

Abstract
Parkinson's disease (PD) is characterized by the selective loss of dopaminergic neurons of the substantia nigra pars compacta (SNc) with motor and nonmotor symptoms. Defective mitochondrial function and increased oxidative stress (OS) have been demonstrated as having an important role in PD pathogenesis, although the underlying mechanism is not clear. The etiopathogenesis of sporadic PD is complex with variable contributions of environmental factors and genetic susceptibility. Both these factors influence various mitochondrial aspects, including their life cycle, bioenergetic capacity, quality control, dynamic changes of morphology and connectivity (fusion, fission), subcellular distribution (transport), and the regulation of cell death pathways. Mitochondrial dysfunction has mainly been reported in various non-dopaminergic cells and tissue samples from human patients as well as transgenic mouse and fruit fly models of PD. Thus, the mitochondria represent a highly promising target for the development of PD biomarkers. However, the limited amount of dopaminergic neurons prevented investigation of their detailed study. For the first time, we established human telomerase reverse transcriptase (hTERT)-immortalized wild type, idiopathic and Parkin deficient mesenchymal stromal cells (MSCs) isolated from the adipose tissues of PD patients, which could be used as a good cellular model to evaluate mitochondrial dysfunction for the better understanding of PD pathology and for the development of early diagnostic markers and effective therapy targets of PD. In this review, we examine evidence for the roles of mitochondrial dysfunction and increased OS in the neuronal loss that leads to PD and discuss how this knowledge further improve the treatment for patients with PD.
Keywords: mitochondrial dysfunction, oxidative stress, PD genes, pathophysiology
INTRODUCTION
Parkinson's disease (PD) is the most common neurodegenerative movement disorder characterized by numerous motor symptoms, including bradykinesia, hypokinesia, rigidity, resting tremor, and postural instability and non-motor symptoms, such as autonomic dysfunction, sleep abnormalities, depression, and dementia [1,2,3]. The motor clinical manifestations are the preferential loss of dopaminergic (DA) neurons of the substantia nigra pars compacta (SNc), resulting in dopamine deletion and derangements of neuronal circuits in the basal ganglia target regions of these neurons [1]. Another pathological hallmark of PD is round eosinophilic intracytoplasmic proteinaceous inclusion bodies that are mainly composed of fibrillar α-synuclein termed Lewy bodies (LBs) and dystrophic neurites (Lewy neurites) present in surviving neurons [4]. The manifestations of non-motor symptoms can contribute considerably to disability, and they usually are not responsive to dopamine replacement therapy [1]. Dementia is a major cause of disability, and currently there is no effective symptomatic treatment and 47% of PD patients show evidence of depression [5].
Aberrant mitochondrial forms and functions are widely accepted pathogenic mechanisms in a subset of people with PD [2,3], which demonstrates that mitochondrial dysfunction and oxidative stress (OS) have a central role in PD pathogenesis. Mitochondrial dysfunction in the dopaminergic neurons of idiopathic and familial PD is well known although the underlying mechanisms are not clear. Mitochondrial dysfunction is mainly characterized by the generation of reactive oxygen species (ROS), a decrease in mitochondrial complex I enzyme activity, cytochrome-c release, ATP depletion and caspase 3 activation. Systemic mitochondrial complex I deficiency has long been implicated in the pathogenesis of idiopathic PD, which may generate additional OS in nigral neurons. Impaired mitochondrial function leads to increased OS and OS would harm the integrity of the neuron so as to accelerate the degeneration of neurons. OS or ROS not only inflict direct cellular damages but also activate signaling pathways leading to cell death [6]. The most direct evidence for disrupted mitochondrial metabolism has come from studies using autopsy tissues and other tissue samples and from in vitro cell cultures derived from human patients with PD [2]. Moreover, mitochondrial abnormalities have been reported in Parkin-deficient mouse and fruit fly models of PD [7,8,9]. Consistently, postmortem studies have highlighted the presence of oxidative damage in the pathogenesis of PD, and particularly oxidative damage to lipids, proteins, and DNA can be observed in the SNc of sporadic PD patients' brains.
In this article, we examined and summarized our recent knowledge in the mitochondria dysfunction in PD pathogenesis and discuss how this knowledge further will improve the treatment for patients with PD.
NEURAL FUNCTION AND PATHOLOGY OF MITOCHONDRIA IN NEURODEGENERATION OF PD
Normal function and pathology of mitochondria
Mitochondria are the intracellular powerhouse in which perform important cellular reactions, including the production of energy through the mitochondrial respiratory chain (RC), the regulation of cell death, calcium metabolism and the production of ROS [10]. Mitochondria are a major source of free radicals in the cell, resulting in OS, but mitochondria are also integral to the OS response. Moreover, the RC comprises four enzymatic complexes (complexes I-IV) embedded in the inner mitochondrial membranes, which catalyze the transfer of reducing equivalents from high-energy compounds produced by the reactions of the Krebs cycle to oxygen, with the ultimate production of an electrochemical gradient through the inner mitochondrial membranes to drive the synthesis of ATP by ATP synthase [10]. Mitochondrial DNA (mtDNA) encodes 13 proteins that are all components of the electron transport chain, and there have been several reports of mtDNA mutations in rare maternally-inherited pedigrees of parkinsonism, including the 12S rRNA gene in one family with parkinsonism, deafness and neuropathy [11].
Mitochondrial dysfunction and OS in PD pathogenesis
Widely accepted pathogenic mechanisms in a subset of people with PD are aberrant mitochondrial form and function. Impaired mitochondrial function leads to increased OS and affects a number of cellular pathways, leading to damage of intracellular components and to cell death. OS is one of pathogenic mechanisms of nigral dopamine cell death in PD. The etiopathogenesis of sporadic PD is complex with variable contributions of environmental factors and genetic susceptibility. Both environmental and genetic factors influence various mitochondrial aspects, including their life cycle, bioenergetic capacity, quality control, dynamic changes of morphology and connectivity (fusion, fission), subcellular distribution (transport), and the regulation of cell death pathways (Fig. 1).
Fig. 1. Mitochondria dysfunction and dopaminergic cell death in PD pathogenesis. Multiple factors, including genetics, aging and environmental toxins, or combinations, have been implicated in the aetiology of PD. Abnormal metabolic function, abnormal morphology, and impaired fission-fusion balance have all been observed in mitochondria in at least some forms of PD. Increased OS can lead to impaired function of the UPS, thereby further affecting cell survival. All these may directly or indirectly affect the mitochondrial function of protein degradation systems, including UPS and ALP, and thereby, cause the death of dopamine neurons.

Environmental factors
Ageing
PD is one of the best examples of an age-related disease. Age influences the clinical progression of PD, which is one of the most important risk factors for sporadic PD. The possible role of ageing in the pathogenesis of PD is suggested by its usual occurrence in late middle age, and by marked increase in its prevalence at older age [5]. Older people may show mild parkinsonian signs, which are associated with reduced function. These may be due to age-related decline in dopaminergic activity, incidental Lewy body disease, degenerative pathologies or vascular pathology [12]. Precisely, several studies have demonstrated that advancing age is associated with a faster rate of motor progression, decreased levodopa responsiveness, more severe gait and postural impairment, and more severe cognitive impairment and the development of dementia in patients with PD [13]. PD may reflect a failure of the normal cellular compensatory mechanisms in vulnerable brain regions, and this vulnerability is increased by ageing.
