples were stored at 4°C wrapped with parafilm. Before image acquisition, PBS was removed and glass coverslips were placed on cells. When using the combination of NG2 and CD15 antibodies, the antibodies were used separately. With primary and secondary antibodies for NG2 applied first, as described in this paragraph, before a further fixing stage in 4% PFA for 5 minutes, the protocol described above was repeated for CD15.
For the immunohistochemistry protocol, patient tumor sections that had not been fixed were defrosted in 4% PFA for 2 minutes. Patient tumor sections were washed before adding 50 μl/cm2 of blocking solution with 0.25% Triton X for 30 minutes. Blocking solution was removed and replaced with the primary antibody at the concentration indicated in Table 1. This was incubated for 12–16 hours at room temperature before being gently washed. Appropriate secondary antibodies from Invitrogen (Thermo Fisher Scientific) were used at a concentration of 1:500, the glial fibrillary acidic protein (GFAP) antibody was used at this stage. Finally Hoechst 33342 was added at a concentration of 1:10,000 for 5 minutes. Sections were washed before being mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, https://www.vectorlabs.com).
For hematoxylin and eosin (H&E) staining, sections of 10 μm were defrosted and fixed in 4% PFA for 3 minutes. Automated staining was performed by Leica Autostainer XL (Leica Microsystems, Buffalo Grove, IL, http://www.leicabiosystems.com). Slides were then coverslipped on the automated Leica CV 500.
Immunofluorescence stainings were imaged on a Zeiss Axioplan 2 confocal microscope with Zeiss software or a Zeiss fluorescent light microscope using Axio vision software as indicated (Carl Zeiss, Jena, Germany, http://www.zeiss.com). H&E staining was imaged using an Olympus BX41 light microscope (Olympus, Tokyo, Japan, http://www.olympusamerica.com).
Fluorescence-Activated Cell Sorting
Cells were incubated with 20 μl/million cells of FITC-conjugated CD15 antibody (mouse monoclonal antibody clone; BD Bioscience, San Jose, CA, https://www.bdbiosciences.com) [23] for 15 minutes in the dark on ice, with frequent gentle shaking. The cells were then washed twice in Hanks’ balanced saline solution (HBSS). After the final wash, 2 ml of medium was added, and the suspension of cells was immediately taken for fluorescence-activated cell sorting (FACS) on ice, with light excluded.
Flow-cytometric analysis and sorting were carried out on a MoFlo high-speed sorter (Beckman Coulter, Pasadena, CA, https://www.beckmancoulter.com). The MoFlo cell sorter was calibrated using fluorospheres (SPHERO Rainbow Fluorescent Particles; Spherotech Inc., Lake Forest, IL, http://www.spherotech.com) to align laser-shaping optics on a daily basis.
Single-line visible light (488 nm) from an argon laser (Coherent, Santa Clara, CA, http://www.coherent.com) was focused onto the stream with spherical beam-shaping optics. Fluorescence was measured with a 530/30 band-pass filter for analysis of FITC-conjugated CD15 antibody fluorescence in front of an H957 photomultiplier tube. The forward scatter signal was used to trigger acquisition. Signals for forward and side scatter were obtained in linear mode, and the fluorescence signal was obtained in logarithmic mode. “Single” sort mode was used to sort cells into 96-wells plates, and “purify” sort mode was used to sort cells into tubes. Sheath pressure was ∼33–34 Ψ, and a 100-μm nozzle was used. Drop drive frequency and amplitude were calibrated each day to ensure optimal drop formation and drop delay.
Analysis was performed using Summit software (versions 4.2 and 4.3.1; Beckman Coulter).
Sorting was performed at room temperature. Control cells were analyzed initially, and gating was set for this population before labeled cells were analyzed and sorted. Most dead cells and debris were excluded on the basis of forward and side scatter. Elimination of doublets and multiplets was performed using pulse width as a selection criterion to ensure that only single cells were sorted. Finally, the fluorescence gating was adjusted for the auto- and background fluorescence signal produced by unlabeled control populations. Only cells with greater fluorescence than the control population were selected. These cells were deemed to be positive for CD15. The cells with approximately half a log scale over basal values of CD15 where selected for experiments.
