

Abstract
AIM: To investigate the prevalence and penetrance of hMSH6 mutations in Spanish HNPCC families that was negative for mutation in hMLH1 or hMSH2.
METHODS: We used PCR-based DGGE assay and direct sequencing to screen for hMSH6 gene in 91 HNPCC families.
RESULTS: we have identified 10 families with germ-line mutations in the DNA sequence. These mutations included two intronic variation, three missense mutation, one nonsense mutation, and four silent mutations. Among the 10 germ-line mutations identified in the Spanish cohort, 8 were novel, perhaps, suggesting different mutational spectra in the Spanish population. Detailed pedigrees were constructed for the three families with a possible pathogenic hMSH6 mutation. The two silent mutations H388H and L758L, detected in a person affected of colorectal cancer at age 29, produce loss of the wild-type allele in the tumor sample. Immunohistochemical analysis showed that expression of MSH6 protein was lost only in the tumors from the carriers of V878A and Q263X mutations.
CONCLUSION: Altogether, our results indicate that disease-causing germ-line mutations of hMSH6 are very less frequent in Spanish HNPCC families.
Keywords: HNPCC, MMR, hMSH6, MSI, IHC, SPANISH
INTRODUCTION
The link between defective DNA mismatch repair and the development of tumors has been firmly established. Inherited mutations in DNA mismatch repair genes are associated with the autosomal-dominant cancer susceptibility syndrome, hereditary non-polyposis colorectal cancer (HNPCC [MIM 114400; MIM 114500]). Patients with HNPCC are at high risk for colorectal and endometrial cancer and a variety of other malignancies. Tumors from HNPCC patients frequently show mutations in repetitive sequences, giving rise to a molecular phenotype referred to as microsatellite instability (MSI). Not surprisingly, a significant fraction of sporadic colorectal and endometrial cancers also show MSI.
The hMSH6 (MIM 600678) protein is a member of the MMR system; it interacts with hMSH2 to form the MutSa heterodimer[1-3]. Besides hMSH6, the hMSH2 protein also interacts with the hMSH3 protein to form the MutSb heterodimer. The MutSa heterodimer is efficient in the repair of both base-base mismatches and insertion-deletion loops, whereas the MutSb heterodimer is mainly active in the repair of insertion-deletion loops and not in the repair of base-base mismatches[4,5]. These data indicate that the hMSH6 and hMSH3 proteins are partially redundant in their DNA repair function. Most families with clinically recognized HNPCC and MSI high, have mutations in either hMLH1 or hMSH2. Mutations in hMSH6 gene, appear to be associated with atypical HNPCC and in particular with the development of endometrial carcinoma[6-9]. The age of onset of cancers in HNPCC kindred’s with hMSH6 mutations is higher than in hMSH2 or hMLH1 mutation carriers, and it has been reported that tumors from affected family members are less likely to have high MSI than tumors from hMSH2 or hMLH1 mutation carriers[10].
The estimate frequency of hMSH6 mutations in patients with colorectal cancer ranges between 0% based on a tumor immunohistochemistry study of a consecutive series and 1.5% based on mutation analysis of a combination of sporadic and familial cases[11,12]. Only a few of these germ-line mutations have been reported in families that fulfill the Amsterdam criteria[12,13]. To date, there have been no studies to determine the frequency of germ-line hMSH6 mutations in Spanish HNPCC families. The aim of this study was to verify the contribution of hMSH6 defects to cancer susceptibility in 132 HNPCC Spanish families that have been previously tested for hMSH2 and hMLH1 mutations[14,15].
MATERIALS AND METHODS
Patients
Families were selected through the clinic for familial cancer at the San Carlos University Hospital in Madrid, and they were Spanish by descent. Informed consent was obtained from each participant. Personal and family cancer histories were obtained from the proband and participating relatives, and cancer diagnoses and deaths were confirmed by reviewing the medical records, pathology reports, or death certificates.
We classified each pedigree by whether they fulfilled the original Amsterdam I criteria (n = 56 families), Amsterdam II (n = 11 families), Bethesda guidelines (n = 37 families), or none of these criteria (n = 28 families)[16-18]. This last category we refer to as “HNPCC-like” where the tumor spectra are reminiscent of HNPCC but not fulfilling Amsterdam I, Amsterdam II, or young age at onset criteria.
All probands, had been previously tested for MSI status and hMLH1/hMSH2 mutations/deletions by either DGGE and direct genomic sequencing and multiplex ligation-dependent probe amplification (MLPA, MRC-Holland).
Control subjects
One hundred unrelated healthy individuals from the Blood Transfusion National Service in Madrid (Spain) were used as controls.