Environmental toxins
Mitochondria exposed to highly oxidative environment, and the ROS was produced in the process of oxidative phosphorylation [2]. Mitochondrial dysfunction plays a major role in the pathogenesis of PD, and in particular, defects of mitochondrial complex-I of the respiratory chain may be the most appropriate cause degeneration of neurons in PD by reducing the synthesis of ATP. Several epidemiological studies have shown that pesticides and other toxins from the environment that inhibits complex-I is involved in the pathogenesis of sporadic PD. First, 1- methyl4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) inhibits complex-I [14], decreases ATP production and increases generation of ROS [15], inhibits mitochondrial complexes III and IV [16], decreases mitochondrial activity and mitochondrial gene expression [17], alters mitochondrial proteins such as chaperones, metabolic enzymes, oxidative phosphorylation-related proteins, inner and outer mitochondrial proteins [18], alters proteins associated with mitochondrial dysfunction, dopamine signaling, ubiquitin system, calcium signaling, OS response and apoptosis [19], and cause the symptoms of PD in humans and animal models. Second, Rotenone reduces complex I activity [20]. Third, paraquat accumulates in mitochondria [21] and acts as a potent redox cycler which converts free radicals that interact with molecular oxygen to superoxide and other ROS [22]. Fourth, maneb inhibits mitochondrial complex III [23].
Genetic factors
Progressive advances in PD genetics have shown the evidence for a vital role of mitochondrial dysfunction in the pathogenesis of the disease. The identification of genes associated with parkinsonism has had a major advance on PD research, showing that most of them can affect or regulate different functional or physiological aspects of mitochondria. Although the majority of PD cases are sporadic, several genes have been linked to familial PD. Familial PD is caused by mutations in genes identified by linkage analyses that are inherited in an autosomal recessive or dominant manner, whereas sporadic PD is considered to be a complex neurodegenerative disease entity with both genetic susceptibility and environmental factors contributing to the etiopathogenesis.
The possibility of mitochondrial DNA mutation related to PD has been mentioned. It has been shown that for the first time that mtDNA mutation levels in SN neurons are significantly elevated in this group of early stage PD and cases of incidental Lewy body disease, which is thought to represent presymptomatic PD [24]. Also, it was investigated that there are no inherited disease specific mtDNA mutations, hence individual homoplasmic mutations or very low grade heteroplasmic mutations in the vicinity of mitochondrial metabolism and OS may contribute to selective neuronal vulnerability in PD [25]. Moreover, it has not been yet proven the hypothesis that the accumulation of somatic mtDNA mutations in SN neurons contribute to the pathogenesis of PD [26].
Contributions of PARK PD-associated genes
Genetic forms of PD are overall rare but, serves as an excellent human model for the much more common idiopathic condition and enables the identification of at-risk individuals in the earliest [27]. In the current PD genetics nomenclature, 18 specific chromosomal regions are termed PARK, and 18 PD-related genetic loci (PARK1-18) were identified in chronological order [27]. Mutations described for these familial forms of PD, include autosomal dominant mutations of SNCA (PARK1, PARK4) [28], UCHL1 (PARK5), LRRK2 (PARK8) [29], HTRA2 (PARK13) [30] or autosomal recessive mutations of Parkin (PARK2) [31], PINK1 (PARK6) [32], DJ-1 (PARK7) [33] and ATP13A2 (PARK9) [34]. A list of the PARK PD-related genes is provided in Table 1. PARK genes are associated with PD. However, the pathogenic mechanisms underlying mitochondrial dysfunction in familial PD require further detailed investigation at the molecular level.
Table 1. Contributions of PARK-designated PD-associated genes.
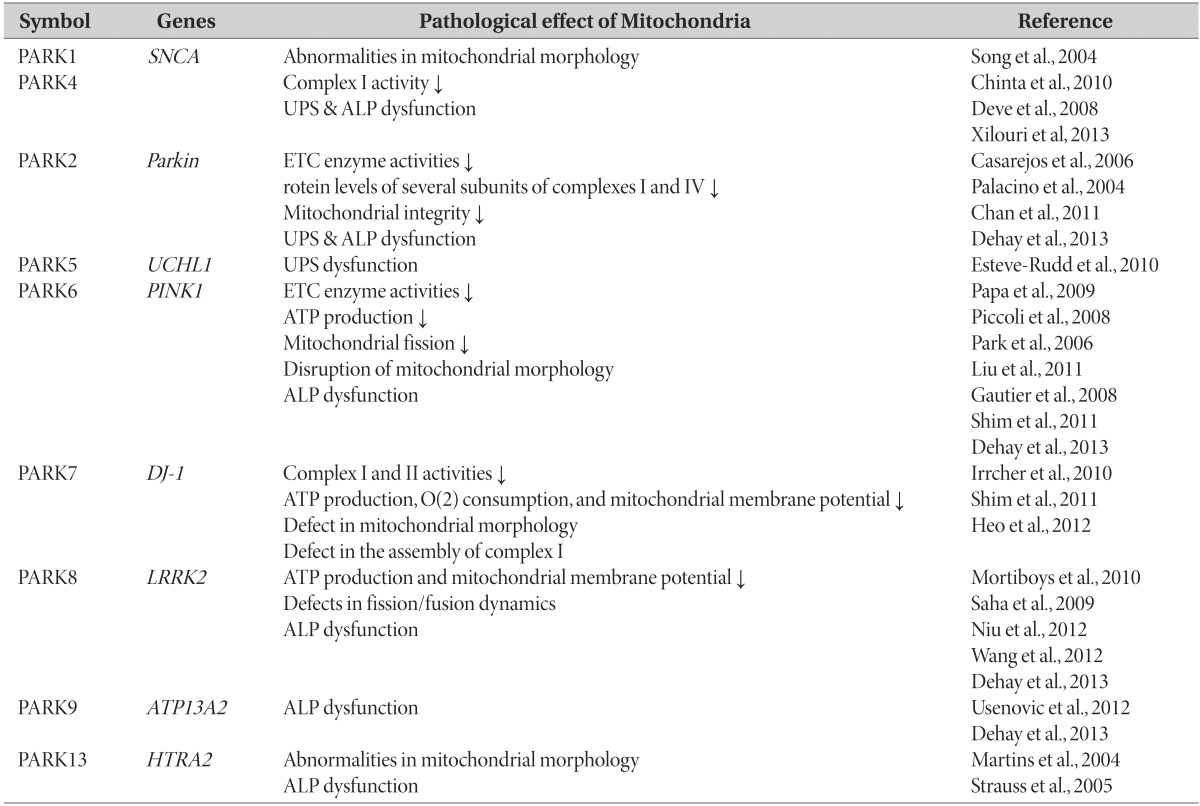
SNCA (PARK1, PARK4)
SNCA was the first gene with mutations to cause autosomal-dominant PD, which can be accumulated in mitochondria in the olfactory bulb, hippocampus, striatum, and thalamus. Mitochondrial abnormalities were observed in transgenic mouse models overexpressing wild type or mutant SNCA: selective oxidation of mitochondria-associated metabolic proteins; degenerating mitochondria containing SNCA; reduced complex IV activity; mitochondrial DNA damage; and increased mitochondrial pathology after treatment with MPTP. Particularily, mutant A53T human SNCA gene in mice caused mtDNA damage and respiratory complex IV impairment and increased sensitivity to MPTP and paraquat [35]. Also, overexpression of mutant A53T or wild type human SNCA in cell lines causes mitochondrial association and leads to cytochrome c release, enhanced mitochondria calcium and nitric oxide, and oxidative modification of mitochondrial components [35]. Moreover, higher accumulation of SNCA and decreased complex I activity were reported in mitochondria from striatum and substantia nigra (SN) of PD patients compared with normal subjects [36].