Cells were sorted as a bulk population into 1.5-ml Eppendorf tubes or sorted as single cells per well into 96-well plates. Single sort mode was used to sort cells into 96-wells plates, and purify sort mode to sort into tubes. Trigger rate was up to ∼1,000–4,000 cells per second and ∼300–900 cells per second for purify and single sort modes, respectively. Between 50,000 and 300,000 sorted cells were collected from each sample into tubes. For clonal experiments, positive and negative cells were sorted onto plates separately using relevant gating strategy based on FITC-conjugated CD15 fluorescence.
DNA and RNA Purification, Single-Nucleotide Polymorphism Array, and Microarray
DNA and RNA were extracted using the Allprep DNA/RNA kit (Qiagen, Venlo, The Netherlands, https://www.qiagen.com), according to the manufacturer’s instructions. Final concentration and purity were assessed using the NanoDrop system (Thermo Fisher Scientific).
Illumina single-nucleotide polymorphism (SNP; HumanCytoSNP-12; Illumina, San Diego, CA, http://www.illumina.com) and gene expression arrays (WG6) were used to interrogate cytogenetic and expression differences between CD15+ and CD15− lines. Array hybridization and data analysis was performed by Cambridge Genomic Services (Cambridge, U.K., http://www.cgs.path.cam.ac.uk). A paired sample comparison was used to run statistical analysis, as reported previously [37]. Array quality control was assessed with Illumina’s GenomeStudio software. R software (lumi Bioconductor package; R Foundation, Vienna, Austria, http://www.r-project.org) was used for processing and to assess differential expression (lumi limma package). Unsupervised hierarchical clustering of gene expression patterns and principal component analysis was performed with the multiexperiment viewer [38].
Animal Transplantation
All animals were housed and maintained in accordance with the U.K. Animal (Scientific Procedures) Act 1986 and the Cambridge University Commission for Animal Health. Tissue was implanted as single-cell suspensions into subcutaneous tissue of male nude mice aged 6–8 weeks. A suspension of 3 × 105 FACS-sorted CD15+ and CD15− GB cells were prepared in 50 μl of HBSS and injected after being mixed with 100–200 μl of ECM into the dermal layer of the hind limbs. CD15+ cells were implanted in the left flank, and CD15− cells were implanted in the right flank from the same cell line at the same time. Animals were sacrificed at 6 months.
Statistical Analysis
Statistical analysis was conducted using Microsoft Excel (Microsoft, Redmond, WA, http://www.microsoft.com) and Social Science Statistics (http://www.socscistatistics.com/tests/mannwhitney/Default.aspx).
Results
We began by examining the proliferation status of CD15 cells in representative, histologically confirmed clinical samples obtained from 10 patients with GB (typical H&E) (Fig. 1A). Immunohistochemistry for CD15, the astrocytic marker GFAP and the proliferation-associated antigen Ki-67, was performed on cryosections. Because CD15 also labels leukocytes, we costained tumor sections with GFAP, which is expressed in GB and astrocytoma cells but not neutrophils [39–42]. An average of 8,000 nucleated GFAP-positive cells were scored in each tumor. Expression of CD15 in our clinical samples was rare: CD15+ cells represented 0.03%–0.24% of GFAP-positive cells. There was a significant difference between the number of cycling cells in the CD15+ and CD15− populations (p < .05). Only 0.003% of CD15+ GFAP+ cells coexpressed Ki-67, a marker of cycling glioma cells [43, 44] (Fig. 1B, 1C), in contrast to 5.49% of cells that were CD15−, GFAP positive, and Ki-67 positive. The scarcity and relative proliferative quiescence of the CD15+ population within GB suggests that it is cycling CD15− cells that drive tumor growth.
Figure 1.

CD15-positive (CD15+) glial fibrillary acidic protein-positive (GFAP+) cells from patient glioblastoma (GB) tumors are quiescent. (A): Representative hematoxylin and eosin staining of S1 patient tumor. Scale bar = 100 μm. (B): Ki-67 expression in CD15+ and CD15-negative cells in 10 representative GB patient tumors. (C): Single slice confocal images of patient tumor S1. Immunohistochemical detection of Hoechst 3342 (blue), GFAP (green), CD15 (red), and Ki-67 (mauve). Scale bar = 10 μm.