DNA isolation
Genomic DNA was isolated from peripheral blood lymphocytes according to the salting out procedure[19]. Paraffin-embedded tumor samples were used to confirm segregation of identified mutations and to study MSI and LOH. In these cases, DNA was extracted from paraffin blocks as described previously by de la Hoya et al[20].
Tumor MSI analysis
One hundred and thirteen paired normal and tumor DNA were analyzed for MSI with the five markers of the Bethesda panel (BAT25, BAT26, D2S123, D5S346, and D17S250). Analysis of microsatellite sequences for instability was performed using an ABI Prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). PCR reactions were performed in a PTC-100 thermocycler (MJ Research, Waltman, MA, USA) and AB/ABI GeneScan software was used for data interpretation. Primer sequences were as described[21,22] and both fluorescent labeled and unlabeled primers were obtained from Applera Hispania SA. PCR reactions were carried out in a 10 mL reaction volume containing 50-100 ng of DNA, 1 PCR buffer (Ecogen SRL, Spain), 2.5 mol/L MgCl2, 200 mmol/L of each deoxynucleotide triphosphate (Promega, Madison, WI, USA), 0.4 mmol/L of each primer, and 1 U Eco Taq DNA polymerase (Ecogen SRL, Spain). The thermocycling conditions were 1 cycle of 10 min 95 °C, followed by 40 cycles consisting of 30 s 98 °C; 1 min 55 °C; 1 min 72 °C, followed by 1 cycle of 7 min 72 °C. Normal and tumor samples were prepared for analysis by pooling 0.5 mL of each reaction, 12 mL of formamide and 0.5 mL GeneScan 500 (Tamra; PE/ABI). The resulting suspension was heated at 95 °C, for 3 min, and then loaded on an ABI 310 sequencer for automated detection. The GeneScan software analyzed the data. Microsatellite stable (MSS) was defined as no observed instability, MSI-H was defined as two unstable loci of the five reference loci.
DNA amplification
The entire MSH6 coding region and the splice-junctions were amplified using the PCR from genomic DNA using 23 primer pairs. All amplicons were subjected to DGGE (below). The sequences of the primers are available upon request.
All DNA amplifications were performed in a 50-mL volume containing 100 ng DNA, 1 PCR buffer (Ecogen SRL, Spain), 0.4 mmol/L of each primer, 200 mmol/L of each deoxynucleotide (Promega, Madison, WI, USA), 1 U Eco Taq DNA polymerase (Ecogen SRL, Spain). Amplification was carried out in a PTC-100 thermocycler (MJ Research, Waltham, MA, USA). After denaturation at 95 °C for 5 min, 35 cycles at 95 °C for 30 s, 55 °C, or 58 °C for 30 s and 72 °C for 60 s were performed, followed by a final extension step of 10 min at 72 °C. Subsequently, PCR fragments were subjected to one round of complete denaturation and renaturation that is 98 °C for 10 min, 55 °C for 30 min, and 37 °C for 30 min to create heteroduplex molecules.
DGGE
DGGE analysis was performed in a denaturing gradient gel electrophoresis D-Code System (Bio-Rad Hercules, CA, USA). A 6-mL aliquot from the PCR reaction volume was mixed with 2 mL of standard dye-loading buffer and then loaded onto a 10% AA/bis-acrylamide (37.5:1) gel (0-60% or 20-80% urea-formamide chemical gradient according to melting profiles of each PCR fragment) in 1 TAE (40 mmol/L Tris-base, 20 mmol/L NaAC, 1 mmol/L EDTA pH 8) for 16 h at 80 V and 60 °C. The DNA fragments were visualized and photo-documented under ultraviolet transillumination after staining with an ethidium bromide solution.
In addition to mutation analysis, we used DGGE to perform DGGE-LOH analysis where appropriate tissue was available, for tumors belonging to families with identified germ-line mutations in hMSH6. All available tumor tissue was sectioned and subjected to hematoxylin-eosin staining to determine the proportion of tumor cells. Only samples with > 95% tumor cells were selected by macrodissection.
DNA sequencing
DNA fragments that displayed an abnormal DGGE pattern were analyzed by cycle sequencing. PCR products were purified with the QIAquick PCR purification Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Cycle sequencing reactions were performed with the ABI Prism dRhodamine Terminator Sequencing Kit (Applied Biosystems, Forest City, CA, USA) and analyzed in the ABI Prism 310 Genetic Analyzer (Applied Biosystems). All mutations were confirmed by two independent sequencing PCR reactions and sequenced in both directions.
hMSH6 immunohistochemistry
Immunohistochemical staining of hMSH6 was performed for the 60 MSS cases and for the 12 MSI-H cases negative for mutation in hMLH1 and hMSH2. The tissue sections were cut at 6 mm and mounted on ChemMate Gap microscopy slides (DAKO A/S, Denmark) and dried at 37 °C. After deparaffinization, slides were steam pretreated in EDTA buffer, pH 8-0, in a Magefesa Handy Steamer Plus (Magefesa, Spain) for 30 min, and then cooled and washed in PBS.
