

Abstract
Growth differentiation factor 15 (GDF15) is, a member of the transforming growth factor β (TGF-β) superfamily of proteins. Although GDF15 is well established as a potent neurotrophic factor for neurons, little is known about its role in glial cells under neuropathological conditions. We monitored GDF15 expression in astrocyte activation after a kainic acid (KA)-induced neurodegeneration in the ICR mice hippocampus. In control, GDF15 immunoreactivity (IR) was evident in the neuronal layer of the hippocampus; however, GDF15 expression had increased in activated astrocytes throughout the hippocampal region at day 3 after the treatment with KA. LPS treatment in astrocytes dramatically increased GDF15 expression in primary astrocytes. In addition, LPS treatment resulted in the decrease of the IκB-α degradation and increase of the phosphorylation level of RelA/p65. These results indicate that GDF15 has a potential link to NF-κB activation, making GDF15 a valuable target for modulating inflammatory conditions.
Keywords: GDF15, Astrocyte, Excitotoxicity, NF kappaB signaling
INTRODUCTION
Growth differentiation factor 15 (GDF15) was discovered as a member of the transforming growth factor β (TGF-β) superfamily of proteins. GDF15 was cloned independently in different laboratories and is therefore known by different names, such as the macrophage inhibitory cytokine-1 (MIC-1) and, nonsteroidal anti-inflammatory drugs (NSAID) activated gene (NAG-1) [1,2]. Research has shown that GDF15 is widely distributed in the central nervous system (CNS) and the peripheral nervous system (PNS) [3]. Low levels of GDF15 are found in all regions of the unlesioned rat and mouse CNS, such as the peripheral nerves, isolated astrocytes, and the dorsal root ganglion cells (DRGs) [4]. GDF15 is a well established and potent neurotrophic factor for dopamingeric neurons [5], cerebellar granular neurons [6], sensory sympathetic neurons, and spinal cord motor neurons [7]. In a cold-induced injury lesion of the cerebral cortex, GDF15 was found to be highly upregulated in regions adjacent to the lesion site [3]. A similar pattern of GDF15 induction was observed in a mouse model of cerebral ischemia [8]. Although GDF15 expression levels in unlesioned neurons and glia are found to be lower, GDF15 is robustly induced in the lesioned neuron and glia, suggesting that the factor may play a role in the lesioned CNS; however, whether GDF15 plays a similar role in astrocytes has not been precisely defined.
Astrocytes play an important role as the essential mediators of the brain's innate immune response to a variety of brain insults. During brain injuries, astrocytes secrete proinflammatory cytokines and express key immune receptors, such as TLRs enabling them to mount a proinflammatory response to a number of signals [9,10]. In addition, astrocytes upregulate the cytoskeletal protein, Glial fibrillary acidic protein (GFAP) and form a physical barrier from infiltrating immune cells in injured brain [11]. Although astrocytes play a critical part in protecting the brain from the inflammatory response, how they respond to anti-inflammatory cytokines and their role in dampening neuroinflammation remains to be determined.
Because Kainic acid (KA) is a glutamate receptor agonist that induces significant excitotoxicity in the hippocampus, an injection of KA results in the hippocampal neuronal cell death and glial activation [12,13]. Therefore, KA-induced brain damage may provide a suitable model for evaluating the role of GDF15 in reactive gliosis during neuroinflammation. In addition, the upregulation of transcription factors, such as NFκB in astrocytes, induce the expression of neuroprotective molecules [14]. Collectively, this study examined the expression of GDF15 in astrocytes after a KA-induction of an excitotoxic lesion in the mouse hippocampus, and the effect of GDF15 on NFκB signaling in primary astrocytes.