Parkin (PARK2)
Homozygous mutations in the Parkin gene were discovered in families with autosomal recessive PD (ARPD). In contrast to SNCA, Parkin mutations are common, and almost half of all cases of ARPD and those before 21 years of age [37]. The Parkin protein functions as a RING-type ubiquitin protein ligase, which possibly impairs the activity of the ubiquitin-proteasomal system (UPS) and antioxidant defenses, and enhances OS. Parkin also has a support role in maintaining mitochondrial function, which is involved in the regulation of mitochondrial morphology and has a hypothesized role in mitochondrial biogenesis in conjunction with PINK1, and overexpression of Parkin enhances transcription and replication of mitochondrial DNA [2,3]. Moreover, mitochondrial impairments are observed in the striatum of Parkin-null fruit flies [38]. Furthermore, Parkin-null mice have decreased levels of protein involved in respiratory chain function with reduced complex activity in the striatum [8]. A complex I deficit has been observed in lymphocytes collected from Parkin mutation positive patients [39].
UCHL1 (PARK5)
Ubiquitin carboxy-terminal hydrolase L1 (UCHL1) is a 223-a.a. protein which is a component of the UPS, which cleaves the carboxy-terminal peptide bond of polyubiquitine chains, working as a deubiquitinating enzyme [40]. It encodes for one of the most abundant proteins in the brain. Mutations in this target were found to be responsible for a genetic form of PD. It is thought a mutation at amino acid position 93 for methionine may decrease UCHL1 hydrolase activity, leading to accumulation of proteins that should have been degraded, and subsequently the progression of PD [41]. Moreover, inactivating oxidative modifications of this protein have been reported in PD post-mortem brains where it correlates with the formation of protein aggregates [42], and UCH-L1 together with parkin and α-synuclein are the major components of LB [43]. Although many studies have investigated the association between UCHL1 and the risk of PD, the results have been ambiguous. It was conducted a large-scale analysis to investigate the age-of-onset effect of the UCHL1 variant in PD among ethnic Chinese. This result demonstrated that UCHL1 variant and the interaction of UCHL1 variant and age at onset were not significantly associated with PD [44]. It was also suggested that UCHL1 S18Y is not a major susceptibility factor for PD in white populations although it cannot be excluded the possibility that the S18Y variant exerts weak effects on risk, particularly in early-onset disease [45]. Moreover, the current meta-analysis suggested no evidence for the association between the UCHL1 S18Y polymorphism and PD risk in the Asian population, especially in subgroups of ethnicity and age at onset [46].
PINK1 (PARK6)
Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) gene encodes a mitochondrially localized serine/threonine kinase and is sub-localized in different regions including inner mitochondrial membrane, intermembrane space and outer mitochondrial membrane. Recessive mutations in PINK1 were found to be responsible for a familial form of early-onset PD [47]. PINK1 knockout mouse and human dopaminergic neurons have abnormalities in mitochondrial morphology, reduced membrane potential, increased ROS generation and high sensitivity to apoptosis. Mutations in the PINK1 genes result in enlarged or swollen mitochondria. Moreover, mice null for Pink1 or Parkin exhibit synaptic dysfunction in neurons projecting to the striatum, and this synaptic dysfunction correlates with progressive loss of mitochondrial function and increased OS in the striatum with age [48]. Also, loss-of-function mutations in PINK1 or Parkin have been associated with mitochondrial dysfunction in cells from patients with familial forms of parkinsonism [48]. PINK1 plays a strong cytoprotective role in maintaining mitochondrial homeostasis via different mechanisms. Overexpression of wild type PINK1 in SH-SY5Y neuroblastoma cells stabilizes respiring mitochondrial networks such as maintenance of mitochondrial membrane potential and suppression of autophagy [49]. Moreover, PINK1 has been shown to phosphorylate the mitochondrial molecular chaperone heat shock protein 75 kDa [50]. Loss of PINK1 leads to severe alterations in mitochondrial homeostasis as evidenced by aberrations in mitochondrial cytoarchitecture, mitochondrial dynamics, calcium homeostasis, biosynthetic pathways, and increased mitochondrial ROS, ultimately inducing a robust increase in mitophagy [51].
DJ-1 (PARK7)
DJ-1 protein is located mostly in the cytosol, and only a fraction is present in the nucleus and mitochondria, where it preferentially partitions to the matrix and intermembrane space of mitochondria and it impairs degradation. Upon OS, DJ-1 rapidly translocates to the mitochondria and, to a lesser extent, to the nucleus, acting as a neuroprotective intracellular redox sensor [48,52]. DJ-1 knockout in fly and mice produced decreased mtDNA levels, respiratory control ratio, and ATP levels, and DJ-1-deficient mice are more sensitive to MPTP-induced loss of dopamine neurons [48,52]. Moreover, DJ-1 and its mutants were found to be strongly associated with Hsp70, and a mitochondria-resident Hsp70 complex in patients with PD. In vitro, association of wild-type DJ-1 with mitochondrial Hsp70 was further increased under OS, indicating that the translocation of DJ-1 to mitochondria may occur by binding to mitochondrial chaperones. DJ-1 inhibits the aggregation and toxicity of α-synuclein by increasing nuclear factor-like 2, a direct interaction is not known; DJ-1 binds to PINK1 and increases PINK1 levels under conditions of PINK1 overexpression; DJ-1 and Parkin interact under conditions of OS. DJ-1 activates transcription of the Mn-SOD gene, which encodes an essential mitochondrial antioxidant enzyme. Furthermore, reduction of DJ-1 was associated with lowered MMP, an increase in mitochondria fragmentation, autophagy and OS, and reduced mitochondrial fusion. DJ-1 was also found to directly bind to the mitochondrial complex I subunits; and loss of function of DJ-1 led to a decrease of mitochondrial complex I activity, but not complex III; whereas overexpression of DJ-1 conferred protection against the complex I inhibitor MPTP [48,52].
LRRK2 (PARK8)
Leucine-rich repeat kinase 2 (LRRK2) pathogenic mutations also cause neurodegeneration. LRRK2 is associated with the mitochondrial outer membrane but the function of LRRK2 in relation to mitochondria is presently unclear. LRRK2 immunoreactivity was shown to partially overlap with mitochondrial and lysosomal markers in the mammalian brain, and ultrastructural analysis revealed that LRRK2 is associated with intracellular membranes, including lysosomes, transport vesicles, and mitochondria [48,53].
