

Using a time-lapse imaging system, a method to determine the growth of human pluripoten stem cells (hPSCs) from nonlabeled, live-cell, phase-contrast images was developed. When the culture conditions became improper, the change in cell growth conditions was detected by analyzing phase-contrast images, providing real-time information on colony growth and cell growth rates without cell damage. This could be useful for basic research on hPSCs and cell processing for hPSC-based therapy.
Keywords: Human pluripotent stem cells, Stem cell culture, Cell quality control, Cell growth, Cell tracking, Time-lapse imaging
Abstract
Cell growth is an important criterion for determining healthy cell conditions. When somatic cells or cancer cells are dissociated into single cells for passaging, the cell numbers can be counted at each passage, providing information on cell growth as an indicator of the health conditions of these cells. In the case of human pluripotent stem cells (hPSCs), because the cells are usually dissociated into cell clumps of ∼50–100 cells for passaging, cell counting is time-consuming. In the present study, using a time-lapse imaging system, we developed a method to determine the growth of hPSCs from nonlabeled live cell phase-contrast images without damaging these cells. Next, the hPSC colony areas and number of nuclei were determined and used to derive equations to calculate the cell number in hPSC colonies, which were assessed on time-lapse images acquired using a culture observation system. The relationships between the colony areas and nuclei numbers were linear, although the equation coefficients were dependent on the cell line used, colony size, colony morphology, and culture conditions. When the culture conditions became improper, the change in cell growth conditions could be detected by analysis of the phase-contrast images. This method provided real-time information on colony growth and cell growth rates without using treatments that can damage cells and could be useful for basic research on hPSCs and cell processing for hPSC-based therapy.
Significance
This is the first study to use a noninvasive method using images to systemically determine the growth of human pluripotent stem cells (hPSCs) without damaging or wasting cells. This method would be useful for quality control during cell culture of clinical hPSCs.
Introduction
Human embryonic stem cells (hESCs) [1] and human induced pluripotent stem cells (hiPSCs) [2] have unique capabilities to proliferate virtually infinitely and to differentiate into almost any cell type. These human pluripotent stem cells (hPSCs) could be a promising new tool for both cell-based regenerative medicine and pharmaceutical research, including drug efficacy and toxicity screening tests. However, it remains challenging to maintain hPSCs in an undifferentiated state because of their differentiation potential and/or genetic instability [3]. Thus, it has been recommended that cultured hPSCs should be routinely characterized [4] to determine whether their phenotype is maintained. Cell growth is one of the important criteria for determining healthy cell conditions.
When somatic cells or cancer cells are passaged, the cells are dissociated into single cells. Therefore, the cell numbers can be counted at each passage, and researchers can monitor cell growth as an indicator of the health conditions of these cells. In the case of hPSCs, the cells are usually dissociated into cell clumps of ∼50–100 cells for passaging, because dissociation into single cells induces apoptotic cell death or triggers their spontaneous differentiation. Next, the cells are dissociated into single cells separately from passaged cells, and the cell numbers are counted. However, it is labor-intensive, time-consuming, and wasteful of the available cells. A Rho-associated coiled-coil forming kinase inhibitor (ROCK inhibitor; Y27632) can be used to rescue hPSCs from apoptotic cell death [5], although its continuous use is currently less common.
Our aim was to monitor the cell growth conditions using imaging analysis without any invasive methods. In the present study, we developed a noninvasive method to determine the cell growth of hiPSCs by analyzing images of live, nonlabeled hiPSCs in culture. We determined the areas of single hiPSC colonies and the number of fluorescently stained nuclei in these colonies to derive equations to assess these variables’ relationships. This analysis showed that these relationships were linear, although the equation coefficients were dependent on the cell lines used, colony morphology (size, either <1 mm2 or >1 mm2; cell morphology, either typical ES-like cells or comparatively flatter cells), and culture conditions. The cell growth rates calculated using these equations were comparable to those when counting the cell numbers using a hemocytometer. This method provided real-time information on the growth of the colonies and cells without damaging or wasting the cells and could be useful for both basic research using hPSCs or hiPSCs derived from patients with rare diseases and cell processing for hPSC-based therapy.
Materials and Methods
Ethics Statement
All experiments using human iPSCs were conducted with the approval of the institutional review board of the National Institute of Biomedical Innovation.
Cell Culture
A Japanese adult human skin fibroblast (JCRB0534: TIG-114)-derived iPSC line [6], iPS-TIG114-4f1 (JCRB1437), which was established by Yamanaka’s group (Center for iPS Cell Research and Application, Kyoto University) [3, 7], and a human lung fibroblast cell MRC-5-derived iPSC line, Tic [8, 9] (JCRB 1331), were obtained from the JCRB Cell Bank (National Institutes of Biomedical Innovation, Health and Nutrition, Osaka, Japan, http://cellbank.nibio.go.jp/english/). These hiPSCs were first expanded to prepare stocks after 5 passages, after which the thawed cells from these stock were maintained for less than 5 months on irradiated-inactivated CF-1 mouse embryo fibroblast (MEF) feeder cells (EMD Millipore, Billerica, MA, http://www.emdmillipore.com) in a human ESC expansion medium (KSR-based medium). The medium consisted of Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium (Life Technologies, Carlsbad, CA, http://www.lifetechnologies.com) supplemented with 20% KnockOut Serum Replacement (KSR; Life Technologies Ltd., Paisley, U.K.), 0.1 mM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com), 2 mM l-glutamine (Gibco, Grand Island, NY, http://www.invitrogen.com), 0.1 mM nonessential amino acids (Gibco), and human recombinant fibroblast growth factor 2 (FGF-2) (Katayama Kagaku Kogyo Ltd., Osaka, Japan, http://www.katayamakagaku.co.jp/en/; iPS-TIG114-4f1, 4 ng/ml; Tic, 10 ng/ml), as previously described [10–12]. The cells were passaged with 1 mg/ml dispase (Roche Applied Science, Penzberg, Upper Bavaria, Germany, https://www.roche-applied-science.com) and split at a 1:5–1:8 ratio every 6 days. For the feeder-free cultures, the hiPSCs were cultured in TeSR-E8 [13] (Stemcell Technologies, Vancouver, BC, Canada, http://www.stemcell.com) on basement membrane extracellular matrix (Matrigel, Corning Life Sciences, Tewksbury, MA, http://www.corning.com) and mechanically passaged with a disposable stem cell passaging tool (EZPassage, Life Technologies). These cells were split at a 1:5–1:8 ratio every 5 days.