We next set out to examine the fate of cells from early passage (passage <10) cultures from 10 tumors representative of the patient samples analyzed above. The optimal method of culturing GB TICs has provoked controversy between those who culture cells in suspension as spheres and those who favor adherent cultures [45–47]. For these experiments, we used a hybrid protocol in which cells are initially cultured as spheres and then grown as a monolayer [19]. This protocol is optimal for these experiments because the fate of individual cells can be followed in adherent cultures. We validated each cell line as TICs by confirming tumorigenicity in vivo [19, 48]. We also showed, using an SNP array, that the primary cells were cytogenetically similar to both the parent tumor and the experimental xenograft derived from the corresponding cell line in two of our TICs (supplemental online Table 1).
Both CD15+ and CD15− cells were present in all TIC lines investigated. A paired sample comparison of the cytogenetic profile of FACS CD15+ and CD15− cells from two of the TIC lines, using whole-genome SNP arrays, confirmed that CD15+ and CD15− populations had no statistically significant cytogenetic differences (Fig. 2A; supplemental online Tables 2, 3), indicating a common clonal heritage. We compared whole-genome expression levels between CD15+ and CD15− cells from one TIC line and failed to reject the null hypothesis (p > .01 after multiple testing correction), thus no differentially expressed genes could be identified between positive and negative cells (Fig. 2B; supplemental online Fig. 1).
Figure 2.

CD15-positive (CD15+) and CD15-negative (CD15−) cells do not have significantly different cytogenetic or gene expression profiles. Both CD15+ and CD15− cells from the S1 cell line have indistinguishable cytogenetic profile. Single-nucleotide polymorphism array results shown by B allele frequency and log R ratio for two example chromosomes. Chromosome 1 (A) and 13 (B). (C): Hierarchical clustering of genes from CD15+ and CD15− cells from the S1 cell line, demonstrated with gene expression array.
To further examine differences between CD15+ and CD15− populations, we investigated the expression of five markers associated with neural stem or progenitor cells to see if these markers could distinguish between CD15+ and CD15− cells in three TIC lines in vitro. We cultured unsorted cells and used immunocytochemistry of a panel of markers and quantified the number of CD15+ and CD15− cells that coexpressed each marker; sample images from the cell line S1 are displayed in Figure 3A. There were high levels of expression of the neural stem cell markers nestin [49] and Sox2 [50] that did not differ between CD15+ and CD15− cells (Fig. 3B). We next looked at three markers of more committed neural progenitors. The transcription factor Olig2 and the cell surface proteoglycan NG2 are widely expressed in both glial progenitors and glial cancers [18, 51, 52] and PDGFRA, one of the earliest markers expressed by cells committed to the oligodendrocyte lineage [53]. We found these markers were similarly expressed in both CD15+ and CD15− cells (Fig. 3B). We were unable to find a significant difference between CD15+ and CD15− cells on the basis of expression of any of the five markers associated with neural stem or progenitor cells that we investigated. We also evaluated the expression of CD133, a putative cancer stem cell marker, but we found lack of coexpression with CD15 and low level of CD133 expression (supplemental online Fig. 2). Given the controversy about the reliability of CD133, we did not pursue any further experiments using it as a marker.
Figure 3.

CD15+ and CD15− cells from glioblastoma cell lines have similar expression of neural stem cell markers and neural progenitor cell markers. (A): Single-slice confocal images of stem cell markers and neural progenitor cell markers in the cell line S1. (B): The proportion of cells expressing neural stem cell and neural progenitor cell markers in three cell lines.
We next evaluated whether CD15+ and CD15− populations could reconstitute one another. Paired CD15+ and CD15− cell populations from the same TIC line were isolated and cultured to determine whether CD15+ cells could be generated from a marker-negative population and vice versa. Using FACS, paired populations of CD15+/− cells were isolated from three TIC lines. We evaluated temporal changes in CD15 expression. At 10 and 20 days after sorting, we observed that the sorted CD15− population began to express CD15 (Fig. 4A). This pattern of CD15 distribution developed slowly, and the CD15+ fraction remained low (Fig. 4A). In contrast, we observed rapid emergence of CD15− cells in isolated CD15+ populations (Fig. 4A). By 30 days after sorting, both sorted cell populations had achieved an identical heterogeneous distribution of both CD15+ and CD15− cells (Fig. 4A) in all TICs investigated. We concluded that cells interconvert between CD15+ and CD15− states.