MATERIALS AND METHODS
Experimental Animals and Lesions
Male imprinting control region (ICR) mice (Samtako, Korea) weighing 23~25 g were used in this study. All animal-related procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Chungnam National University (CNU-00151). KA (Sigma, MO, USA) was prepared as a stock solution at 5 mg/ml in sterile 0.1 M PBS; aliquots were stored at -20℃ until required. Briefly, KA was injected at right lateral cerebral ventricle (anteroposterior, -0.4 mm; mediolateral, 1 mm; dorsoventral, -2.3 mm relative to bregma) using a 50-µl Hamilton microsyringe fitted with a 26-gauge needle inserted to a depth of 2.4 mm (0.1 µg/5 µl in PBS, i.c.v.). Control mice received an equal volume of saline. KA-injected animals (n=6~8 per group) and saline-injected control animals (n=6/group) were allocated. At 1, 3, and 7 days after KA or saline injection, mice were anesthetized with sodium pentobarbital (50 mg/kg i.p.), and perfused transcardially with heparinized PBS, followed by perfusion with 4% paraformaldehyde in PBS. Their brains were removed, immersed in the same fixative for 4 h, and then cryoprotected in a 30% sucrose solution. They were embedded in tissue freezing medium and then frozen rapidly in 2-methyl butane precooled to its freezing point with liquid nitrogen. Frozen coronal sections (40 µl thick) were obtained using a Leica cryostat.
Immuonohistochemistry and Double Immunofluorescence
Parallel free-floating sections were subjected to endogenous peroxidase blocking with 1% H2O2 in PBS, followed by treatment with blocking buffer (1% fetal bovine serum [FBS] in PBS and 0.3% Triton X-100 for 30 min) and incubation with primary anti-GDF15 (1:100, #E2430, Spring bioscience) overnight. Immunohistochemical staining of the tissue sections were performed using the avidin-biotin peroxidase complex (ABC) method described previously [15,16]. In order to simultaneously demonstrate a pair of antigens, in the same section, goat GDF15 was used with glial fibrillary acidic protein (GFAP, 1:1000, #AM020, Biogenex). Free-floating sections were immunoreacted for GDF15 and Cy3-conjugated anti-goat secondary antibody. Sections were then further processed for GFAP and Cy2-conjugated anti-mouse secondary antibody. Nucleus staining was performed with DAPI. Double-stained sections were analyzed with Zeiss Axiophot microscope.
Primary Astrocyte Culture
Rat primary cerebral astrocytes were purified from neonatal rats according to standard procedures [15]. Briefly, a postnatal day 1 (P1) Sprague-Dawley rat pup (Samtako, Korea) was decapitated in an ice-chilled dish and the brain was removed. After removal of the meninges the cerebral cortex was dissected and dissociated in Hanks' balanced salt solution (HBSS; Invitrogen) supplemented with 5.5 mM glucose, 20.4 mM sucrose, and 4.2 mM sodium bicarbonate. After centrifugation, the cells were seeded into poly-L-lysine-coated T75 flasks and maintained in Minimal Essential Medium (MEM) containing 20% fetal bovine serum (FBS), 100 µM non-essential amino acid solution, 2 mM L-glutamine, and antibiotics. After 7 days, the flasks were agitated on an orbital shaker for 12 h at 200 rpm at 37℃, and the nonadherent oligodendrocytes and microglial cells were removed. The flasks were then trypsinized and expanded in Dulbecco's Modified Eagle Medium (DMEM) growth media containing 10% FBS, 2 mM L-glutamine, and 1 mM sodium pyruvate. Under these conditions, the purity of the astrocyte population was 95% as determined by Immunofluorescence analysis using anti-OX-42 to detect microglial cells, anti-CNPase to detect oligodendrocyte contamination, and GFAP to identify astrocytes. For lipopolysaccharide (LPS) treatment, primary astrocytes were trypsinized and seeded in a 60-mm dish at 70% confluence. Cell lysates were processed for immunoblot analysis for indicated times after LPS treatment.
Western Blotting
For western blot, the cells were incubated for 24 h prior to 4 h serum-starvation and stimulated with LPS (100 ng/mL) for the indicated times. Cultured astrocytes were collected by scraping, and the pellet was solubilized in lysis buffer using PRO-PREP reagent (Intron Biotechnology, Sungnam, Korea) with a protease inhibitor cocktail (Sigma P5726). Following normalization of protein content in each sample, 30 µg of the total cellular fraction of each sample was separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transblotted onto nitrocellulose membranes. The blot was probed with primary antibodies, e.g. GDF15, P-p65 (Cell Signaling, #3033S), IκB-α (Santa Cruz, #sc-371), beta-actin (T308, #2965, Cell Signaling, CA, USA) in blocking solution. Membranes were washed for 3 times for 10 min in TBST, and incubated for 1 h with peroxidase labeled secondary antibody (Vector) diluted 1:2000 in TBST. After three further washes, immunolabeled proteins were detected by chemiluminescence using a Supersignal ECL kit (Pierce Chemical) and Biomax Light-1 films (Kodak,USA).