To detect the changes in cell growth, the cells were cultured in an improper medium, E5 medium. E5 medium, in which insulin, FGF-2, and transforming growth factor-β (TGF-β) were excluded from TeSR-E8, consisted of DMEM/F12 medium supplemented with 64 mg/ml l-ascorbic acid-2-phosphate magnesium, 14 μg/ml sodium selenium, 543 mg/ml sodium hydrogen carbonate, and 10.7 mg/ml apo-transferrin (Sigma-Aldrich for all). To repress cell proliferation, the cells were treated with an inhibitor of phosphatidylinositol 3 (PI3) kinase of LY294002 at a concentration of 20 μM (Cell Signaling Technology, Inc., Danvers, MA, http://www.cellsignal.com) in the TeSR-E8 culture medium from 3 days after seeding.
Live Cell Imaging
The cells were seeded into 3 wells of a 6-well plate to acquire the results cost-effectively and cultured in a culture observation system (BioStation CT, Nikon Corp., Tokyo, Japan, http://www.nikon.com) at 37°C in a fully humidified atmosphere of 5% carbon dioxide in air and monitored using time-lapse live imaging. Phase-contrast images were automatically acquired every 12 hours at a magnification of ×4 and a resolution of 2 μm per pixel. To detect nuclei in live cells, the cell nuclei were stained with a cell-permeable SYTO 24 green fluorescent dye (0.5 µM; Life Technologies), which exhibits bright green fluorescence on binding to DNA rather than RNA. The excitation and emission of SYTO 24 is 490 and 515, which can be detected by BioStation CT, which was used in our study. Fluorescent images of the phase-contrast images and stained nuclei were acquired with the culture observation system and analyzed using imaging analysis software, CL-Quant, version 3.1 (Nikon Corp.), using a program we devised. This program allowed the detection of individual colonies, measuring the areas of hiPSC colonies, and determining the number of nuclei in the detected hiPSC colonies on the phase-contrast and fluorescent images.
Conventional Cell Counting
Immediately after acquiring green fluorescent images, the cells were dissociated into single cells using 0.025% trypsin and 0.01% EDTA in phosphate-buffered saline (Life Technologies). Cell numbers were counted using a disposable hemocytometer, OneCell Counter (BioMedical Science, Ltd., Tokyo, Japan http://www.bmsci.com/bmscure.php) under a microscope (Nikon Corp.).
Image Analysis Procedures
Cell images for more than 70 colonies were analyzed using the CL-Quant software (Nikon Corp.). The algorithm used to determine the growth of hiPSCs in culture is shown in Figure 1. The scatter graphs were prepared from the colony areas and nuclei numbers, and analysis was performed using the statistical software GraphPad Prism, version 6.0, for Windows (GraphPad Software, San Diego, CA, http://www.graphpad.com) to determine the regression line using a tool of linear fitting. From the regression line, the equation coefficients for the relationship between the colony areas and nuclei numbers were determined.
Figure 1.

Algorithm for determining cell growth using a culture observation system. This algorithm was developed for the noninvasive method to determine the growth of hiPS cells in culture. Abbreviations: hiPS, human induced pluripotent stem; iPS, induced pluripotent stem.
Immunocytochemical Staining
Immunocytochemical staining was performed as previously described [11, 14]. Anti-Oct-3/4 antibody (N-19, sc-8628; Santa Cruz Biotechnology, Santa Cruz, CA, http://www.scbt.com), anti-vimentin antibody (V6630; Sigma-Aldrich), Alexa Fluor 647 conjugated chicken anti-goat IgG (H+L) secondary antibody, and Alexa Fluor 594 conjugated chicken anti-mouse IgG (H+L) secondary antibody (Life Technologies) were used for immunostaining. Nuclei were stained with Hoechst 33342 (Life Technologies). The fluorescent and phase contrast images were obtained using BioStation CT (Nikon Corp.) and were analyzed using CL-Quant, version 3.1 (Nikon Corp.) to evaluate the fluorescent intensity per pixel of the colony areas (pixels).
Statistical Analysis
All experiments were biologically performed at a minimum of three replicates. The results for triplicate determinations are shown in the figures and tables.
Results
Image Analysis Procedures
The algorithm for developing the noninvasive method to determine the growth of hiPSCs in culture is shown in Figure 1. The cells were stained with a fluorescent nuclei staining solution, and fluorescent images of the cells acquired using the culture observation system were analyzed with image analysis software, CL-Quant, version 3.1, to count the number of nuclei. The hiPSC colony coverage areas extracted from the fluorescent images using the colony extraction program of CL-Quant were overlaid on the fluorescent images to remove the nuclei of feeder cells or scattered differentiated cells from these images. The hiPSC colony coverage areas extracted from the phase-contrast images using the colony extraction recipe of CL-Quant were grouped according to cell morphology and colony size. Next, equations were derived using GraphPad Prism, version 6.0, for Windows (GraphPad Software) to calculate the cell numbers from the colony coverage areas according to the relationships between the colony sizes and the number of nuclei counted. The cell numbers were calculated using these equations. From these cell numbers, the cell growth rates were then calculated using the following formula:
where T is the incubation time, N1 is the cell numbers at the beginning of the incubation time, and N2 is the cell numbers at the end of the incubation time.