The antibody to hMSH6 (clone 44; 1/100; catalog number G70220; Transduction Laboratories, Becton Dickinson, Lexington, UK) is a mouse mAb generated with an NH2-terminal fragment (codons 225-333) of the hMSH6 protein. For the immunohistochemical analysis, avidin-biotin complex immunoperoxidase technique was performed by using a commercial ChemMate detection kit (DAKO A/S) in a Techmate automate machine. Endogenous peroxidase was blocked by incubation in hydrogen peroxide with methanol. Incubation with no immune horse serum was followed by incubation with primary antibody. The sections were then incubated in biotinylated second antibody and peroxidase-labeled avidin-biotin complex. All dilutions were made in PBS (pH 7.2). The stainings were visualized with diaminobenzidine tetrahydrochloride solution. The sections were counterstained in Mayer’s hematoxylin, rinsed with water, and mounted in an aqueous mounting media (Aquamount, BDH, Poole, UK). The percentage of positive nuclei was evaluated by two pathologists. Cases with more than 10% of nuclei staining were considered as positive protein expression. Normal colonic epithelium and lymphocytes exhibited strong nuclear staining for hMSH6 and, thus, served as positive internal controls for the staining of this protein.
RESULTS
MSI testing was carried out in 113 of the 132 families enrolled in our study. Of these 113 families, 60 had tumors that exhibited a non-MSI-high (MSS) phenotype and 53 had tumors that exhibited an MSI-high (MSI-H) phenotype. Nineteen families were not typed for MSI. All families were tested for mutations in hMLH1 and hMSH2, 24 families had a mutation in hMLH1 and 17 had a mutation in hMSH2 and were not studied further (Table 1). The remaining families were studied for mutations in hMSH6. In the 91 index cases tested for mutations in hMSH6, we identified 14 DNA variants. Of these, one was a novel definite deleterious mutation, nine were unclassified variants, six of them not described previously (Table 2) and four were polymorphisms described previously (Table 3). None of the unclassified variants were found in 100 control subjects.
Table 1.
Classification of colorectal cancer families, frequencies of pathogenic mutations in hMLH1 and hMSH2 genes and MSI phenotype
| Clinical criteria | Number of families | hMLH1 mutations | MSH2 mutations | MSI-H | MSS | MSI untyped |
| Amsterdam I | 56 | 17 | 15 | 42 | 13 | 1 |
| Amsterdam II | 11 | 2 | 2 | 2 | 7 | 2 |
| Bethesda | 37 | 5 | 0 | 6 | 20 | 11 |
| HNPCC-like | 38 | 0 | 0 | 3 | 20 | 5 |
| Total | 132 | 24 | 17 | 53 | 60 | 19 |
Table 2.
hMSH6 mutations in Spanish HNPCC families
| Family | Clinical criteria | Dx aged index patient (y) | No. of tumors in the family | Exon/intron | Nucleotide change | Amino acid change | Predicted consequence | Nfdht data base |
| 3 | Bethesda | 42 | 1E, 1G, 1M, 1H, 1F, and 1A | Ex. 8 | c.3725 G > T | R1242L | Inconclusive | No |
| 11 | HNPCC-like | 58 | 1G, 2C, and 1B | Ex. 4 | c.2319 C > T | L773L | Inconclusive | No |
| 68 | Bethesda | 29 | 1L, and 1C | Ex. 4 | c.1164 C > T | H388H | Inconclusive | No |
| Ex. 4 | c.2272 C>T | L758L | ||||||
| 81 | HNPCC-like | 50 | 4C | Int. 5 | c.3439?6 C > T | NA | Inconclusive | No |
| 94 | Amsterdam II | 45 | 3 P, 2E, 1G, and 3C | Ex. 4 | c.2633 T > C | V878A | Pathogenic | Yes |
| 100 | Amsterdam II | 54 | 4C, 1P, and 3G | Ex. 4 | c.706 C > T | Q236X | Truncation | No |
| 73 | Amsterdam | 29 | 2SM, 1G, 3C, and 1L | Ex. 4 | c.2633 T > C | V878A | Pathogenic | Yes |
| 121 | HNPCC-like | 58 | 3C, 1G, and 1B | Int. 5 | c.3439?6 C > T | NA | Inconclusive | No |
| 138 | Bethesda | 43 | 2C and 1 ureter in the same patient | Ex. 4 | c.1677 C > T | C559C | Inconclusive | No |
E, endometrial; G, glioma; M, meningioma; H, hemangioma; F, fibrosarcoma; A, adenoma; G, gastric; C, colon; B, breast; L, larynx; P, prostate; B, brain; SM, small intestine; y, years.