RESULTS
Enhancement of GDF15 expression in Astrocytes Following KA-Induced Excitotoxicity
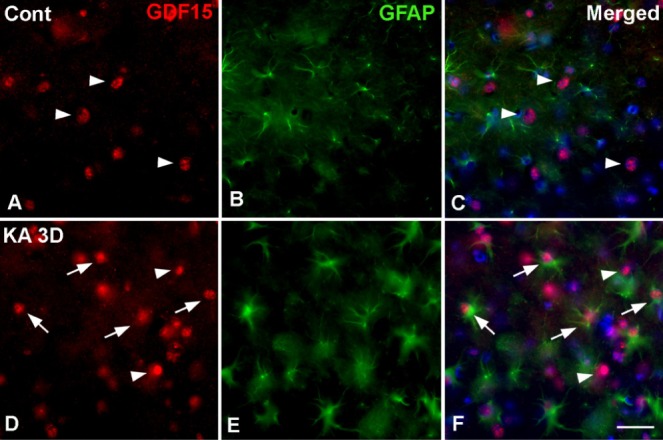
The intracerebroventricular injection of KA is a well established excitotoxicity model that promotes seizures in mice and selective hippocampal cell death [17,18]. Previously, we showed a time-dependent pyramidal cell loss and glial activation in this ICR mice model as measured by cresyl violet staining [15]. In this study, a typical loss of pyramidal neurons in the CA3 regions of the ipsilateral hippocampus was apparent on day1 post-injection of KA. Moreover, glial activation had increased in the hippocampus from day 1 to day 3 (Fig. 1A). To evaluate the involvement of GDF15 in KA-induced excitotoxicity, we measured GDF15 expression in the ipsilateral hippocampus of KA-treated mice. GDF15 immunoreactivity (IR) was evident in the neuronal layer of the hippocampus, including the CA1, CA3, and the dentate gyrus in the control mice (Fig. 1B); however, GDF15 IR in the small nuclei was increased markedly throughout the CA3 region on day 3 post-treatment with KA (Fig. 1Bc, g). To further confirm the cell type in which GDF15 was found, we employed double staining with the anti-GDF15 and anti-GAFP (astrocytic marker) primary antibodies. GDF15 was found to be localized to astrocytic nuclei, indicating that GDF15 expression increased in the activated astrocytes in the hippocampi of KA-treated mice (Fig. 2).
Fig. 1. Representative images with Cresyl violet staining and immunohistochemical staining with anti-GDF15 primary antibodies in kainic acid (KA) treated ICR mice. (A) In the KA-group, cresyl violet-positive cells were decreased in the striatum pyramidal (SP) of the hippocampal CA3 region (asterisk in Ab). In addition to pyramidal cell loss, small glial cell immunoreactivity was evident (Ac, d). (B) GDF15 immunohistochemistry in the hippocampal CA3 region of the control and KA groups at 1, 3 and 7 days after KA injection. GDF15 immunoreactivity was markedly increased in the KA group (Aa~d). Higher magnification images of the rectangular area Aa-d in the hippocampus show sequential changes in the GDF15 expression (Ae~h). GDF15 immunoreactivity was found in relative small nuclei than those in control (arrowheads). SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bars=100 µm in A, 200 µm in Ba~d, 20 µm in Be~h.
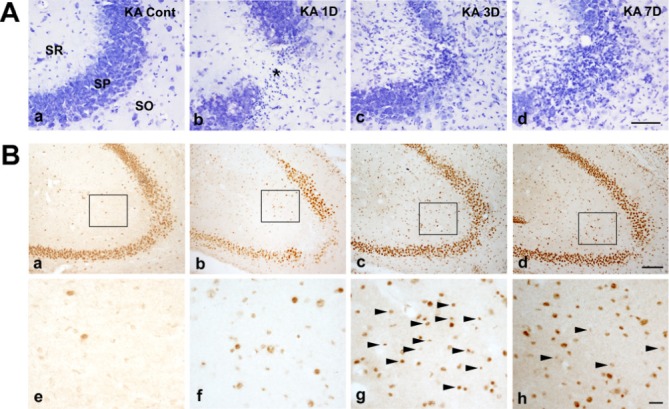
Fig. 2. Double immunofluorescence staining for the identification of GDF15-positive cells in the hippocampus of control (A~C) and KA-treated mice at 3 days after treatment (D, E). GDF15 was colocalized with the glial fibrillary acidic protein (GFAP) in the nuclei of positive cells in the CA3 region of KA-treated hippocampi (arrows), but not in neurons-like cells (arrowheads). Nuclear counterstaining was performed with DAPI (blue). Scale bar=20 µm.