hiPSC Colony Areas During Culture
To analyze each hiPSC colony during culture, time-lapse phase-contrast images of live hiPSCs grown in a 6-well plate were acquired using the culture observation system every 12 hours. Two hiPSC lines were used under two culture conditions: an hiPSC line derived from the human lung fibroblast MRC-5 cell line, Tic cells (JCRB1331) [14–16], grown on inactivated MEFs in KSR-based medium (Tic on-feeder); Tic cells grown on Matrigel in TeSR-E8 (Tic feeder-free); another hiPSC line derived from Japanese skin fibroblast TIG114 cells (JCRB0534), derived iPS-TIG114-4f1 cells (JCRB1437) [3, 7] grown on MEF in KSR-based medium (iPS-TIG114-4f1 on-feeder); and iPS-TIG114-4f1 cells grown on Matrigel in TeSR-E8 (iPS-TIG114-4f1 feeder-free). An analysis of the scanned images using the hiPSC colony detection program of the imaging analysis software CL-Quant, version 3.1, revealed a diversity in the colony areas, in the range of ∼104–107 μm2 (Fig. 2A, 2B; supplemental online Fig. 1A, 1B). For Tic feeder-free cells (Fig. 2A, 2C) and Tic on-feeder cells (supplemental online Fig. 1A, 1C), the detected colonies became larger during a culture period of 120 hours. In contrast, for the iPS-T114-4f1 feeder-free (Fig. 2B, 2D) and iPS-TIG114-4f1 on-feeder (supplemental online Fig. 1B, 1D) cells, small colonies (<25 μm2), which were not detected at 48 hours, began to be noted every 12 hours and resulted in an increase in the total colony numbers, although some of the large colonies merged into larger colonies. These results indicated that colony growth was dependent on the cell line used.
Figure 2.

Colony areas and numbers during culture. Human induced pluripotent stem cell (hiPSC) lines or Tic or iPS-TIG114-4f1 cells were seeded using feeder-free culture conditions in 6-well-plates and cultured for 5 days. After the cell clumps had settled on the plate surfaces (48 hours), phase-contrast images of these cells were acquired every 12 hours and analyzed using the culture observation system and software. Cell clumps areas of >25 μm2 were recognized as undifferentiated hiPSC colonies for analysis. (A): Colony areas for Tic feeder-free cell culture. (B): Total colony numbers for Tic feeder-free cell culture. (C): Colony areas for iPS-TIG114-4f1 feeder-free cell culture. (D): Total colony numbers for iPS-TIG114-4f1 feeder-free cell culture. The plots are representative of three wells from independent experiments. Abbreviation: iPS, induced pluripotent stem.
Relationships Between hiPSC Colony Areas and Single hiPSC Areas
In general, within a few days after passage, the morphologies of the hESCs/hiPSCs were comparatively flatter and larger than those of typical undifferentiated hESCs/hiPSCs, and spaces between the cells were detected in small colonies, as previously described [17]. For example, phase-contrast images of iPS-TIG114-4f1 on-feeder cells showed that a small colony consisted of flatter cells at 84 hours after passage. In contrast, a larger colony had tightly packed, round cells without spaces in between the cells and consisted of cells with large nuclei and notable nucleoli at 108 hours after passage (Fig. 3A). From these images, we assumed that the cell areas in the hiPSC colonies might have decreased during culture and that the number of cells in these colonies might depend on how large these colony areas were. Thus, the total areas of hiPSC colonies might not be directly proportional to the numbers of hiPSCs in these colonies.
Figure 3.

Cell areas in colonies with different morphology types. Phase-contrast images of iPS-TIG114-4f1 cells cultured on feeder cells were acquired for a period of 120 hours. Different colony morphology types appeared during this culture period. (A): Two representative images of small colonies consisting of flatter cells, which appeared at 84 hours or 96 hours after passage, and two representative images of larger colonies consisting of tightly packed, round cells without spaces in between, which appeared at 96 hours or 108 hours after passage. (B): Cells were stained using an anti-E-cadherin antibody. The cell sizes of the representative cells (outlined in red) of representative large and small colonies were analyzed using image analysis software. The results of the analyzed cell sizes are shown in supplemental online Table 1.
An analysis of representative colonies using staining with an anti-E-cadherin antibody using the image analysis software showed that the areas of single cells were from 75 µm2 to ∼1,500 µm2 (Fig. 3B; supplemental online Table 1). The averages of the single cell areas in a larger colony were from 136 μm2 (area A) to 390 μm2 (area B). The averages of the single cell areas in a smaller colony were from ∼491 μm2 (area C) to 840 μm2 (area D). From these results, we hypothesized that if the relationship between the colony areas and cell numbers was determined, we could calculate the cell numbers from live cell colony images.
Counts for Fluorescently Stained Nuclei
To determine the cell numbers in colonies, we created a program for the image analysis software to detect intact nuclei and disregard fragmented nuclei in live hiPSCs stained with the fluorescent dye SYTO 24, following the instructions of fluorescent image analysis of the imaging analysis software. We confirmed that SYTO 24 at 0.5 μM (a concentration lower than that given in the manufacturer’s instruction) could be used to stain live hiPSCs and that the stained cells could survive at least 4 hours, although the cells tended to die after 12 hours (supplemental online Fig. 2). Thus, both the fluorescently stained nuclei and colony areas consisting of packed nuclei were detected individually and then overlaid to count the number of fluorescently stained nuclei only in the extracted colony areas (supplemental online Fig. 3A). For this purpose, each colony area was extracted from a fluorescent image. For comparisons of the number of counted fluorescent nuclei, after acquiring the fluorescent images, the cells were immediately dissociated by trypsin/EDTA for cell counts using the hemocytometer 4 hours after the cells were stained with SYTO 24.