Approximately 10% of wild-type and mutant LRRK2 were present in the mitochondrial fraction in cells overexpressing the proteins. Patients with a G2019S mutation had a decrease in MMP and ATP. Overexpression of G2019S mutant LRRK2 in differentiated human neuroblastoma cells caused neurite retraction and shortening, which correlated with increased autophagy. LRRK2 G2019S mutation increases the kinase activity of LRRK2 and others are not clear yet. Overexpression of either wild-type or mutant (R1441C or G2019S) LRRK2 caused mitochondrial fragmentation, reduced mitochondrial fusion, and increased Dynamin related protein 1 (Drp1) recruitment to mitochondria by direct interaction with LRRK2 in vitro [48,53].
ATP13A2 (PARK9)
Autosomal recessive mutations in lysosomal type 5 P-type ATPase (ATP13A2) were first identified in 2006 in a Chilean family and are associated with a juvenile-onset, levodopa-responsive type of parkinsonism called Kufor-Rakeb syndrome (KRS): c.3057delC (p.1019GfsX1021), c.1306+5G>A (p.G399_L435del) and c.1632_ 1653dup22 (p.Leu552fsX788) [54].
Mutant ATP13A2 proteins are retained in the endoplasmic reticulum and degraded by the proteasome [34]. Recently, it has been reported that ATP13A2 suppresses α-synuclein and manganese toxicity in primary neuron cultures [54].
HTRA2 (OMI, PARK13)
HtrA serine peptidase 2 (HTRA2) is consisted of 458 amino acids localized to mitochondria, released during mitochondrial membrane permeabilization in programmed cell death [30,56]. Cells overexpressing HTRA2 mutant with G399S have shown mitochondrial morphological changes followed by dysfunction and increased susceptibility against OS [30,55]. Moreover, wild type HTRA2 activates autophagy through digestion of Hax-1, a Bcl-2 family related protein that represses autophagy via Beclin-1 inhibition, suggesting an insufficient protein degradation system may play a key role [56].
However, PARK3, PARK10, and PARK12 are uncertain and not confirmed [27]. Sequencing of the genes within the PARK16 locus in a British cohort of 182 pathologically proven PD cases revealed the presence of two novel mutations; in one patient in the RAB7L1 (K157R) and in another patient in the SLC41A1 (A350V) gene [57]. Furthermore, mutations in GYGYF2 (PARK11), PLA2G6 (PARK14), FBXO7 (PARK15), VPS35 (PARK17), EIF4G1 (PARK18), DNAJC6 (PARK19), SYNJ1 (PARK20) and recently reported causative genes as RAB39B and CHCHD2 cause PD that is an inconsistent or only a minor feature of a more complex phenotype or are a very rare cause of PD [27,58,59,60].
The pathophysiology of mitochondrial dysfunction
Mitochondrial ubiquitin-proteasome system (UPS) dysfunction
The UPS is responsible for a highly selective degradation of short-lived intracellular and plasma membrane proteins under basal metabolic conditions, as well as misfolded or damaged proteins in the cytosol, nucleus or endoplasmic reticulum. The system involves the targeting of susceptible proteins by ubiquitin and only the unfolded ubiquitinated proteins can pass through the narrow pore of the proteasome barrel. Dysfunction of the UPS and the resultant accumulation of misfolded proteins have been strongly implicated in the pathogenesis of PD, which in combination with oxidative danger will lead to the death of dopaminergic neurons. Increased OS can lead to impaired function of the UPS, thereby affecting cell survival. PINK1, Parkin and DJ-1 would regulate the normal function of mitochondria, whereas α-synuclein, Parkin, UCHL1 are involved in maintaining the UPS [61]. Parkin mutations in autosomal recessive juvenile parkinsonism showed a decrease in ubiquitin-ligase enzymatic activity in the substantia nigra, which also support the hypothesis that failure of the UPS leads to the neurodegeneration underlying PD [62]. Evidence from cell and animal models suggests that α-synuclein aggregation can elicit mitochondrial damage, therefore ultimately leads to cellular damage. Monomeric α-synuclein may also influence mitochondrial activity and dynamics. Aggregation of α-synuclein clearly decreased from complex-I inhibition, increased OS and ATP synthesis deficit, thus all of which can interfere with the normal function of the UPS [62]. This observation leads to a large degree of cross correlation between mitochondria and UPS, and disease-related mutations in these genes will lead to the degeneration of DA neurons.
Mitochondrial dynamics - fusion, fission and mitophagy
Mitochondria were once thought to be rigidly structured, but these organelles fuse and divide and undergo regulated turnover. These dynamic processes include fusion, fission, and mitophagy [63]. Mitochondrial dynamics is important for neurotransmission, synaptic maintenance and neuronal survival, which demonstrated that neuronal dysfunction can ensure if mitochondrial dynamics are disrupted [64]. Fusion seems to be required for a normal mitochondrial function as this process is likely to protect function by providing a chance for mitochondria to mix their contents, thus enabling protein complementation, mtDNA repair and equal distribution of metabolites [65]. Fission seems to be crucial for mitochondrial function as it facilitates equal segregation of mitochondria into daughter cells in cellular division and to enhance distribution of mitochondria along cytoskeletal tracks. Also, it plays a key role in the targeting of damaged segments of mitochondria to the autophagic process, playing a housekeeping role in the cell. The mitochondrial fragmentation induction by toxins used to study PD was demonstrated that both rotenone and MPP+ induced mitochondria fission in rat dopaminergic cell line N27 [66].
In addition, the autophagy lysosomal pathway (ALP) can be divided by macroautophagy (generally referred to as autophagy), microautophagy and chaperone-mediated autophagy (CMA). Autophagy-mediated protein degradation is an important component of the protein quality control system in the cell that is mobilized upon the accumulation of misfolded, damaged, or unnecessary proteins. Autophagy can be induced within short periods of nutrient deprivation, and CMA can be induced after prolonged nutrient deprivation, while microautophagy is not activated by nutritional deprivation or stress. In contrast to the UPS, the ALP plays an important role in maintaining cellular homeostasis by degrading bulky cytoplasmic material including damaged organelles and misfolded and accumulated proteins. This degradation pathway appears crucial for clearance of aggregated proteins that represent a pathological hallmark of several neurodegenerative disorders including PD. In addition to the UPS, α-synuclein is also cleared by autophagy, which supports the hypothesis that impaired autophagic degradation of α-synuclein is an important mechanism of neurodegeneration in PD [67]. Furthermore, other PD-related genes, such as LRRK2, Parkin, and PINK1, have been mechanistically linked to alterations in ALP. PD-related mutations/deficiency in the ATP13A2 also lead to a general lysosomal impairment characterized by lysosomal membrane instability, impaired lysosomal acidification, decreased processing of lysosomal enzymes, reduced degradation of lysosomal substrates, and diminished clearance of autophagosomes, collectively contributing to α-synuclein accumulation and cell death [68]. Apoptosis contributes to DA neuronal loss in the SNc of PD patients as well as in neurotoxin models, whereas autophagy has been suggested as an alternative mechanism of cell death in neurotoxin models [69], in the familial PD gene mutant model [70] and in human PD brains [71]. Decreased expression of genes that regulate autophagy can cause neurodegenerative diseases in which deficient quality control results in inflammation and in the death of neuronal cell populations. One feature of the mammalian target of rapamycin (mTOR) kinase inhibition is autophagy. Thus, a combination of mitochondrial dysfunction and insufficient autophagy may contribute to PD pathogenesis [72].