Table 3.
hMSH6 polymorphisms in Spanish HNPCC families
| Location | Nucleotide change | Amino acid change | Previously reported | Frequency1 |
| Exon 1 | 116 G→A | G 39 E | Yes | 0.40 |
| Exon 2 | 276 A→G | P 92 P | Yes | 0.38 |
| Exon 3 | 540 T→C | D 180 D | Yes | 0.24 |
| Exon 4 | 642 C→T | Y 214 Y | Yes | 0.25 |
1Patients harboring/patients screened.
The deleterious mutation Q236X, was found in an Amsterdam II family FC-100; change of cytosine for thymine at position c.706 leads to the formation of a termination codon at amino acid residue 236 that produces an 235 amino acid product instead of the normal 1 360 amino acid product. This mutation was found in a patient with two tumors, one colorectal and one endometrial (Figure 1). MSI testing was carried out in both tumors, colorectal and endometrial, and they exhibited a MSS phenotype; the immunohistochemical analysis of MSH6 showed an absence of staining (Table 4). In this family, we could test atleast two affected members, her father affected of colorectal cancer who was a carrier of the mutation and her sister also affected of colorectal cancer that resulted not to be a carrier. Both members had MSS tumors and MSH6 protein staining was absent in the carrier and was present in the non carrier.
Figure 1.
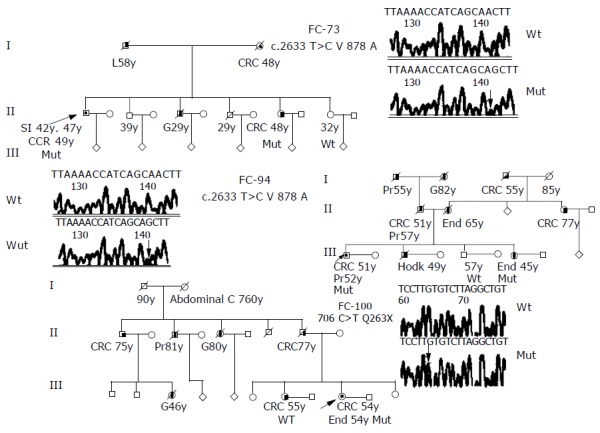
Pedigrees and DNA nucleotide sequence of the three families with disease-causing mutations of hMSH6 gene. Squares, males; circles, females; diagonal bars, deceased; unblackened symbols, no tumor; semi-blackened symbols, patients with histological-verified carcinomas; arrows, probands; mut, carries the indicated mutation. Abbreviations indicating type of tumor: CRC, colorectal; SI, small intestine; G, gastric; H, Hodgkin; End, endometrium; Pr, prostate; L, liver. Number after abbreviation indicates age at which tumor was diagnosed. Electropherograms of the wild type and mutant nucleotide sequences for V878A and Q263X mutations located in exon 4 of the hMSH6 gene.
Table 4.
Molecular alterations of tumors from persons carrying mutations of hMSH6 gene
| Family/member | Tumor type | MSI status |
Immunohistochemistry |
hMSH6 mutation | Other MMR gene mutation | ||
| MLH1 | MSH2 | MSH6 | |||||
| 3/270 | CRC | MSI-H | P | P | P | R1242L | V716M (hMLH1) |
| 11/310 | CRC | MSI-H | P | P | P | L773L | NO |
| 68/675 | CRC | MSS | P | P | P | H388H and L758L | NO |
| 73/526 | 3 CRCs | MSI-H | P | P | N | V878A | NO |
| 81/757 | CRC | MSS | P | P | P | c.3439?6 C > T | K618A (hMLH1) |
| 94/854 | UTERO | MSI-H | P | P | N | V878A | K618A (hMLH1) |
| 100/940 | CRC | MSS | P | P | N | Q236X | NO |
| 121/1041 | CRC | MSS | ND | ND | ND | c.3439?6 C > T | NO |
| 138/1165 | 2 CRCs | MSI-H | P | P | P | C559C | P350P (hMLH1) |
CRC, colorectal cancer; MSI-H, high microsatellite instability; MSS, negative microsatellite instability; P, positive for MMR protein; N, negative for MMR protein; ND, not done MMR expression.