The number of fluorescent nuclei counted using the culture observation system software was comparable to the number counted using the hemocytometer when the numbers were greater than the hemocytometer measurable limit: 104 cells (r = .9359; supplemental online Fig. 3B). We also compared the accuracy of colony area detection between the fluorescent images and phase-contrast images. A strong correlation was found between the colony areas detected by the fluorescent images and those detected by the phase-contrast images (r = .9877; supplemental online Fig. 3C). From these results, for the following experiments, the number of nuclei was considered to indicate the cell number, and we used the phase-contrast images to detect the colony area.
Relationships Between hPSC Colony Areas and Cell Numbers
To determine the relationships between the hiPSC colony areas and cell numbers, phase-contrast and fluorescent images of Tic cells and iPS-TIG114-4f1 cells in a 6-well plate stained with SYTO 24 were acquired using the culture observation system every 12 hours. Next, the cell numbers were plotted against the colony areas to generate equations to determine the relationship between these variables. When the coefficients in these equations were set to constant values, the errors for the calculated numbers compared with the nuclei numbers were ∼50% (data not shown). Thus, we considered that the single cell size was changed during culture.
Phase-contrast images showed that there were two types of colonies. One type consisted of comparatively flatter cells and was designated the type A colony. The second type consisted of small compact cells and was designated the type B colony (supplemental online Fig. 4). Next, the detected colonies in Tic feeder-free cell culture were divided into these two types (supplemental online Table 2) and used in plots against the cell numbers. These plots showed that the relationships between the colony areas and cell numbers were linear, although the equation coefficients were different between the type A (Fig. 4A) and type B colonies (Fig. 4B) for the smaller colonies (<1 mm2). No type A colonies found in the larger colonies (>1 mm2). The equation coefficients for the relationships between the areas and cell numbers with the larger colonies were greater than those for the smaller colonies (Fig. 4C). The numbers determined from these equations were compared with those counted from the fluorescent images, which showed that the error ranges were from −8.9% to +25.0% for the larger colonies; for the smaller colonies, the error ranges were comparatively greater (from −57.5% to +23.6%; Fig. 4D).
Figure 4.

Relationships between colony areas and cell numbers. Graphs show cell numbers (y-axes) versus colony areas (x-axes) for Tic feeder-free (A–D): and iPS-TIG114-4f1 (E–H) feeder-free cells. For type A colonies, the equations derived for the cell numbers versus the colony areas were y = 2,461x for Tic cells and y = 2,328x for iPS-TIG114-4f1 cells. For type B colonies of <1 mm2, these equations were y = 2,538x for Tic cells and y = 3,597x for iPS-TIG114-4f1 cells. For type B colonies of >1 mm2, these equations were y = 3,630x − 1,092 for Tic cells and y = 6,458x − 2,501 for iPS-TIG114-4f1 cells. (D, H): The errors for the cell numbers are shown. Open circles indicate type A colonies; open triangles, type B colonies of <1 mm2; and open squares, type B colonies of >1 mm2. The results in each graph are for three independent experiments. Abbreviation: iPS, induced pluripotent stem.
For iPS-TIG114-4f1 feeder-free cells, the relationship between colony area and cell number in the type A colonies in the smaller colonies was similar to that of the Tic feeder-free cells. In contrast, the equation coefficients for the relationships between the colony areas and cell numbers in the type B colonies for both smaller and larger colonies were greater than those for the Tic feeder-free cells (Fig. 4E–4G). The errors for the cell numbers from these equations were comparatively larger with the smaller colonies (from −57.5% to +23.6%), but the errors were comparably smaller (from −8.0% to +25.0%) with the larger colonies (Fig. 4H). For Tic on-feeder cells, the relationships between colony areas and cell numbers were linear for all types of colonies, although the equation coefficients were different depending on the colony type (supplemental online Fig. 5A–5C). The errors for the cell numbers were comparatively greater than those for the Tic feeder-free cells (supplemental online Fig. 5D). Because the images revealed that iPS-TIG114-4f1 on-feeder underwent spontaneous differentiation, we eliminated the results of the iPS-TIG114-4f1 cells on-feeder from the cell growth analysis. The equation coefficients were different depending on the cell line used and the culture conditions. The average errors were <10% with Tic feeder-free and on-feeder, although those for iPS-TIG114-4f1 feeder-free cells were comparatively greater (15%) (supplemental online Table 3).
hiPSC Doubling Time Calculated According to Colony Areas
Using the equations that we derived, the doubling times of the cells during the culture period were determined from the time-lapse phase-contrast images of the two hiPSC lines (Table 1). For the feeder-free culture conditions, the growth rates determined using the equations were not much different from those determined from the hemocytometer counts. For the feeder culture conditions, the cell growth rates during 48–96 hours were not much different from those determined from the hemocytometer counts. These results showed that it was possible to calculate the cell growth from the time-lapse images of the hPSC cultures without damaging the cells.
Table 1.