Mitophagy, the selective autophagy process of mitochondria, is a critical quality control mechanism required to eliminate damaged or excess mitochondria [73]. Defective mitophagy has been suggested as one of the major pathological mechanisms underlying mitochondrial dysfunction in autosomal recessive forms of PD, including those caused by mutations in PINK1 and Parkin [73]. The PINK1-Parkin signaling model has become a new paradigm for priming damaged mitochondria for further degradation by acting as a sensor and effector pair [74,75]. Narendra et al. [73] observed that parkin is targeted to dysfunctional mitochondria and induces their degradation by mitophagy. When CCCP (carbonyl cyanide 3-chlorophenylhydrazone), an uncoupling agent that dissipates the mitochondrial membrane potential, was added to HeLa cells, a robust recruitment of overexpressed parkin to uncoupled mitochondria was observed after 1 h of treatment. Strikingly, upon prolonged exposure to CCCP, mitochondria were cleared from parkin-expressing cells within 24 h. In a next step, the role of PINK1 in parkin-mediated mitochondrial degradation was analyzed. Several groups found that expression of PINK1 is essential for the recruitment of parkin to dysfunctional mitochondria. Downregulation of PINK1 by RNA interference resulted in decreased parkin recruitment to mitochondria following CCCP treatment. Also, parkin targeting to mitochondria was completely abolished in fibroblasts from PINK1 KO mice and could be rescued by reintroduction of wild-type PINK1. Thus, in PINK1-deficient cells, parkin cannot induce mitophagy after CCCP treatment [76]. Moreover, overexpression of PINK1 or targeting of PINK1 to mitochondria via a heterologous mitochondrial membrane anchor derived from Tom20 or OPA3 was shown to be sufficient for the mitochondrial recruitment of parkin [75]. In addition, the stabilization of PINK1 at mitochondria is thought to trigger parkin translocation to uncoupled mitochondria, and therefore, it has been of great interest to identify the protease(s) mediating PINK1 cleavage. Studies in Drosophila have shown a genetic link between PINK1, parkin, and Rhomboid-7, the fly homolog of the presenilin-associated rhomboid-like protease (PARL), suggesting that PARL mediates PINK1 cleavage [77]. Fibroblasts derived from PARL deficient mice show an altered PINK1 cleavage pattern that can be reconstituted by reintroducing wild-type PARL, but not by a catalytically inactive PARL mutant [78]. However, PARL seems not to be the only PINK1-cleaving enzyme, as processed PINK1 isoforms are present in fibroblasts from PARL KO mice [78]. PARL is a transmembrane protein at the inner mitochondrial membrane catalyzing intramembrane proteolysis, while PINK1 seems to be anchored to the outer mitochondrial membrane; thus, the exact molecular mechanism of PARL-induced PINK1 cleavage remains to be demonstrated.
Mitochondrial α-synuclein fibrillisation and aggregation
Mutation, nitration, DA modification or abnormal accumulation of α-synuclein can lead to the formation of protofibrils. Natively unfolded or disordered α-synuclein monomers form β-sheet rich oligomers that comprise a transient population of protofibrils of heterogenous structure that may include spheres, chains, or rings. The protofibrils may give rise to more stable amyloid-like fibrils, which is called as α-synuclein fibrillisation. α-synuclein fibrils eventually aggregate and precipitate to form Lewy bodies (LBs) in vivo. α-synuclein protofibrils can induce leakage of DA, and inhibit complex I in the mitochondria and proteasome in the UPS. α-synuclein protofibrils can also increase permeability of mitochondria which can result into the leakage of pro-apoptotic molecules that can promote neurodegeneration [79].
Mitochondria, calcium and cell death
Mitochondria sequester calcium when intracellular calcium levels rise during the excitotoxic process. The threshold for excitotoxicity might decrease if mitochondrial ATP production is impaired. Mitochondria also have a pivotal role in apoptotic cell death. Mitochondrial release of cytochrome c and other 'pro-apoptotic factors' such as apoptosis-initiating factor into the cytoplasm triggers a cascade of events, culminating in cell death [2].
USE OF SURROGATE TISSUES IN STUDYING PD PATHOGENESIS
So far, the limited availability of dopaminergic cell lines has prevented investigation of mitochondrial abnormalities in PD. In order to overcome this problem, surrogate tissues could be useful in studying PD pathology. Recent study employed an initial survey of primary skin fibroblasts from PARK6 patients with microarray technology to define the molecular events resulting from the loss-of-function mutations of the mitochondrial kinase PINK1 [80]. This information indicates that loss of function of PINK1 lead to mitochondrial dysfunction by the loss of phosphorylation of mitochondrial proteins and following oxidative and cellular stress triggers the upregulation of α-synuclein expression. Elevated expression of α-synuclein might underlie the formation of "Lewy bodies" in the brain of most PD variants, representing a central pathogenic event for PD. Also, to investigate the cytoprotective physiological function of PINK1, primary fibroblasts from three patients stably overexpressing GFP-tagged wild type PINK1 were used. This result indicate that PINK1 plays an important and specific physiological role in protecting cells from proteasomal stress, and suggest that PINK1 might exert its cytoprotective effects upstream of mitochondria engagement [80]. Previously, transcriptome surveys of PD patient SN genes demonstrated the importance of cell adhesion and extracellular matrix in the pathogenesis of PD [81]. This skin fibroblast data are in good agreement with the observation from brain tissues in PD patients in that some impairment of synaptic integrity accompanies α-synuclein upregulation.
Multipotent MSCs are fibroblast-like plastic adherent cells that can be isolated from a variety of tissues, such as bone marrow, periosteum, trabecular bone, adipose tissue, synovium, skeletal muscle, dental pulp, and other tissues. MSCs are defined by their potential to differentiate into osteoblasts, chondrocytes, neurons, skeletal muscle cells, endothelial cells and vascular smooth muscle cells in vitro [82] and undergo differentiation in vivo [83]. Human adipose tissue is a rich source of MSCs, providing an abundant and accessible source of adult stem cells [84]. Adipose tissue can be obtained by less invasive methods and in larger quantities than bone marrow cells. Moreover, the advantage of adipose tissue as a source of multi-lineage cells is its relative abundance and ease of procurement by local excision or suction-assisted liposuction as well as appealing source of donor tissue for autologous cell replacement [84]. Therefore, MSCs from human adipose tissue represent a useful source of progenitor cells for cell therapy and tissue engineering [83]. Here, we isolated human adipose tissue-derived mesenchymal stromal cells (hAD-MSCs) of patients with idiopathic PD, named as "PD", with Parkin deficient PD, named as "Parkin" and with pituitary adenoma, named as "non-PD" shortly. Based on the previous studies, we hypothesize that an approach of carrying out a transcriptome microarray analysis using early-passage adipose tissue-derived mesenchymal stromal cell cultures from human adult patients with early-onset hereditary Parkin deficient PD as well as late-onset idiopathic PD has potential benefit for understanding of brain pathology in PD patients. We also propose that the expression changes described might potentially lead to better understanding of PD pathology and development of early diagnosis and effective therapy targeted their human biomarkers. Moreover, for the first time, to overcome the cellular senescence of primary cultured AD-MSCs from PD patients, we established hTERT-immortalized wild type, idiopathic and Parkin deficient mesenchymal stromal cell lines isolated from the adipose tissues of PD patients [85]. The pGRN145 plasmid containing hTERT was introduced to establish telomerase immortalized cells. The established hTERT-immortalized cell lines showed chromosomal aneuploidy sustained stably over two-years. The morphological study of mitochondria in the primary and immortalized hAD-MSCs showed that the mitochondria of the non-PD were normal; however, those of the PD and Parkin were gradually damaged. A striking decrease in mitochondrial complex I, II, and IV activities was observed in the hTERT-immortalized cells from the patients with idiopathic and Parkin-defect PD. Comparative Western blot analyses were performed to investigate the expressions of PD specific marker proteins in the hTERT-immortalized cell lines. Then, we used these immortalized cell lines to investigate mitochondrial abnormalities in the idiopathic and Parkin-deficient PD cell lines compared to the wild type cell line. This study suggests that the hTERT-immortalized hAD-MSC cell lines established from patients with idiopathic and familial Parkin-deficient PD could be good cellular models to evaluate mitochondrial dysfunction to better understand the pathogenesis of PD and to develop early diagnostic markers and effective therapy targets for the treatment of PD.