The missense mutation V878A was detected in two Amsterdam families, FC-73 and FC-94, this mutation has been reported previously and had been considered as possibly pathogenic[6]. FC-94 (Figure 1) is also a carrier of the unclassified variant K618A in the MLH1 gene (Table 4). Both mutations (V878A in MSH6 and K618A in MLH1) were found in two affected members of the family, the proband (affected with colorectal and prostate cancer at ages 51 and 52) and his sister (affected with endometrial cancer at age 45). MSI study in tumors from carriers showed a MSI-H phenotype and the immunohistochemical analysis of hMSH6 showed absence of staining. Mutation V878A was also detected in FC-73 (Figure 1). This mutation was found in four affected members of the family, the proband was affected of three different cancers (two small intestine cancer and one colorectal cancer at ages 42, 47, and 49), his mother (affected of colorectal cancer at age 48), his brother (affected with gastric cancer at age 29) and his sister (affected with colorectal cancer at age 48). MSI study in tumors from carriers showed a MSI-H phenotype and the immunohistochemical analysis of hMSH6 showed absence of staining (Figure 2).
Figure 2.
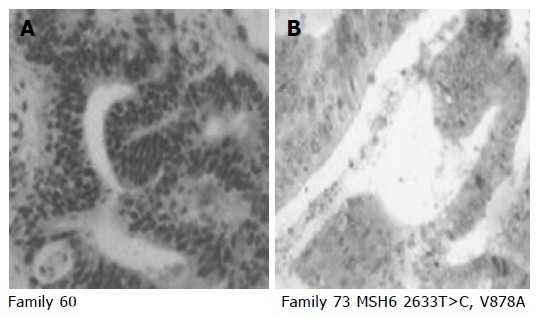
Representative examples of positive (A) and negative (B) immunohistochemical staining for hMSH6 protein (clone 44, Transduction laboratories/Becton Dickinson, Lexington, UK). A: Tumor from family 60, exhibited positive nuclear staining for hMSH6; B: tumor from family 73 carrier of V878A mutation, exhibited loss of hMSH6 expression.
The missense mutation R1242L was detected in FC-3 (Table 2); it is located in a conserved amino acid in exon 8 of the gene and causes substitution of a polar amino acid arginine to a non-polar amino acid leucine. This family also carries the mutation V716M in hMLH1 gene. Both mutations (R1242L in hMSH6 and V716M in hMLH1) were found in two members of the family, in the proband (affected with glioma, colorectal adenoma, meningioma, fibrosarcoma, and colorectal cancer at ages 42, 43, 52, 52, and 53), and in his father who was healthy. The MSI status of the colorectal cancer showed a MSI-H phenotype and the MLH1 and MSH6 staining were present (Table 4).
The silent mutation L773L was detected in FC-11 (Table 2). This mutation was found in an HNPCC-like criteria family with two members affected with colorectal cancer (ages 58 and 64), one affected with gastric cancer (age 60) and one with breast cancer (age 60). The mutation was found only in the proband, the other affected members were of wild type. The MSI status of the colorectal cancer showed a MSI-H phenotype and the hMSH6 staining was present (Table 4).
The silent mutation C559C was detected in FC-138 (Table 2), the proband was affected of two colorectal and one ureter cancers and the first age at diagnostic was at 43 years. The MSI status of the colorectal cancer showed an MSI-H phenotype and the hMSH6 staining was present (Table 4). Mutations H388H and L758L were detected in FC-68 (Table 2). These mutations were found in a patient with early onset colorectal cancer (age 29), the MSI status of the tumor was MSS and the hMSH6 staining was present (Table 4). The intron five mutation IVS 5-16 C > T was detected in two families FC-81 and FC-121 (Table 2). FC-81 has four members affected by colorectal cancer in two generations. The mutation was found in two affected members of the family, the proband (affected with colorectal cancer at age 66), and his brother (affected with colorectal cancer at age 61); the two other members of the family, his father who was not tested and his other brother (affected with colorectal cancer at age 50) was wild type. The two members who carry the hMSH6 mutation, were also carriers of K618A in the MLH1 gene. The MSI status of the tumors from the three affected brothers was MSS. The hMSH6 staining was present in the member wild type and in the two carriers of hMSH6 mutation. The MLH1 staining was present in the tumors of the carriers and the non-carriers of K618A (Table 4). FC-121 has three members affected of colorectal cancer (ages 60, 75, and 55), one with gastric cancer (age 73) and one with brain cancer (age 70, Table 2). We could only test the proband (affected with colorectal cancer at age 60). The MSI status of the tumor was MSS and the MSH6 staining was not determined (Table 4).