Cell doubling times
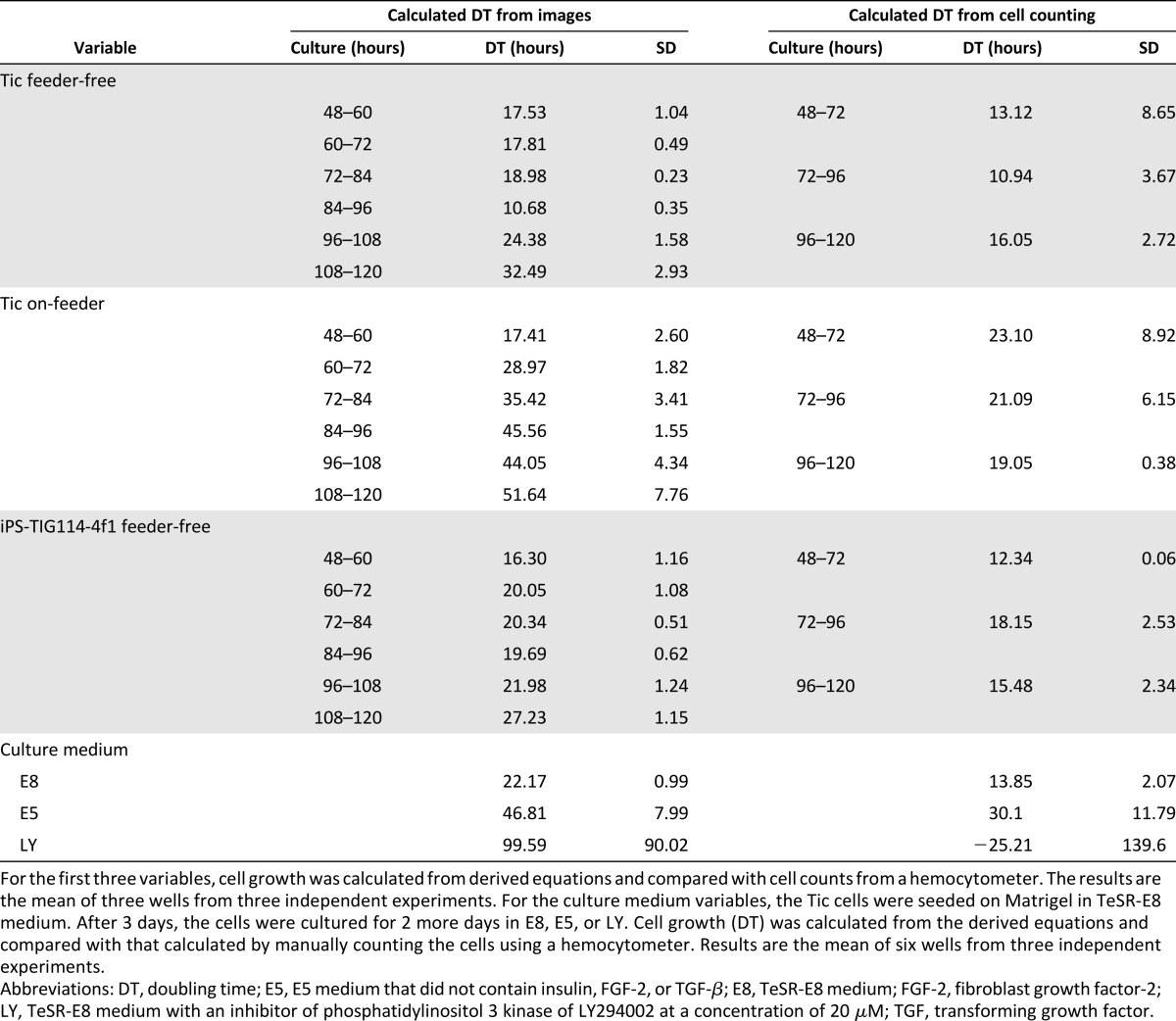
When the cells were counted using the hemocytometer, feeder cells were not counted. However, differentiated cells could not be removed from the dissociated cell suspensions. In contrast, differentiated cells could be removed from the images to estimate cell growth.
Detection of Changes in Growth by the Imaging System
To examine whether our imaging system could detect changes in cell growth, the Tic cells were seeded on Matrigel in TeSR-E8 medium. After 3 days, the cells were cultured for 2 more days in E5 medium, which did not contain insulin, FGF-2, TGF-β, or TeSR-E8 medium with an inhibitor of PI3 kinase of LY294002 at a concentration of 20 μM. After acquiring the phase-contrast images, the cells were dissociated to count the cells or were stained with anti-Oct-4 or anti-vimentin antibodies. Image analysis revealed that when the cells were cultured in TeSR-E8 medium, type B colonies increased from day 4 to day 5 (Fig. 5A, 5B, 5G; supplemental online Table 4). Because the cells did not grow sufficiently in E5 medium or with LY294002 to count the cell numbers when the cells were seeded at the usual cell density, the cells were seeded at a higher density. Many colonies in TeSR-E8 medium overgrew at day 5 (Fig. 5B). In contrast, when the cells were cultured in E5 medium, or in TeSR-E8 medium with LY294002, the type B colonies rarely increased (Fig. 5C, 5D–5F, 5J, 5M; supplemental online Table 4). Using the equations we derived, the cell numbers (Fig. 5H, 5K, 5N; supplemental online Table 5) and doubling time (Table 1) were calculated and compared with the numbers of live and dead cells counted using the hemocytometer (Fig. 5I, 5L, 5O). The calculated cell numbers of the cells cultured in TeSR-E8 medium at day 4 were comparable with the cell numbers counted using the hemocytometer. However, the calculated cell numbers at day 5 were approximately one half of those counted using the hemocytometer. Furthermore, the calculated cell numbers of the cells cultured in E5 medium were comparable to those counted using the hemocytometer. The calculated cell numbers of the cells cultured in TeSR-E8 medium with LY294002 were twice that of the cells counted by the hemocytometer. These results might be caused by the possibility that the hemocytometer creates errors with the smaller cell numbers and the imaging analysis system creates errors with the larger cell numbers. However, the calculation of the doubling time demonstrated that both methods could detect the change in cell growth, although the calculated doubling time from the image analysis was slower than that calculated from the counting using the hemocytometer (Table 1).
Figure 5.