GDF15 is associated with NF-κB activity in Astrocytes Activation
Because it has been well established that NF-κB plays an essential role in the LPS-induced expression of proinflammatory mediators such as CCL2 and, CXCL2 in astrocytes [19,20,21], we next examined whether GDF15 influences LPS-induced NF-κB activity in these cells. Consistently, LPS treatment in astrocytes dramatically increased GDF15 expression in a time-dependent manner (Fig. 3, upper panel). To determine the molecular mechanism involved in the NF-κB signaling pathway, we examined the degradation of IκB-α and the phosphorylation of RelA/p65 in LPS-stimulated astrocytes. When the astrocytes were treated with LPS, the phosphorylation level of RelA/p65 began to increase at 1 h after the treatment, and continued to increase for 4 h. LPS treatment also resulted in the decrease of the IκB-α degradation (Fig. 3, middle and lower panel). These results suggest that GDF15 play an important role in NF-κB activity during astrocyte activation induced by LPS.
Fig. 3. LPS induced GDF15 expression and phosphorylation of p65, and degradation IκBα. Serum-starved astrocytes were treated with LPS (100 ng/ml) for the indicated time period and western blot analysis was performed using the specific antibodies anti-GDF15, anti P-p65; phosphorylated p65 subunit, and anti- IκBα. Anti- actin antibody was used as loading control. Relative density was obtained by densitometry analysis of the corresponding immunoblot data. Statistical differences were determined by comparing the values for actin in each lane. Data are expressed as optical densities and represent means±SEM of three independent experiments. ***p < 0.001 vs. control.

DISCUSSION
We have shown that GDF15 expression increased in the activated astrocytes in the hippocampi of KA-treated mice. Previously, GDF15 expression was investigated in several CNS lesions. In a cold-induced lesion of the cerebral cortex, GDF-15 mRNA was found to be highly upregulated in the majority of lesioned neurons, a minority of microglial cells, and not at all in the astroglial cells [3]. Unsicker groups indicate that GDF- 15 IR, while hardly detectable in unlesioned brain areas, was prominently upregulated in neurons, such as neurons within the hippocampus, and while moderate numbers of microglial cells were also stained, astrocytes were consistently GDF15 negative [4]. They also note that GDF15 is robustly induced in lesioned neurons and scattered microglia, and can be visualized by in situ hybridization and immunocytochemistry; however, they observed that astrocytes in the unlesioned and lesioned CNS are devoid of GDF15, in contrast to cultured astrocytes [4,5]. Conversely, Schwann cells seem to be the most prominent source for GDF15 in the PNS [7]. Studies also show that GDF15 expression in glioblastoma cell lines was significantly lower than that in benign glioma cells and normal human astrocytes [22]. The discrepancy between previous finding on GDF15 expression and our results may be due to the sensitivity of the primary antibody. Unsicker groups mention that GDF15 is hardly detectable in unlesioned brain areas. However, we demonstrate that GDF15 IR was evident in the neuronal layer of the hippocampus, including the CA1, CA3, and the dentate gyrus of the control mice.
TGF-β signaling in astrocytes is highlighted because TGF-β is a master regulatory and primarily anti-inflammatory cytokine that is universally increased during CNS injury [23,24]. Astrocytic TGF-β signaling limits the inflammation and reduces neuronal damage in CNS toxoplasma infection [25]. In addition, astrocytic TGF-β signaling after stroke decreases subacute neuroinflammation and preserves neuronal function [26]. This emerging evidence suggests that astrocytic TGF-β signaling may be a key pathway for limiting brain inflammation during CNS infection and injury. Astrocytes respond to CNS injury by producing proinflammatory cytokines and chemokines. For example, astrocytes produce the chemokines CCL2, CXCL1 and CXCL2 after ischemia or spinal cord injury [27,28], and CCL2 and CCL5 after traumatic brain injury [29]. These pro-inflammatory chemokines may play a beneficial role by attracting the immune cells, which have been shown to reduce tissue damage by clearing dead cells debris [30]. In contrast, excessive inflammation can be harmful by inducing free-radical damage to the neurons [31] and increasing cerebral edema, which can cause brain herniation and further tissue damage [32].