DGGE-LOH analysis was done to further characterize the role of these variants. Figure 3 shows a typical DGGE analysis of different hMSH6 exons amplified from tumor and peripheral blood DNA. Examples of missense mutations R1242L, V878A, and nonsense mutation K263X are shown. The silent mutations H388H and L758L were identified in the same individual. LOH analysis showed loss of the wild-type allele in both informative loci, indicating that these changes were located in the same chromosome. Taking into account our results, no evidence for LOH acting as a second-hit in the hMSH6 locus was found.
Figure 3.
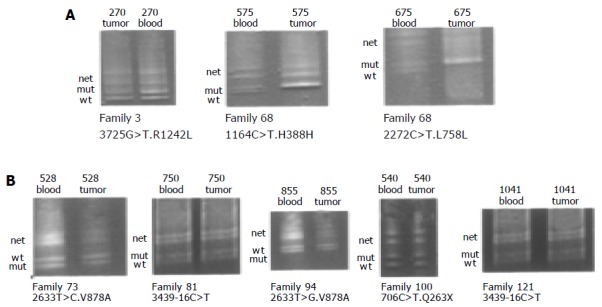
DGGE-LOH analysis of different hMSH6 exons, amplified from both tumor and blood DNA. Analysis of 7 patients (270, 675, 526, 750, 855, 940, and 1 041) harboring the mutations R1242L, H388L, L758L, V878A, 3439-16 C > T, and Q263X. Bands corresponding to wild-type (Wt), mutant (Mut) alleles and the heteroduplex (Het) are indicated in all cases. Panel A shows LOH of the mutant allele in patient 270 and LOH of wild-type allele in patient 675. Panel B shows retention of both alleles (wt and mut) in all patients.
DISCUSSION
Carrier frequencies of germ-line hMSH6 mutations in selected HNPCC families have been extensively described[6,10,12,23,24]. However, there is no evidence in the literature on this point regarding the Spanish population. We screened 91 HNPCC families (24 Amsterdam I, 7 Amsterdam II, 32 Bethesda guidelines, and 28 HNPCC-like) for the presence of germ-line mutations in the hMSH6 gene and identified 14 DNA changes. Seven of them were previously unreported DNA changes, including the intronic change at -16 base position of intron 5 (intronic mutation) found in two HNPCC-Like families, 1 amino acid substitution, 1 protein truncation mutation, and 4 silent mutations (Tables 2 and 4). The remaining seven DNA changes were reported previously, four of which were attested polymorphisms, one was the silent mutation H388L and the other 2 were V878A substitution that have been described as a possible disease causing mutation by other authors[6,9,25]. In our study, the Val878Ala substitution was detected in all affected members tested of both families, FC-73 (Amsterdam I) and FC-94 (Amsterdam II), suggesting that the hMSH6 germ-line mutation segregated with the disease. The MSI study of the tumors revealed an MSI-H phenotype. Moreover, immunohistochemical analysis revealed loss of hMSH6 protein in tumors from these patients. Our data suggest that this alteration may be a disease-causing mutation. However, we cannot discard that another mutation not detected could be linked to V878A in our population. Peterlongo et al[24] has found the V878A in 4 out of 190 controls (from 18000 volunteers of varying ethnic backgrounds who live in the New York metropolitan area), suggesting that this variant may not be linked to the disease in their population.
From the study presented here, we propose the following interpretations concerning the role of each previously unreported DNA change in the susceptibility to CRC. We interpret the hMSH6 3 435-16 C > T mutation found in two HNPCC-like families (FC-81 and FC-121) as a non disease-causing mutation for the following reasons: the 3 435-16 C > T did not segregate in the families, the tumors showed a MSS phenotype and the immunohistochemical analysis showed normal expression of the hMSH6; moreover, the wild type and mutant alleles were both retained in the tumor (LOH study, Figure 3). The hMSH6 c.706 C > T mutation produces a stop codon, Q236X, and presumably causes the synthesis of a truncated hMSH6 protein of 235 amino acids (instead of 1 360), the immunohistochemical analysis showed lack of expression of hMSH6 and reduced expression of hMSH2 proteins from cancer cells. Reduced expression of hMSH2 was observed in CRC and endometrial cancer cases with a germ-line mutation in hMSH6[26,28]. We could study three affected members of the family, and we have seen that the mutation segregate with the disease because the father of the index case is also a carrier. Her sister, diagnosed of colorectal cancer at age 55, resulted not to be a carrier, this case could be a phenocopy. We can conclude that the mutation could be responsible for the susceptibility to cancer in this family. Mutation c.3725 G > T (R1242L) was found in a patient with multiple tumors and he is also a carrier of K618A in hMLH1 gene, his father is also a carrier of both mutations but he is still healthy. The immunohistochemical study showed expression of all MMR proteins and the study of LOH in the tumor cells showed loss of mutant allele. We could not study any other member of the family because the pedigree was very short (no more members, the index case was alone). Mutations c.1164 C > T and c.2272 C > T occur in the same chromosome in a patient from a short family (the index case diagnosed of colorectal cancer at 29, her brother was healthy, and her father was diagnosed of larynx cancer at 50 years). The hMSH6 protein was present in the tumor cells and the LOH study showed retention of mutant allele and perhaps, this fact could produce chromosomal instability, and therefore, mutational events that led to hyperproliferation. At the end, the silent mutation c.1677 C > T (C559C) was found in a patient with two CRCs at age 43 and one urothelial cancer at age 47. This patient was also a carrier of another silent mutation in hMLH1 gene (P350P) and both colorectal tumors showed a MSI-H phenotype and the immunohistochemical study showed expression of hMLH1 and hMSH6 proteins. With these results, we cannot conclude if the silent mutations are responsible for the tumors in our patient.