Figure 4.

CD15-positive (CD15+) and CD15-negative (CD15−) populations from glioblastoma (GB) sorted by fluorescence-activated cell sorting (FACs) established a phenotypic equilibrium that recapitulated the original GB cell line phenotype. (A): The proportion of CD15+ cells in a population of GB cells over time, the initial populations were sorted by FACS as CD15+ and CD15−. (B): Single CD15+ and CD15− cells were sorted into 96-well plates and allowed to proliferate for at least 6 weeks, after which CD15 expression was analyzed by FACS. The table shows the results of this experiment in 10 tumor-initiating cell lines. (C): Colony-forming ability of single CD15+ and CD15− cells from 10 GB cell lines after an 8-week time course. (D): Average clone size with error bars showing standard deviation of the data set. The variety of clone sizes was wide at each time point. The table shows the results of Mann-Whitney values at each time point. Abbreviations: FACS, fluorescence-activated cell sorting; GB, glioblastoma; W, week.
To determine whether the phenotypic behavior of CD15+/− cells was a function of interaction with the cellular microenvironment, we isolated single CD15+ or CD15− cells from the same TIC population of 10 different lines. We then evaluated their ability to form new colonies and to interconvert between the CD15+ and CD15− states. Single CD15+ and CD15− cells were sorted and grown in 96-well plates for at least 6 weeks before being analyzed for CD15 expression (Fig. 4B). Analysis by flow cytometry demonstrated that both single CD15+ and CD15− cells were able to form CD15+ progeny (Fig. 4B). These data from population studies and single-cell studies indicate that CD15+ and CD15− cells in GB do not display a unidirectional cellular hierarchy.
In parallel, we evaluated the ability of single CD15+ or CD15− cells to form new colonies in 96-well plates from each of our 10 cell lines. There was no significant difference in colony-forming ability, as demonstrated by the formation of spheres, for single CD15+ and CD15− cells in any of the TIC lines (p > .5) (Fig. 4C). This demonstrates that CD15+ cells do not have a colony-forming advantage in vitro when compared with CD15− cells.
We were next interested in a more detailed investigation of single CD15+ and CD15− clones at early time points. We isolated single CD15+ or CD15− cells from the same TIC population and tracked cell proliferation of cells, grown as a monolayer, over a 12-day time course for each specific clone. We studied 192 clones per cell line. We found no significant difference between clone sizes in CD15+ and CD15− cells on days 2, 4, 7, and 12 after sorting in either of the two cell lines investigated (Fig. 4D; supplemental online Fig. 3).
Our observations of clinical samples coupled with the identical pattern of phenotypic behavior of CD15+/− cells under defined experimental conditions in vitro led us to hypothesize that CD15 expression would not distinguish distinct GB cell populations in vivo. To test this hypothesis, we implanted 300,000 CD15+ and CD15− cells subcutaneously into the flanks of nude mice to observe any tumor formation. We used two TIC lines cultured from two unique patient GB samples and tested each cell line in triplicate. Animals were culled after 40 weeks or when tumors reached 15 mm, whichever came first. By 40 weeks, 5 of 6 paired implants had generated tumors. In each case, both CD15+ and CD15− cells generated tumors. We did not observe any cases in which CD15+ GB cells generated a tumor but the paired CD15− cells did not (Fig. 5).
Figure 5.

CD15-positive (CD15+) and CD15-negative (CD15−) populations from glioblastoma sorted by fluorescence-activated cell sorting (FACS) are equally tumorigenic in nude mice. Tumor-initiating cell lines were sorted by FACs for CD15+ and CD15− cells, which were implanted into nude mice. Examples of tumors with corresponding histology and the results of tumor forming assays.