CLINICAL MITOCHONDRIAL THERAPEUTICS IN PD
The mitochondria represent a highly promising target for the development of PD biomarkers. Biochemical detection methods of potential biomarkers in PD are classified as genetic screening, mitochondrial complex I measurement, and α-synuclein levels and isoforms in blood [2,3]. Commercial genetic testing is available for mitochondrial gene mutations such as Parkin, PINK1 and SNCA gene mutations [2,3]. Moreover, our molecular study has established the utility of the hAD-MSCs of patients with idiopathic PD, with Parkin deficient PD and with non-PD for the high-throughput microarray analysis, and that identified differentially expressed groups of genes in idiopathic and Parkin deficiency PD compared with non-PD patients [86]. Hence, these newly identified genes that are regulated in non-PD, PD and Parkin, are likely to help to develop valuable molecular tools to understand PD pathology, to facilitate early diagnosis of PD, and eventually to come up with human potential biomarkers for PD.
So far, some compounds with neuroprotective potential, including a few ordinary mitochondrial modulators such as creatine (N-aminoiminomethyl-N-methylglycine) and coenzyme Q10 (CoQ10), have been currently being investigated in clinical trials of PD. Creatine is an endogenous nitrogenous guanidine compound involved in supplying energy to all cells in the body, especially muscle and nerve cells [87]. Several groups have reported the neuroprotective effect of creatine supplementation in an MPTP-induced mouse model of PD [88]. However, creatine treatment has not been shown to have a significant effect on Unified Parkinson's Disease Rating Scale scores in human clinical trials [89]. Also, CoQ10 is a lipid-soluble electron carrier in the electron transport chain (ETC), and the reduced form of CoQ10, ubiquinol, is an important antioxidant in mitochondrial and lipid membranes [90]. Administration of CoQ10 has significantly protective effects on dopaminergic neuron in that it inhibits dopamine depletion and α-synuclein aggregation in acute and chronic MPTP-induced mice models of PD. The efficacy of CoQ10 is still controversial, with varying outcomes depending on dosage [91]. The applications of mitochondria-targeted antioxidant compounds (MitoQ, Mito PBN, MitoVitE) and peptides (SS-31, SS-20) to PD are in the early stages [92]. Therefore, it is still required to identify novel therapeutic targets in dysfunctional mitochondria in PD.
CONCLUSIONS
Numerous studies conducted using various genetic and toxin models of PD have contributed that mitochondria dysfunction play a crucial role in pathophysiology. Mitochondrial function is regulated by a complex network of sensors and effectors, by which regulatory mitochondrial function, such as fusion and fission, mitophagy and the mitochondrial UPR, have recently been elucidated. The investigation of mitochondrial homeostatic processes as a model of PD is useful for proposing the therapeutic interventions aiming for a cure to PD.
ACKNOWLEDGEMENTS
This study was supported by the Korea Institute of Planning & Evaluation for Technology in Food, Agriculture, Forestry, and Fisheries, Republic of Korea (311011-05-3-SB020), by the Korea Healthcare Technology R&D Project (HI10C14110400, HI12C02050101, HI11C21100200) funded by Ministry of Health & Welfare, Republic of Korea, and by the Technology Innovation Program (10050154, Business Model Development for Personalized Medicine Based on Integrated Genome and Clinical Information) funded By the Ministry of Trade, industry & Energy (MI, Korea).
References
- 1.Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038–3062. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 3.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 4.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayeux R, Denaro J, Hemenegildo N, Marder K, Tang MX, Cote LJ, Stern Y. A population-based investigation of Parkinson's disease with and without dementia. Relationship to age and gender. Arch Neurol. 1992;49:492–497. doi: 10.1001/archneur.1992.00530290076015. [DOI] [PubMed] [Google Scholar]
- 6.Engelhardt JF. Redox-mediated gene therapies for environmental injury: approaches and concepts. Antioxid Redox Signal. 1999;1:5–27. doi: 10.1089/ars.1999.1.1-5. [DOI] [PubMed] [Google Scholar]
- 7.Shim JH, Yoon SH, Kim KH, Han JY, Ha JY, Hyun DH, Paek SH, Kang UJ, Zhuang X, Son JH. The antioxidant Trolox helps recovery from the familial Parkinson's disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion. 2011;11:707–715. doi: 10.1016/j.mito.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 9.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 11.Thyagarajan D, Bressman S, Bruno C, Przedborski S, Shanske S, Lynch T, Fahn S, DiMauro S. A novel mitochondria 12SrRNA point mutation in parkinsonism, deafness, and neuropathy. Ann Neurol. 2000;48:730–736. [PubMed] [Google Scholar]
- 12.Hindle JV. Ageing, neurodegeneration and Parkinson's disease. Age Ageing. 2010;39:156–161. doi: 10.1093/ageing/afp223. [DOI] [PubMed] [Google Scholar]
- 13.Levy G. The relationship of Parkinson disease with aging. Arch Neurol. 2007;64:1242–1246. doi: 10.1001/archneur.64.9.1242. [DOI] [PubMed] [Google Scholar]
- 14.Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- 15.Fabre E, Monserrat J, Herrero A, Barja G, Leret ML. Effect of MPTP on brain mitochondrial H2O2 and ATP production and on dopamine and DOPAC in the striatum. J Physiol Biochem. 1999;55:325–331. [PubMed] [Google Scholar]
- 16.Desai VG, Feuers RJ, Hart RW, Ali SF. MPP(+)-induced neurotoxicity in mouse is age-dependent: evidenced by the selective inhibition of complexes of electron transport. Brain Res. 1996;715:1–8. doi: 10.1016/0006-8993(95)01255-9. [DOI] [PubMed] [Google Scholar]
- 17.Piao Y, Kim HG, Oh MS, Pak YK. Overexpression of TFAM, NRF-1 and myr-AKT protects the MPP(+)-induced mitochondrial dysfunctions in neuronal cells. Biochim Biophys Acta. 2012;1820:577–585. doi: 10.1016/j.bbagen.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Burté F, De Girolamo LA, Hargreaves AJ, Billett EE. Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. J Proteome Res. 2011;10:1974–1986. doi: 10.1021/pr101211k. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Zhou JY, Chin MH, Schepmoes AA, Petyuk VA, Weitz KK, Petritis BO, Monroe ME, Camp DG, Wood SA, Melega WP, Bigelow DJ, Smith DJ, Qian WJ, Smith RD. Region-specific protein abundance changes in the brain of MPTP-induced Parkinson's disease mouse model. J Proteome Res. 2010;9:1496–1509. doi: 10.1021/pr901024z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 21.Cochemé HM, Murphy MP. Complex I is the major site of mitochondrial superoxide production by paraquat. J Biol Chem. 2008;283:1786–1798. doi: 10.1074/jbc.M708597200. [DOI] [PubMed] [Google Scholar]