Detection of change in cell growth by the imaging system. Tic cells were seeded on Matrigel in TeSR-E8 medium. After 3 days, the cells were cultured for more 2 days in TeSR-E8 medium (A, B, G–I), E5 medium that did not contain insulin, fibroblast growth factor-2 or transforming growth factor-β or TeSR-E8 medium with an inhibitor of phosphatidylinositol 3 kinase of LY294002 at a concentration of 20 μM (E, F, M–O). (A–F): The phase contrast images were analyzed using the imaging analysis system. Type A colonies were recognized as blue, and type B colonies as yellow. (G, J, M): The colony areas were extracted from the images and analyzed for type A or type B colonies of <1 mm2, or type B of >1 mm2. (H, K, N): The cell numbers calculated by the equation with the ratio of type A or type B colonies. (I, L, O): The cells dissociated by trypsin/EDTA were counted by the hemocytometer. Scale bars = 2 mm.
The phase-contrast images showed that the cells cultured in TeSR-E8 seemed intact (Fig. 6A), but the cells cultured in E5 medium seemed flat (Fig. 6E), and those cultured with LY294002 seemed unhealthy (Fig. 5I). Immunostaining showed that most cells were positive for Oct-4 in all culture conditions (Fig. 5C, 5D, 5G, 5H, 5K, 5L). In contrast, the vimentin-positive cells were localized in super-peripheral areas of the colonies in TeSR-E8 medium (Fig. 6C, 6D). In E5 medium, the vimentin-positive cells were localized in the peripheral and central areas of the colonies (Fig. 6G, 6H). With LY294002, the vimentin-positive cells were increased in the colonies (Fig. 6K, 6L). With respect to caspase 3/7(supplemental online data), the localization was similar to that of vimentin (supplemental online Fig. 6). To compare the result analyzed using phase-contrast image analysis system, the vimentin-positive areas of the colony areas were determined using the fluorescence imaging software (supplemental online Fig. 7). The vimentin-positive areas (pixels), for which the fluorescence intensity was more than eight per pixel, were calculated according to the areas (pixel) of the individual colonies. The number of colonies with >3% vimentin-positive areas per colony area was higher in E5 medium or LY294002 than in TeSR-E8 medium, just as the number of type A colonies was higher in E5 medium or TeSR-E8 medium with LY294002 than in TeSR-E8 medium. These finding indicated that although vimentin was not a direct marker of type A colonies, diagnosis by analyzing the type A or B colonies reflected the differentiated state of hiPSCs.
Figure 6.

Immunocytochemical staining for Oct-4 and vimentin. Tic cells were cultured in the same culture conditions (described in Fig. 5). The cells were cultured in TeSR-E8 medium (A–D), E5 medium (E–H), and TeSR-E8 medium with LY294002 (I–L) at day 4. A representative phase-contrast image in each culture condition (A, E, I) analyzed by the imaging analysis system for type A (blue) and type B (green) is shown. The cells stained with anti-Oct-3/4 or anti-vimentin antibody were individually visualized with Alexa Fluor 647 (green) or Alexa Fluor 594 (red) (C, D, G, H, K, L). Scale bars = 500 μm (A–C, E–G, I–K) and 100 μm (D, H, L).
Discussion
In the present study, we developed an image analysis method using a culture observation system to determine hiPSC growth without using any invasive treatments. By determining the relationships between the areas of hiPSC colonies and the number of fluorescently stained nuclei, equations were derived to calculate the cell numbers from colony images acquired using the culture observation system for two hiPSC lines under two culture conditions.
Cells that are typically cultured in monolayers, such as fibroblasts, keratinocytes, or cancer cells, can generally be recognized individually under a microscope. In contrast, because undifferentiated hESCs/hiPSCs are tightly packed within colonies [2, 18], it is challenging to recognize single cell areas owing to the difficulty with adapting image segmentation approaches. Furthermore, hESCs/hiPSCs tend to differentiate during culture, which makes it quite difficult to accurately identify and count the number of undifferentiated hESCs/hiPSCs.
We had first hypothesized that the number of hiPSCs would increase linearly with the increase in colony areas. However, the relationship between the hiPSC colony areas and cell numbers could not be expressed with an equation for a straight line, because the single hiPSC areas changed during culture. In practice, stem cell researchers have found that the morphologies of hESCs/hiPSCs usually changed at 3–4 days after passage from comparatively flatter shapes to small, packed, round shapes, or so-called mature cells. Our measurements under a microscope showed that the size of the iPS-TIG114-4f1 cells decreased from ∼1,500 μm2 to 75 μm2 during culture after passage and that the average size of the single hiPSCs in a “mature colony” was ∼136 μm2. The size of these hiPSCs was greater than that observed in a previous study [18].
Separately, we attempted to measure the size of hES H9 cells on phase-contrast images. The average size of single hESCs in a “mature colony” was ∼315 μm2 (data not shown). These findings suggested that the size of hESCs/hiPSCs will be different among cell lines. Other investigators have argued that relationships exist among the cell sizes, DNA contents, and metabolic activities [19, 20]. One recent study reported that cancer cell lines exhibited specific cell volumes that correlated with the protein content, DNA content, and protein synthesis rates [21]. These findings implied that the activities in hPSCs might dynamically change during culture, which might cause morphological changes.