Discussion
The application of the cancer stem cell hierarchical model to GB remains controversial. No cell surface marker has been found that will reliably discriminate a tumor-initiating cell population from an epigenetically stable nontumorigenic population. Studies on cell populations defined by the expression of CD133 and CD44 have not reliably isolated tumor-initiating cells [28, 54–58].
We were unable to replicate previous data suggesting that CD15+ cells were more clonogenic or tumor forming or had higher expression of stem cell-associated markers Sox2 and Olig2 than their CD15− counterparts [23]. We found that paired CD15+/− cells were equally tumorigenic and clonogenic. Possible explanations for these differences in results include the effects of different experimental systems and conditions on colony-forming and tumor-forming assays [12]. We used different mouse model systems and different locations for xenograft compared with previous studies [23]. These previous studies used orthotopic xenogeneic implantation into neonatal SCID mice; in comparison, we used heterotopic xenogenic subcutaneous implantation into nude mice. We chose this model because it allowed us to visualize tumor formation directly, even for small tumors that the resolution of small animal imaging would struggle to detect in orthotopic models. Although heterotopic implants are not exposed to organ-specific tumor-stroma interactions, it is recognized that analysis of any tumor-host interactions using a xenogeneic model of any kind must be interpreted with caution. For these reasons, we have confined our in vivo analysis to comparisons of paired CD15+ and CD15− cell populations exposed to the same environmental conditions in vivo to show that in each pair both CD15+ and CD15− cells were similarly tumorigenic. These data support our findings in patient tumor samples, in which we found that CD15+ cells were not more regularly cycling than their CD15− counterparts. It also supports our in vitro findings that CD15+ clones did not have a survival advantage or early proliferative advantage compared with matched CD15− clones.
The number of CD15 cells in our tissue analyses was low compared with previous reports [23], raising the possibility that these studies included additional cell subpopulations in their CD15+ samples. CD15 is widely expressed on macrophages and neutrophils, on which it mediates phagocytosis and chemotaxis [59], and in cancers including lymphoma, leukemia, gastric cancer, and hepatocellular carcinoma [60, 61]. High-grade glioma displays a heterogeneous parenchymal cell population including peripheral immune cells and microglia [62]. Consequently, we excluded leukocytic cell populations by costaining for GFAP, which is not expressed by leukocytes, to restrict CD15 expression to GB cells in this study, possibly explaining why CD15 cell counts appear lower in our tissue samples.
Although CSCs and TICs have been well established in some cancers including leukemia [4, 5], germ cell tumors [6, 7], and breast [8] and colon carcinomas [9, 10], their existence seems unlikely in other cancers including melanoma [11, 12] and lymphoma [13]. Data identifying the glycoprotein CD133 as a putative CSC marker in glioblastoma [20–22] have subsequently been challenged [23–26], and the case for a cellular hierarchical organization in GBM remains unproven. Our data challenge the use of CD15 as a marker of a hierarchy in GB. We found that single cells, both CD15+ and CD15−, were able to generate mixed CD15+ and CD15− populations in vitro.
Increasingly molecular genetic data indicate that in cancer disease, complexity develops as a result of a branched pattern of evolution [63–66], resulting in previously unrecognized levels of intratumor heterogeneity in which genetically diverse cancer cell clones coexist with associated variegated phenotypes. Neuroepithelial malignancies including medulloblastoma and GB demonstrate similar evolutionary complexity [65–69]. The clonal diversity and rapidly branching evolution of GB [70] represent a challenging environment in which to identify an epigenetically stable common clonal ancestor consistent with the cancer stem cell hypothesis [71].
We were not able to find a difference in the phenotypic or genomic characteristics of CD15+ cells compared with the CD15− population. Sorted CD15+ and CD15− cells were capable of reconstituting a heterogeneous population containing both CD15+ and CD15− cells over time, suggesting that, in vitro, cells were able to interconvert between CD15+ and CD15− states. Our observations that sorted CD15+/− cells gave rise to positive and negative cells in a nonhierarchical manner suggest that the term “cancer stem cell” might represent a dynamic state. Consistent with this concept, clonal heterogeneity does not necessarily depend on cellular hierarchy because genetically identical cells can exhibit phenotypic diversity due to stochastic variability in gene expression and associated signaling pathways [72]. Consequently, expression of CD15 may be transient, reflecting an intrinsic functional state during progression through the cell cycle or a response to microenvironmental selection pressures. This may explain the vanishingly small number of cycling CD15 cells we observed in clinical specimens compared with the relative ease with which CD15-expressing cell lines could be generated in vitro.