- 22.Thiruchelvam M, Prokopenko O, Cory-Slechta DA, Buckley B, Mirochnitchenko O. Overexpression of superoxide dismutase or glutathione peroxidase protects against the paraquat + maneb-induced Parkinson disease phenotype. J Biol Chem. 2005;280:22530–22539. doi: 10.1074/jbc.M500417200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Fitsanakis VA, Gu G, Jing D, Ao M, Amarnath V, Montine TJ. Manganese ethylene-bis-dithiocarbamate and selective dopaminergic neurodegeneration in rat: a link through mitochondrial dysfunction. J Neurochem. 2003;84:336–346. doi: 10.1046/j.1471-4159.2003.01525.x. [DOI] [PubMed] [Google Scholar]
- 24.Lin MT, Cantuti-Castelvetri I, Zheng K, Jackson KE, Tan YB, Arzberger T, Lees AJ, Betensky RA, Beal MF, Simon DK. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol. 2012;71:850–854. doi: 10.1002/ana.23568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter G, Sonnenschein A, Grünewald T, Reichmann H, Janetzky B. Novel mitochondrial DNA mutations in Parkinson's disease. J Neural Transm. 2002;109:721–729. doi: 10.1007/s007020200060. [DOI] [PubMed] [Google Scholar]
- 26.Clark J, Dai Y, Simon DK. Do Somatic Mitochondrial DNA Mutations Contribute to Parkinson's Disease? Parkinsons Dis. 2011;2011:659694. doi: 10.4061/2011/659694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klein C, Westenberger A. A Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 29.Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- 30.Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, Abuin A, Grau E, Geppert M, Livi GP, Creasy CL, Martin A, Hargreaves I, Heales SJ, Okada H, Brandner S, Schulz JB, Mak T, Downward J. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizuno Y, Hattori N, Kubo S, Sato S, Nishioka K, Hatano T, Tomiyama H, Funayama M, Machida Y, Mochizuki H. Progress in the pathogenesis and genetics of Parkinson's disease. Philos Trans R Soc Lond B Biol Sci. 2008;363:2215–2227. doi: 10.1098/rstb.2008.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56:424–427. doi: 10.1002/ana.20251. [DOI] [PubMed] [Google Scholar]
- 33.Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, Brice A, van Duijn CM, Oostra B, Meco G, Heutink P. DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci. 2003;24:159–160. doi: 10.1007/s10072-003-0108-0. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 35.Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog Neurobiol. 2013;106-107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010;486:235–239. doi: 10.1016/j.neulet.2010.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lücking CB, Dürr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denèfle P, Wood NW, Agid Y, Brice A French Parkinson's Disease Genetics Study Group; European Consortium on Genetic Susceptibility in Parkinson's Disease. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 38.Han JY, Kim JS, Son JH. Mitochondrial homeostasis molecules: regulation by a trio of recessive Parkinson's disease genes. Exp Neurobiol. 2014;23:345–351. doi: 10.5607/en.2014.23.4.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schapira AH. Mitochondrial dysfunction in Parkinson's disease. Cell Death Differ. 2007;14:1261–1266. doi: 10.1038/sj.cdd.4402160. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 41.Contu VR, Kotake Y, Toyama T, Okuda K, Miyara M, Sakamoto S, Samizo S, Sanoh S, Kumagai Y, Ohta S. Endogenous neurotoxic dopamine derivative covalently binds to Parkinson's disease-associated ubiquitin C-terminal hydrolase L1 and alters its structure and function. J Neurochem. 2014;130:826–838. doi: 10.1111/jnc.12762. [DOI] [PubMed] [Google Scholar]
- 42.Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem. 2004;279:13256–13264. doi: 10.1074/jbc.M314124200. [DOI] [PubMed] [Google Scholar]
- 43.Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol. 2002;160:1655–1667. doi: 10.1016/S0002-9440(10)61113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan EK, Lu CS, Peng R, Teo YY, Wu-Chou YH, Chen RS, Weng YH, Chen CM, Fung HC, Tan LC, Zhang ZJ, An XK, Lee-Chen GJ, Lee MC, Fook-Chong S, Burgunder JM, Wu RM, Wu YR. Analysis of the UCHL1 genetic variant in Parkinson's disease among Chinese. Neurobiol Aging. 2010;31:2194–2196. doi: 10.1016/j.neurobiolaging.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Hutter CM, Samii A, Factor SA, Nutt JG, Higgins DS, Bird TD, Griffith A, Roberts JW, Leis BC, Montimurro JS, Kay DM, Edwards KL, Payami H, Zabetian CP. Lack of evidence for an association between UCHL1 S18Y and Parkinson's disease. Eur J Neurol. 2008;15:134–139. doi: 10.1111/j.1468-1331.2007.02012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun S, Zhao Y, Jin G, Kang H. Lack of association between UCHL1 S18Y gene polymorphism and Parkinson's disease in the Asian population: a meta-analysis. Neurol Sci. 2014;35:1867–1876. doi: 10.1007/s10072-014-1973-4. [DOI] [PubMed] [Google Scholar]
- 47.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 48.Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342:619–630. doi: 10.1124/jpet.112.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Exner N, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Krüger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lev N, Roncevic D, Ickowicz D, Melamed E, Offen D. Role of DJ-1 in Parkinson's disease. J Mol Neurosci. 2006;29:215–225. doi: 10.1385/jmn:29:3:215. [DOI] [PubMed] [Google Scholar]
- 53.Martin I, Kim JW, Dawson VL, Dawson TM. LRRK2 pathobiology in Parkinson's disease. J Neurochem. 2014;131:554–565. doi: 10.1111/jnc.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Müller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Krüger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 56.Li B, Hu Q, Wang H, Man N, Ren H, Wen L, Nukina N, Fei E, Wang G. Omi/HtrA2 is a positive regulator of autophagy that facilitates the degradation of mutant proteins involved in neurodegenerative diseases. Cell Death Differ. 2010;17:1773–1784. doi: 10.1038/cdd.2010.55. [DOI] [PubMed] [Google Scholar]
- 57.Tucci A, Nalls MA, Houlden H, Revesz T, Singleton AB, Wood NW, Hardy J, Paisán-Ruiz C. Genetic variability at the PARK16 locus. Eur J Hum Genet. 2010;18:1356–1359. doi: 10.1038/ejhg.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonifati V. Genetics of Parkinson's disease--state of the art, 2013. Parkinsonism Relat Disord. 2014;20(Suppl 1):S23–S28. doi: 10.1016/S1353-8020(13)70009-9. [DOI] [PubMed] [Google Scholar]