Next, we hypothesized that if a relationship between the hiPSC colony areas and cell numbers could be determined, this relationship could provide cell growth. We applied several different types of equations to determine this relationship and concluded that the relationship was linear, although the equation coefficients were dependent on the cell line used, cell morphology, colony size, and culture conditions. The cell growth rates were also different with the different culture conditions. These results are consistent with those from our previous study of a growth factor-defined serum-free medium for culturing hESCs. That study showed that the growth of hESCs cultured under feeder-free culture conditions was different from that of hESCs cultured on feeders [22]. When the cells overgrew in TeSR-E8 or the cell growth was inhibited in E5 medium or LY294002, the calculated cell numbers using these equations were different from the cell numbers counted using the hemotocytometer. In contrast, the changes in cell growth could be detected. Both phase-contrast images of live cells and fluorescent images of fixed nuclei stained with Hoechst 33342 showed that largeness of the cells and nuclei seemed different among these culture conditions (supplemental online Fig. 8). Considering that cell size might be related to DNA content or metabolic activities, the equation coefficients for the relationships between the hPSC colony areas and nuclei numbers could provide valuable biological information by reflecting the different hPSC states. We are currently investigating whether the method we have developed is applicable to hESCs. Analysis with other hESC/hiPSC lines in proper and/or improper culture conditions should be performed in future studies.
To the best of our knowledge, our is the first study to use a noninvasive method using images to systemically determine the growth of hPSCs. When the culture conditions are not suitable for the cells, the cell growth rates are delayed. In the present study, our results demonstrated that when the culture conditions became improper, our image analysis system could detect the changes in cell growth without any invasive treatment. Previous studies have demonstrated that “culture-adapted” cells tend to have an increased growth rate or reduced apoptosis, with karyotypic changes [23–28]. Frequent monitoring by determining cell growth is required to maintain or expand hPSCs. Although the method detailed in the present study requires determining the primary data for the relationship between the colony areas and cell numbers for each hPSC line, its utility should outweigh the labor involved. Furthermore, we are now preparing a method to detect the morphologies of hPSCs as a quality control measure. Monitoring cell growth noninvasively could be used to detect the healthy condition of hPSCs during culture. It would be also be more convenient if images could be acquired using a standard microscope. However, both tracking and acquiring images are indispensable using the culture observation system with a high-quality field lens, which ensures a constant light intensity, such as the CFI Plan Apo λ4× (numerical aperture, 0.2; red LED; center wavelength, 620 nm; half bandwidth, 10 nm; and a high image resolution, such as 1,000 × 1,000 pixels), to remove feeder cells or differentiated cells from the images and accurately detect the colony areas. The common microscope with low resolution or spotty light intensity can detect the hiPSC colony areas using the analysis software (supplemental online Fig. 9). However, it cannot accurately distinguish between type A and type B colonies. After the high-quality microscope has been used to determine the calculation of the growth of a cell line cultured under culture conditions, the common microscope will be able to approximate the cell numbers, although the variation errors will increase.
In recent years, automated culture systems for clinical human stem cells, including mesenchymal stem cells (MSCs), have attracted attention because large-scale human stem cell culture is required for cell-based medical or pharmaceutical applications [29]. An automated observation system is required for an automated culture system. In particular, a monitoring system for hPSC growth would be imperative. When images are captured every 12 hours, 30 of φ10-cm dishes or 75-cm2 flasks are capable of culturing in BioStation CT. Because our cell counting system can detect changes in growth rates by the growth conditions, this system could be used to monitor the cell quality and culture environment, such as improper culture medium in the automated culture system. Recent studies reported that cell movements were important for monitoring the healthy conditions of hPSCs and for detecting any mutant cells [28, 30]. The method we have developed in the present study could be applied for cell movement analysis and to detect mutant cells. Additional verification using different hESC/hiPSC lines needs to be conducted in the future.
For MSCs, monitoring their differentiation potential is useful, rather than simply monitoring for maintenance of an undifferentiated state. We previously developed a noninvasive prediction method for osteogenic differentiation of human MSCs [31–33]. When the differentiation protocols for hPSCs for specific cell lineages are established, noninvasive monitoring of differentiation will be required for clinical applications. Our noninvasive analysis system using a culture observation system would be useful for quality control during cell culture of clinical hPSCs, culturing of hiPSCs derived from the rare patients suffering from rare diseases, or for high-throughput screening analysis in pharmaceutical research.
Conclusion
Human pluripotent stem cell growth was determined using an image analyzer. This method provided real-time information on colony growth and cell growth rates without using treatments that can damage the cells and could be useful for basic research on hPSCs and cell processing for hPSC-based therapy.
Supplementary Material
Acknowledgments
This study was supported by grants-in-aid from the New Energy and Industrial Technology Development Organization of Japan to Y.K. and M.K.F and the Ministry of Health, Labor and Welfare of Japan to M.K.F. We thank Dr. Ryuji Kato (Nagoya University) for discussions and Akiko Hamada for excellent technical support.
Author Contributions
M.S.: collection and/or assembly of data, data analysis and interpretation, performance of experiments, manuscript writing; H.K.: collection and/or assembly of data, data analysis and interpretation; K.N.: data analysis and interpretation; Y.K.: conception/design, financial support, data analysis and interpretation; M.K.F.: conception and design, financial support, data analysis and interpretation, manuscript writing.
Disclosure of Potential Conflicts of Interest
H.K., K.N., and Y.K. are compensated employees of Nikon Corporation. The other authors indicated no potential conflicts of interest.