Conclusion
We conclude that CD15 does not represent a robust cell-surface marker able to prospectively distinguish a rapidly proliferating, tumorigenic, or stem-like population in GB. We suggest that the several markers, including CD15, that have been reported to identify a cancer stem cell in GB simply reflect clonal and functional heterogeneity within each GB. Indeed, stochastic and hierarchical models have been previously considered alternative mechanisms of cancer progression [73]. An alternative hypothesis is that they are part of the same process because clonal diversity is likely to be generated and sustained by genetically distinct CSCs, which provide the units for evolutionary selection [74, 75]. Evidence in support of this hypothesis exists in leukemia [63, 76] and it has been suggested that GBs similarly harbor genetically diverse tumor cell populations [77]. Such insights are important for understanding treatment failure because it is the resistant disease emerging from a clonally heterogeneous population that ultimately kills patients.
Supplementary Material
Acknowledgments
This project was funded by the Medical Research Council (studentship for E.K.-H.), the Isaac Newton Trust and the Institute of International Education Fellowship (T.A.-M.), the NIHR Cambridge Biomedical Research Centre (C.W.), the Evelyn Trust, and the Royal College of Surgeons of Edinburgh, U.K.
Author Contributions
E.K.-H. and T.A.-M.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; S.G.M.P.: collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; J.F.: collection and/or assembly of data, final approval of manuscript; I.S.: data analysis and interpretation, final approval of manuscript; P.J.: conception and design, financial support, final approval of manuscript; C.W.: conception and design, financial support, provision of study material or patients, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 3.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 5.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 6.Kleinsmith LJ, Pierce GB., Jr Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964;24:1544–1551. [PubMed] [Google Scholar]
- 7.Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci USA. 1976;73:549–553. doi: 10.1073/pnas.73.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 10.O'Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 11.Quintana E, Shackleton M, Foster HR, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510–523. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quintana E, Shackleton M, Sabel MS, et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly PN, Dakic A, Adams JM, et al. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 14.Heywood RM, Marcus HJ, Ryan DJ, et al. A review of the role of stem cells in the development and treatment of glioma. Acta Neurochir (Wien) 2012;154:951–969; discussion 969. doi: 10.1007/s00701-012-1338-9. [DOI] [PubMed] [Google Scholar]
- 15.Piccirillo SG, Binda E, Fiocco R, et al. Brain cancer stem cells. J Mol Med (Berl) 2009;87:1087–1095. doi: 10.1007/s00109-009-0535-3. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, McKay RM, Parada LF. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell. 2012;149:36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piccirillo SG, Dietz S, Madhu B, et al. Fluorescence-guided surgical sampling of glioblastoma identifies phenotypically distinct tumour-initiating cell populations in the tumour mass and margin. Br J Cancer. 2012;107:462–468. doi: 10.1038/bjc.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Mayhani MT, Grenfell R, Narita M, et al. NG2 expression in glioblastoma identifies an actively proliferating population with an aggressive molecular signature. Neuro Oncol. 2011;13:830–845. doi: 10.1093/neuonc/nor088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fael Al-Mayhani TM, Ball SL, Zhao JW, et al. An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J Neurosci Methods. 2009;176:192–199. doi: 10.1016/j.jneumeth.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 20.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 21.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 22.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 23.Son MJ, Woolard K, Nam DH, et al. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly JJ, Stechishin O, Chojnacki A, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27:1722–1733. doi: 10.1002/stem.98. [DOI] [PubMed] [Google Scholar]