Our data indicate that the frequency of hMSH6 mutations is low in Spanish HNPCC, they can be found either in families with MSI-H or MSS tumors, which on average had an older age at diagnosis of cancer and are associated with other HNPCC tumors-overall gastric and endometrium. These data are consistent with the results of other published studies[24,27-29].
Thus, we can conclude from our study, that the two possible pathogenic mutations of hMSH6 were found in three families, in two of them the patients had tumors exhibiting a MSI-H phenotype and had been tested negative for mutations in hMLH1 and hMSH2. The remaining family was also negative for mutations in hMLH1 and hMSH2 but the tumors of the two carriers were MSS. The frequency of hMSH6 mutations in our series was low, as they were found only in 3 out of 91 families studied (1 of 19 Amsterdam I and 2 of 8 Amsterdam II). The mean age of dx cancer was higher than in hMLH1- and hMSH2-related families. On the other hand, we have not found these variants in site-specific CRC families but in families with a broad spectrum of cancer types. Therefore, we suggest that hMSH6 testing should be pursued in Amsterdam families that have been negative for mutation in hMLH1 and hMSH2 genes or, as the literature suggests, in families with a history of endometrial, gastric, or other cancers in addition to a history of CRC.
ACKNOWLEDGMENTS
We thank the families for participating in the study. We thank C Corbacho, Dr. J Sanz Garcia and Dr. C Saez for their technical work.
Footnotes
Science Editor Guo SY Language Editor Elsevier HK
Supported by the Instituto Nacional Carlos III (RTICC C03/10); Fondo de Investigación Sanitaria (FIS 04/0957) and Sanofi-Synthelabo
References
- 1.Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909–1912. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 2.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D'Arrigo A, Truong O, Hsuan JJ, Jiricny J. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 3.Papadopoulos N, Nicolaides NC, Liu B, Parsons R, Lengauer C, Palombo F, D'Arrigo A, Markowitz S, Willson JK, Kinzler KW. Mutations of GTBP in genetically unstable cells. Science. 1995;268:1915–1917. doi: 10.1126/science.7604266. [DOI] [PubMed] [Google Scholar]
- 4.Das Gupta R, Kolodner RD. Novel dominant mutations in Saccharomyces cerevisiae MSH6. Nat Genet. 2000;24:53–56. doi: 10.1038/71684. [DOI] [PubMed] [Google Scholar]
- 5.Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- 6.Berends MJ, Wu Y, Sijmons RH, Mensink RG, van der Sluis T, Hordijk-Hos JM, de Vries EG, Hollema H, Karrenbeld A, Buys CH, et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet. 2002;70:26–37. doi: 10.1086/337944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–272. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 8.Wagner A, Hendriks Y, Meijers-Heijboer EJ, de Leeuw WJ, Morreau H, Hofstra R, Tops C, Bik E, Bröcker-Vriends AH, van Der Meer C, et al. Atypical HNPCC owing to MSH6 germline mutations: analysis of a large Dutch pedigree. J Med Genet. 2001;38:318–322. doi: 10.1136/jmg.38.5.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wijnen J, de Leeuw W, Vasen H, van der Klift H, Møller P, Stormorken A, Meijers-Heijboer H, Lindhout D, Menko F, Vossen S, et al. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet. 1999;23:142–144. doi: 10.1038/13773. [DOI] [PubMed] [Google Scholar]
- 10.Wu Y, Berends MJ, Mensink RG, Kempinga C, Sijmons RH, van Der Zee AG, Hollema H, Kleibeuker JH, Buys CH, Hofstra RM. Association of hereditary nonpolyposis colorectal cancer-related tumors displaying low microsatellite instability with MSH6 germline mutations. Am J Hum Genet. 1999;65:1291–1298. doi: 10.1086/302612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunningham JM, Kim CY, Christensen ER, Tester DJ, Parc Y, Burgart LJ, Halling KC, McDonnell SK, Schaid DJ, Walsh Vockley C, et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am J Hum Genet. 