References
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 3.International Stem Cell Initiative. Amps K, Andrews PW, et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adewumi O, Aflatoonian B, Ahrlund-Richter L, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25:803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe K, Ueno M, Kamiya D, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- 6.Kondo H, Yonezawa Y. Fetal-adult phenotype transition, in terms of the serum dependency and growth factor requirements, of human skin fibroblast migration. Exp Cell Res. 1995;220:501–504. doi: 10.1006/excr.1995.1342. [DOI] [PubMed] [Google Scholar]
- 7.Koyanagi-Aoi M, Ohnuki M, Takahashi K, et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc Natl Acad Sci USA. 2013;110:20569–20574. doi: 10.1073/pnas.1319061110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagata S, Toyoda M, Yamaguchi S, et al. Efficient reprogramming of human and mouse primary extra-embryonic cells to pluripotent stem cells. Genes Cells. 2009;14:1395–1404. doi: 10.1111/j.1365-2443.2009.01356.x. [DOI] [PubMed] [Google Scholar]
- 9.Makino H, Toyoda M, Matsumoto K, et al. Mesenchymal to embryonic incomplete transition of human cells by chimeric OCT4/3 (POU5F1) with physiological co-activator EWS. Exp Cell Res. 2009;315:2727–2740. doi: 10.1016/j.yexcr.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Amit M, Carpenter MK, Inokuma MS, et al. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000;227:271–278. doi: 10.1006/dbio.2000.9912. [DOI] [PubMed] [Google Scholar]
- 11.Kinehara M, Kawamura S, Mimura S, et al. Protein kinase C-induced early growth response protein-1 binding to SNAIL promoter in epithelial-mesenchymal transition of human embryonic stem cells. Stem Cells Dev. 2014;23:2180–2189. doi: 10.1089/scd.2013.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohnuma K, Fujiki A, Yanagihara K, et al. Enzyme-free passage of human pluripotent stem cells by controlling divalent cations. Sci Rep. 2014;4:4646. doi: 10.1038/srep04646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen G, Gulbranson DR, Hou Z, et al. Chemically defined conditions for human iPSC derivation and culture. Nat Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinehara M, Kawamura S, Tateyama D, et al. Protein kinase C regulates human pluripotent stem cell self-renewal. PLoS One. 2013;8:e54122. doi: 10.1371/journal.pone.0054122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishino K, Toyoda M, Yamazaki-Inoue M, et al. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011;7:e1002085. doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toyoda M, Yamazaki-Inoue M, Itakura Y, et al. Lectin microarray analysis of pluripotent and multipotent stem cells. Genes Cells. 2011;16:1–11. doi: 10.1111/j.1365-2443.2010.01459.x. [DOI] [PubMed] [Google Scholar]
- 17.Amit M, Itskovitz-Eldor J. Morphology of human embryonic and induced pluripotent stem cell colonies cultured with feeders. In: Amit M, Itskovitz-Eldor J, editors. Atlas of Human Pluripotent Stem Cells: Derivation and Culturing. New York: Humana Press; 2012. pp. 15–39. [Google Scholar]
- 18.Wakao S, Kitada M, Kuroda Y, et al. Morphologic and gene expression criteria for identifying human induced pluripotent stem cells. PLoS One. 2012;7:e48677. doi: 10.1371/journal.pone.0048677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozłowski J, Konarzewski M, Gawelczyk AT. Cell size as a link between noncoding DNA and metabolic rate scaling. Proc Natl Acad Sci USA. 2003;100:14080–14085. doi: 10.1073/pnas.2334605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savage VM, Allen AP, Brown JH, et al. Scaling of number, size, and metabolic rate of cells with body size in mammals. Proc Natl Acad Sci USA. 2007;104:4718–4723. doi: 10.1073/pnas.0611235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolfi SC, Chan LL, Qiu J, et al. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013;1:20. doi: 10.1186/2049-3002-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furue MK, Na J, Jackson JP, et al. Heparin promotes the growth of human embryonic stem cells in a defined serum-free medium. Proc Natl Acad Sci USA. 2008;105:13409–13414. doi: 10.1073/pnas.0806136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Draper JS, Smith K, Gokhale P, et al. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- 24.Harrison NJ, Baker D, Andrews PW. Culture adaptation of embryonic stem cells echoes germ cell malignancy. Int J Androl. 2007;30:275–281; discussion 281. doi: 10.1111/j.1365-2605.2007.00762.x. [DOI] [PubMed] [Google Scholar]
- 25.Närvä E, Autio R, Rahkonen N, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28:371–377. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- 26.Enver T, Soneji S, Joshi C, et al. Cellular differentiation hierarchies in normal and culture-adapted human embryonic stem cells. Hum Mol Genet. 2005;14:3129–3140. doi: 10.1093/hmg/ddi345. [DOI] [PubMed] [Google Scholar]
- 27.Hyka-Nouspikel N, Desmarais J, Gokhale PJ, et al. Deficient DNA damage response and cell cycle checkpoints lead to accumulation of point mutations in human embryonic stem cells. Stem Cells. 2012;30:1901–1910. doi: 10.1002/stem.1177. [DOI] [PubMed] [Google Scholar]
- 28.Barbaric I, Biga V, Gokhale PJ, et al. Time-lapse analysis of human embryonic stem cells reveals multiple bottlenecks restricting colony formation and their relief upon culture adaptation. Stem Cell Reports. 2014;3:142–155. doi: 10.1016/j.stemcr.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kami D, Watakabe K, Yamazaki-Inoue M, et al. Large-scale cell production of stem cells for clinical application using the automated cell processing machine. BMC Biotechnol. 2013;13:102. doi: 10.1186/1472-6750-13-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Na J, Baker D, Zhang J, et al. Aneuploidy in pluripotent stem cells and implications for cancerous transformation. Protein Cell. 2014;5:569–579. doi: 10.1007/s13238-014-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuoka F, Takeuchi I, Agata H, et al. Morphology-based prediction of osteogenic differentiation potential of human mesenchymal stem cells. PLoS One. 2013;8:e55082. doi: 10.1371/journal.pone.0055082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuoka F, Takeuchi I, Agata H, et al. Characterization of time-course morphological features for efficient prediction of osteogenic potential in human mesenchymal stem cells. Biotechnol Bioeng. 2014;111:1430–1439. doi: 10.1002/bit.25189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki H, Takeuchi I, Okada M, et al. Label-free morphology-based prediction of multiple differentiation potentials of human mesenchymal stem cells for early evaluation of intact cells. PLoS One. 2014;9:e93952. doi: 10.1371/journal.pone.0093952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.