- 25.Joo KM, Kim SY, Jin X, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808–815. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 26.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 27.Andolfo I, Liguori L, De Antonellis P, et al. The micro-RNA 199b-5p regulatory circuit involves Hes1, CD15, and epigenetic modifications in medulloblastoma. Neuro Oncol. 2012;14:596–612. doi: 10.1093/neuonc/nos002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patru C, Romao L, Varlet P, et al. CD133, CD15/SSEA-1, CD34 or side populations do not resume tumor-initiating properties of long-term cultured cancer stem cells from human malignant glio-neuronal tumors. BMC Cancer. 2010;10:66. doi: 10.1186/1471-2407-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward RJ, Lee L, Graham K, et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 2009;69:4682–4690. doi: 10.1158/0008-5472.CAN-09-0342. [DOI] [PubMed] [Google Scholar]
- 30.Mao XG, Zhang X, Xue XY, et al. Brain tumor stem-like cells identified by neural stem cell marker CD15. Transl Oncol. 2009;2:247–257. doi: 10.1593/tlo.09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fox N, Damjanov I, Martinez-Hernandez A, et al. Immunohistochemical localization of the early embryonic antigen (SSEA-1) in postimplantation mouse embryos and fetal and adult tissues. Dev Biol. 1981;83:391–398. doi: 10.1016/0012-1606(81)90487-5. [DOI] [PubMed] [Google Scholar]
- 32.Solter D, Knowles BB. Monoclonal antibody defining a stage-specific mouse embryonic antigen (SSEA-1) Proc Natl Acad Sci USA. 1978;75:5565–5569. doi: 10.1073/pnas.75.11.5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomperts M, Garcia-Castro M, Wylie C, et al. Interactions between primordial germ cells play a role in their migration in mouse embryos. Development. 1994;120:135–141. doi: 10.1242/dev.120.1.135. [DOI] [PubMed] [Google Scholar]
- 34.Capela A, Temple S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron. 2002;35:865–875. doi: 10.1016/s0896-6273(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 35.Capela A, Temple S. LeX is expressed by principle progenitor cells in the embryonic nervous system, is secreted into their environment and binds Wnt-1. Dev Biol. 2006;291:300–313. doi: 10.1016/j.ydbio.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 36.Guilfoyle MR, Weerakkody RA, Oswal A, et al. Implementation of neuro-oncology service reconfiguration in accordance with NICE guidance provides enhanced clinical care for patients with glioblastoma multiforme. Br J Cancer. 2011;104:1810–1815. doi: 10.1038/bjc.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du P, Kibbe WA, Lin SM. lumi: A pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 38.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 39.Hirato J, Nakazato Y, Ogawa A. Expression of non-glial intermediate filament proteins in gliomas. Clin Neuropathol. 1994;13:1–11. [PubMed] [Google Scholar]
- 40.Rushing EJ, Sandberg GD, Horkayne-Szakaly I. High-grade astrocytomas show increased Nestin and Wilms's tumor gene (WT1) protein expression. Int J Surg Pathol. 2010;18:255–259. doi: 10.1177/1066896909338596. [DOI] [PubMed] [Google Scholar]
- 41.Lolait SJ, Harmer JH, Auteri G, et al. Expression of glial fibrillary acidic protein, actin, fibronectin and factor VIII antigen in human astrocytomas. Pathology. 1983;15:373–378. doi: 10.3109/00313028309085162. [DOI] [PubMed] [Google Scholar]
- 42.McComb RD, Burger PC. Pathologic analysis of primary brain tumors. Neurol Clin. 1985;3:711–728. [PubMed] [Google Scholar]
- 43.McKeever PE, Ross DA, Strawderman MS, et al. A comparison of the predictive power for survival in gliomas provided by MIB-1, bromodeoxyuridine and proliferating cell nuclear antigen with histopathologic and clinical parameters. J Neuropathol Exp Neurol. 1997;56:798–805. [PubMed] [Google Scholar]