Cell death resulting from excitotoxicity has been associated with different brain disorders implicating an inflammatory response in affected regions [33,34]. Glial cells, as mediators of the inflammatory response, also have an important role in the course of kainic acid (KA)-induced hippocampal neurodegeneration. To mimic the inflammatory response after KA treated hippocampal neuronal death, we used LPS for in vitro study. In this study, we examined the effect of GDF15, one of the diverse members of the TGF-β superfamily, on the activity of NF-κB, since it has been well established that NF-κB plays an essential role for the expression of proinflammatory mediators such as CCL2, CXCL2 induced by LPS in astrocytes [19,20,21]. Our results are consistent with the model in which astrocytes use TGFβ signaling to upregulate TGFβ1 and its activator thrombospondin-1 in the peri-infarct cortex during the subacute time window after stroke [26]. We found that astrocytic GDF15 signaling is associated with NF-κB activity in KA-induced neuroinflammation in astrocytes, but found no evidence that it affects pro- or anti-inflammatory cytokine release in these cells. Additionally, it remains with open question whether upregulation of GDF15 in astrocytes modulated immune cell polarization around the injury site, thus exhibiting a neuroprotective function. Because TGFβ is universally upregulated in brain injury, it may represent a common pathway that mediates the endogenous immunoregulatory functions of astrocytes. The anti-inflammatory and neuroprotective functions of astrocytic TGFβ signaling may therefore encompass interactions with other glial cells, such as the microglia, and finally extend their protective effects to the neuron.
ACKNOWLEDGEMENT
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2013R1A1A 1A05006966) and in part by research fund of Chungnam National University (2014).
References
- 1.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A. 1997;94:11514–11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol. 2001;59:901–908. [PubMed] [Google Scholar]
- 3.Schober A, Bottner M, Strelau J, Kinscherf R, Bonaterra GA, Barth M, Schilling L, Fairlie WD, Breit SN, Unsicker K. Expression of growth differentiation factor-15/ macrophage inhibitory cytokine-1 (GDF-15/MIC-1) in the perinatal, adult, and injured rat brain. J Comp Neurol. 2001;439:32–45. doi: 10.1002/cne.1333. [DOI] [PubMed] [Google Scholar]
- 4.Unsicker K, Spittau B, Krieglstein K. The multiple facets of the TGF-beta family cytokine growth/differentiation factor-15/macrophage inhibitory cytokine-1. Cytokine Growth Factor Rev. 2013;24:373–384. doi: 10.1016/j.cytogfr.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Strelau J, Sullivan A, Bottner M, Lingor P, Falkenstein E, Suter-Crazzolara C, Galter D, Jaszai J, Krieglstein K, Unsicker K. Growth/differentiation factor-15/macrophage inhibitory cytokine-1 is a novel trophic factor for midbrain dopaminergic neurons in vivo. J Neurosci. 2000;20:8597–8603. doi: 10.1523/JNEUROSCI.20-23-08597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramaniam S, Strelau J, Unsicker K. Growth differentiation factor-15 prevents low potassium-induced cell death of cerebellar granule neurons by differential regulation of Akt and ERK pathways. J Biol Chem. 2003;278:8904–8912. doi: 10.1074/jbc.M210037200. [DOI] [PubMed] [Google Scholar]
- 7.Strelau J, Strzelczyk A, Rusu P, Bendner G, Wiese S, Diella F, Altick AL, von Bartheld CS, Klein R, Sendtner M, Unsicker K. Progressive postnatal motoneuron loss in mice lacking GDF-15. J Neurosci. 2009;29:13640–13648. doi: 10.1523/JNEUROSCI.1133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schindowski K, von Bohlen und Halbach O, Strelau J, Ridder DA, Herrmann O, Schober A, Schwaninger M, Unsicker K. Regulation of GDF-15, a distant TGF-beta superfamily member, in a mouse model of cerebral ischemia. Cell Tissue Res. 2011;343:399–409. doi: 10.1007/s00441-010-1090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol. 2012;12:623–635. doi: 10.1038/nri3265. [DOI] [PubMed] [Google Scholar]