2001;69:780–790. doi: 10.1086/323658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, Wahlberg S, Fox EA, Peel D, Ziogas A, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 13.Plaschke J, Kruppa C, Tischler R, Bocker T, Pistorius S, Dralle H, Rüschoff J, Saeger HD, Fishel R, Schackert HK. Sequence analysis of the mismatch repair gene hMSH6 in the germline of patients with familial and sporadic colorectal cancer. Int J Cancer. 2000;85:606–613. doi: 10.1002/(sici)1097-0215(20000301)85:5<606::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Caldes T, Godino J, de la Hoya M, Garcia Carbonero I, Perez Segura P, Eng C, Benito M, Diaz-Rubio E. Prevalence of germline mutations of MLH1 and MSH2 in hereditary nonpolyposis colorectal cancer families from Spain. Int J Cancer. 2002;98:774–779. doi: 10.1002/ijc.10240. [DOI] [PubMed] [Google Scholar]
- 15.Godino J, de La Hoya M, Diaz-Rubio E, Benito M, Caldés T. Eight novel germline MLH1 and MSH2 mutations in hereditary non-polyposis colorectal cancer families from Spain. Hum Mutat. 2001;18:549. doi: 10.1002/humu.1240. [DOI] [PubMed] [Google Scholar]
- 16.Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34:424–425. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 17.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, Lynch H, Perucho M, Smyrk T, Sobin L, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89:1758–1762. doi: 10.1093/jnci/89.23.1758. [DOI] [PubMed] [Google Scholar]
- 19.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de La Hoya M, Díaz-Rubio E, Caldés T. Denaturing gradient gel electrophoresis-based analysis of loss of heterozygosity distinguishes nonobvious, deleterious BRCA1 variants from nonpathogenic polymorphisms. Clin Chem. 1999;45:2028–2030. [PubMed] [Google Scholar]
- 21.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 22.Hoang JM, Cottu PH, Thuille B, Salmon RJ, Thomas G, Hamelin R. BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res. 1997;57:300–303. [PubMed] [Google Scholar]
- 23.Buttin BM, Powell MA, Mutch DG, Babb SA, Huettner PC, Edmonston TB, Herzog TJ, Rader JS, Gibb RK, Whelan AJ, et al. Penetrance and expressivity of MSH6 germline mutations in seven kindreds not ascertained by family history. Am J Hum Genet. 2004;74:1262–1269. doi: 10.1086/421332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peterlongo P, Nafa K, Lerman GS, Glogowski E, Shia J, Ye TZ, Markowitz AJ, Guillem JG, Kolachana P, Boyd JA, et al. MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer. 2003;107:571–579. doi: 10.1002/ijc.11415. [DOI] [PubMed] [Google Scholar]
- 25.Charames GS, Millar AL, Pal T, Narod S, Bapat B. Do MSH6 mutations contribute to double primary cancers of the colorectum and endometrium? Hum Genet. 2000;107:623–629. doi: 10.1007/s004390000417. [DOI] [PubMed] [Google Scholar]
- 26.de Leeuw WJ, Dierssen J, Vasen HF, Wijnen JT, Kenter GG, Meijers-Heijboer H, Brocker-Vriends A, Stormorken A, Moller P, Menko F, et al. Prediction of a mismatch repair gene defect by microsatellite instability and immunohistochemical analysis in endometrial tumours from HNPCC patients. J Pathol. 2000;192:328–335. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH701>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Parc YR, Halling KC, Wang L, Christensen ER, Cunningham JM, French AJ, Burgart LJ, Price-Troska TL, Roche PC, Thibodeau SN. HMSH6 alterations in patients with microsatellite instability-low colorectal cancer. Cancer Res. 2000;60:2225–2231. [PubMed] [Google Scholar]
- 28.Plaschke J, Krüger S, Pistorius S, Theissig F, Saeger HD, Schackert HK. Involvement of hMSH6 in the development of hereditary and sporadic colorectal cancer revealed by immunostaining is based on germline mutations, but rarely on somatic inactivation. Int J Cancer. 2002;97:643–648. doi: 10.1002/ijc.10097. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Lasset C, Desseigne F, Saurin JC, Maugard C, Navarro C, Ruano E, Descos L, Trillet-Lenoir V, Bosset JF, et al. Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2, and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum Genet. 1999;105:79–85. doi: 10.1007/s004399900064. [DOI] [PubMed] [Google Scholar]