- 44.Onda K, Davis RL, Shibuya M, et al. Correlation between the bromodeoxyuridine labeling index and the MIB-1 and Ki-67 proliferating cell indices in cerebral gliomas. Cancer. 1994;74:1921–1926. doi: 10.1002/1097-0142(19941001)74:7<1921::aid-cncr2820740716>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 45.Woolard K, Fine HA. Glioma stem cells: Better flat than round. Cell Stem Cell. 2009;4:466–467. doi: 10.1016/j.stem.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 46.Reynolds BA, Vescovi AL. Brain cancer stem cells: Think twice before going flat. Cell Stem Cell. 2009;5:466–467; author reply 468–469. doi: 10.1016/j.stem.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 47.Pollard SM, Yoshikawa K, Clarke ID, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 48.Kenney-Herbert EM, Ball SL, Al-Mayhani TM, et al. Glioblastoma cell lines derived under serum-free conditions can be used as an in vitro model system to evaluate therapeutic response. Cancer Lett. 2011;305:50–57. doi: 10.1016/j.canlet.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 49.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 50.Komitova M, Eriksson PS. Sox-2 is expressed by neural progenitors and astroglia in the adult rat brain. Neurosci Lett. 2004;369:24–27. doi: 10.1016/j.neulet.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 51.Rowitch DH, Lu QR, Kessaris N, et al. An ‘oligarchy’ rules neural development. Trends Neurosci. 2002;25:417–422. doi: 10.1016/s0166-2236(02)02201-4. [DOI] [PubMed] [Google Scholar]
- 52.Nishiyama A, Chang A, Trapp BD. NG2+ glial cells: A novel glial cell population in the adult brain. J Neuropathol Exp Neurol. 1999;58:1113–1124. doi: 10.1097/00005072-199911000-00001. [DOI] [PubMed] [Google Scholar]
- 53.Hack MA, Saghatelyan A, de Chevigny A, et al. Neuronal fate determinants of adult olfactory bulb neurogenesis. Nat Neurosci. 2005;8:865–872. doi: 10.1038/nn1479. [DOI] [PubMed] [Google Scholar]
- 54.Prestegarden L, Svendsen A, Wang J, et al. Glioma cell populations grouped by different cell type markers drive brain tumor growth. Cancer Res. 2010;70:4274–4279. doi: 10.1158/0008-5472.CAN-09-3904. [DOI] [PubMed] [Google Scholar]
- 55.Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12:49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 57.Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, et al. TGF-β receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 58.Merzak A, Koocheckpour S, Pilkington GJ. CD44 mediates human glioma cell adhesion and invasion in vitro. Cancer Res. 1994;54:3988–3992. [PubMed] [Google Scholar]
- 59.Kerr MA, Stocks SC. The role of CD15-(Le(X))-related carbohydrates in neutrophil adhesion. Histochem J. 1992;24:811–826. doi: 10.1007/BF01046353. [DOI] [PubMed] [Google Scholar]
- 60.Shen M, Hu P, Donskov F, et al. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS One. 2014;9:e98259. doi: 10.1371/journal.pone.0098259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hammer RD, Collins RD, Ebrahimi S, et al. Rapid immunocytochemical analysis of acute leukemias. Am J Clin Pathol. 1992;97:876–884. doi: 10.1093/ajcp/97.6.876. [DOI] [PubMed] [Google Scholar]
- 62.Charles NA, Holland EC, Gilbertson R, et al. The brain tumor microenvironment. Glia. 2011;59:1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 63.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 64.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sottoriva A, Spiteri I, Piccirillo SG, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Piccirillo SG, Spiteri I, Sottoriva A, et al. Contributions to drug resistance in glioblastoma derived from malignant cells in the sub-ependymal zone. Cancer Res. 2015;75:194–202. doi: 10.1158/0008-5472.CAN-13-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu X, Northcott PA, Dubuc A, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482:529–533. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Northcott PA, Shih DJ, Peacock J, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 70.Piccirillo SG, Colman S, Potter NE, et al. Genetic and functional diversity of propagating cells in glioblastoma. Stem Cell Reports. 2015;4:7–15. doi: 10.1016/j.stemcr.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Altschuler SJ, Wu LF. Cellular heterogeneity: Do differences make a difference? Cell. 2010;141:559–563. doi: 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shackleton M, Quintana E, Fearon ER, et al. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 74.Greaves M. Cancer stem cells as ‘units of selection’. Evol Appl. 2013;6:102–108. doi: 10.1111/eva.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 76.Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469:362–367. doi: 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- 77.Piccirillo SG, Combi R, Cajola L, et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene. 2009;28:1807–1811. doi: 10.1038/onc.2009.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.