- 11.Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faherty CJ, Xanthoudakis S, Smeyne RJ. Caspase-3-dependent neuronal death in the hippocampus following kainic acid treatment. Brain Res Mol Brain Res. 1999;70:159–163. doi: 10.1016/s0169-328x(99)00143-6. [DOI] [PubMed] [Google Scholar]
- 13.Jarrard LE. Use of excitotoxins to lesion the hippocampus: update. Hippocampus. 2002;12:405–414. doi: 10.1002/hipo.10054. [DOI] [PubMed] [Google Scholar]
- 14.Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi MH, Lee YS, Kang JW, Kim SJ, Oh SH, Kim YM, Lee YH, Lee SD, Kim DW. NFAT5-dependent expression of AQP4 in astrocytes. Cell Mol Neurobiol. 2013;33:223–232. doi: 10.1007/s10571-012-9889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee YS, Kang JW, Lee YH, Kim DW. ID4 mediates proliferation of astrocytes after excitotoxic damage in the mouse hippocampus. Anat Cell Biol. 2011;44:128–134. doi: 10.5115/acb.2011.44.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi MH, Kwon K, Zhang E, Seo JH, Kang SS, Son CG, Kang JW, Kim DW. RhoGDI2 Expression in Astrocytes After an Excitotoxic Lesion in the Mouse Hippocampus. Cell Mol Neurobiol. 2014 doi: 10.1007/s10571-014-0108-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi MH, Zhang E, Kang JW, Shin YN, Byun JY, Oh SH, Seo JH, Lee YH, Kim DW. Expression of CD200 in alternative activation of microglia following an excitotoxic lesion in the mouse hippocampus. Brain Res. 2012;1481:90–96. doi: 10.1016/j.brainres.2012.08.053. [DOI] [PubMed] [Google Scholar]
- 19.Thompson WL, Van Eldik LJ. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes [corrected] Brain Res. 2009;1287:47–57. doi: 10.1016/j.brainres.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laureys G, Gerlo S, Spooren A, Demol F, De Keyser J, Aerts JL. beta(2)-adrenergic agonists modulate TNF-alpha induced astrocytic inflammatory gene expression and brain inflammatory cell populations. J Neuroinflammation. 2014;11:21. doi: 10.1186/1742-2094-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi K, Ni L, Jonakait GM. Fas ligation and tumor necrosis factor alpha activation of murine astrocytes promote heat shock factor-1 activation and heat shock protein expression leading to chemokine induction and cell survival. J Neurochem. 2011;116:438–448. doi: 10.1111/j.1471-4159.2010.07124.x. [DOI] [PubMed] [Google Scholar]
- 22.Yoshioka H, Kamitani H, Watanabe T, Eling TE. Nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) expression is increased by the histone deacetylase inhibitor trichostatin A. J Biol Chem. 2008;283:33129–33137. doi: 10.1074/jbc.M805248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFbeta signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:62. doi: 10.1186/1742-2094-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM. TGF-beta 1 is an organizer of responses to neurodegeneration. J Cell Biochem. 1993;53:314–322. doi: 10.1002/jcb.240530408. [DOI] [PubMed] [Google Scholar]
- 25.Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, Koshy AA, Buckwalter MS. Astrocytic TGF-beta signaling limits inflammation and reduces neuronal damage during central nervous system Toxoplasma infection. J Immunol. 2014;193:139–149. doi: 10.4049/jimmunol.1303284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014;62:1227–1240. doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pineau I, Sun L, Bastien D, Lacroix S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav Immun. 2010;24:540–553. doi: 10.1016/j.bbi.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23:7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown GC. Nitric oxide and neuronal death. Nitric Oxide. 2010;23:153–165. doi: 10.1016/j.niox.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Heiss WD. The ischemic penumbra: how does tissue injury evolve? Ann N Y Acad Sci. 2012;1268:26–34. doi: 10.1111/j.1749-6632.2012.06668.x. [DOI] [PubMed] [Google Scholar]
- 33.Cortes-Canteli M, Luna-Medina R, Sanz-Sancristobal M, Alvarez-Barrientos A, Santos A, Perez-Castillo A. CCAAT/enhancer binding protein beta deficiency provides cerebral protection following excitotoxic injury. J Cell Sci. 2008;121:1224–1234. doi: 10.1242/jcs.025031. [DOI] [PubMed] [Google Scholar]
- 34.Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130:1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]