- 59.Wilson GR, Sim JC, McLean C, Giannandrea M, Galea CA, Riseley JR, Stephenson SE, Fitzpatrick E, Haas SA, Pope K, Hogan KJ, Gregg RG, Bromhead CJ, Wargowski DS, Lawrence CH, James PA, Churchyard A, Gao Y, Phelan DG, Gillies G, Salce N, Stanford L, Marsh AP, Mignogna ML, Hayflick SJ, Leventer RJ, Delatycki MB, Mellick GD, Kalscheuer VM, D'Adamo P, Bahlo M, Amor DJ, Lockhart PJ. Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am J Hum Genet. 2014;95:729–735. doi: 10.1016/j.ajhg.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Funayama M, Ohe K, Amo T, Furuya N, Yamaguchi J, Saiki S, Li Y, Ogaki K, Ando M, Yoshino H, Tomiyama H, Nishioka K, Hasegawa K, Saiki H, Satake W, Mogushi K, Sasaki R, Kokubo Y, Kuzuhara S, Toda T, Mizuno Y, Uchiyama Y, Ohno K, Hattori N. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol. 2015;14:274–282. doi: 10.1016/S1474-4422(14)70266-2. [DOI] [PubMed] [Google Scholar]
- 61.Heo JM, Rutter J. Ubiquitin-dependent mitochondrial protein degradation. Int J Biochem Cell Biol. 2011;43:1422–1426. doi: 10.1016/j.biocel.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ebrahimi-Fakhari D, Wahlster L, McLean PJ. Protein degradation pathways in Parkinson's disease: curse or blessing. Acta Neuropathol. 2012;124:153–172. doi: 10.1007/s00401-012-1004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi: 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McInnes J. Insights on altered mitochondrial function and dynamics in the pathogenesis of neurodegeneration. Transl Neurodegener. 2013;2:12. doi: 10.1186/2047-9158-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santos D, Cardoso SM. Mitochondrial dynamics and neuronal fate in Parkinson's disease. Mitochondrion. 2012;12:428–437. doi: 10.1016/j.mito.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 66.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xilouri M, Brekk OR, Stefanis L. α-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47:537–551. doi: 10.1007/s12035-012-8341-2. [DOI] [PubMed] [Google Scholar]
- 68.Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E. Lysosomal impairment in Parkinson's disease. Mov Disord. 2013;28:725–732. doi: 10.1002/mds.25462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- 70.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 72.Choi KC, Kim SH, Ha JY, Kim ST, Son JH. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J Neurochem. 2010;112:366–376. doi: 10.1111/j.1471-4159.2009.06463.x. [DOI] [PubMed] [Google Scholar]
- 73.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schapira AH. Targeting mitochondria for neuroprotection in Parkinson's disease. Antioxid Redox Signal. 2012;16:965–973. doi: 10.1089/ars.2011.4419. [DOI] [PubMed] [Google Scholar]
- 75.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pilsl A, Winklhofer KF. Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson's disease. Acta Neuropathol. 2012;123:173–188. doi: 10.1007/s00401-011-0902-3. [DOI] [PubMed] [Google Scholar]
- 77.Whitworth AJ, Lee JR, Ho VM, Flick R, Chowdhury R, McQuibban GA. Rhomboid-7 and HtrA2/Omi act in a common pathway with the Parkinson's disease factors Pink1 and Parkin. Dis Model Mech. 2008;1:168–174. doi: 10.1242/dmm.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shi G, Lee JR, Grimes DA, Racacho L, Ye D, Yang H, Ross OA, Farrer M, McQuibban GA, Bulman DE. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson's disease. Hum Mol Genet. 2011;20:1966–1974. doi: 10.1093/hmg/ddr077. [DOI] [PubMed] [Google Scholar]
- 79.Bandopadhyay R, de Belleroche J. Pathogenesis of Parkinson's disease: emerging role of molecular chaperones. Trends Mol Med. 2010;16:27–36. doi: 10.1016/j.molmed.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 80.Hoepken HH, Gispert S, Azizov M, Klinkenberg M, Ricciardi F, Kurz A, Morales-Gordo B, Bonin M, Riess O, Gasser T, Kögel D, Steinmetz H, Auburger G. Parkinson patient fibroblasts show increased alpha-synuclein expression. Exp Neurol. 2008;212:307–313. doi: 10.1016/j.expneurol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 81.Grünblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm. 2004;111:1543–1573. doi: 10.1007/s00702-004-0212-1. [DOI] [PubMed] [Google Scholar]
- 82.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 83.Bruder SP, Jaiswal N, Ricalton NS, Mosca JD, Kraus KH, Kadiyala S. Mesenchymal stem cells in osteobiology and applied bone regeneration. Clin Orthop Relat Res. 1998:S247–S256. doi: 10.1097/00003086-199810001-00025. [DOI] [PubMed] [Google Scholar]
- 84.Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–4295. doi: 10.1091/mbc.E02-02-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moon HE, Yoon SH, Hur YS, Park HW, Ha JY, Kim KH, Shim JH, Yoo SH, Son JH, Paek SL, Kim IK, Hwang JH, Kim DG, Kim HJ, Jeon BS, Park SS, Paek SH. Mitochondrial dysfunction of immortalized human adipose tissue-derived mesenchymal stromal cells from patients with Parkinson's disease. Exp Neurobiol. 2013;22:283–300. doi: 10.5607/en.2013.22.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moon HE, Park HW, Shin HY, Paek SL, Kim DG, Son JH, Paek SH. Genetic profiling in human adipose tissue-derived mesenchymal stromal cells from the idiopathic and familial parkin-deficient patients of parkinson's disease in comparison with non-PD patients. Tissue Eng Regen Med. 2010;7:237–247. [Google Scholar]
- 87.Chaturvedi RK, Beal MF. Mitochondria targeted therapeutic approaches in Parkinson's and Huntington's diseases. Mol Cell Neurosci. 2013;55:101–114. doi: 10.1016/j.mcn.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 88.Chaturvedi RK, Beal MF. Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci. 2008;1147:395–412. doi: 10.1196/annals.1427.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bender A, Samtleben W, Elstner M, Klopstock T. Long-term creatine supplementation is safe in aged patients with Parkinson disease. Nutr Res. 2008;28:172–178. doi: 10.1016/j.nutres.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 90.Burchell VS, Gandhi S, Deas E, Wood NW, Abramov AY, Plun-Favreau H. Targeting mitochondrial dysfunction in neurodegenerative disease: Part I. Expert Opin Ther Targets. 2010;14:369–385. doi: 10.1517/14728221003652489. [DOI] [PubMed] [Google Scholar]
- 91.Storch A. Coenzyme Q10 in Parkinson's disease. Symptomatic or neuroprotective effects? Nervenarzt. 2007;78:1378–1382. doi: 10.1007/s00115-007-2285-1. [DOI] [PubMed] [Google Scholar]
- 92.Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res. 2011;8:393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]