

Abstract
Objective
Ketone bodies (KB) are products of fatty acid oxidation and serve as essential fuels during fasting or treatment with the high-fat anti-seizure ketogenic diet (KD). Despite growing evidence that KB exert broad neuroprotective effects, their role in seizure control has not been firmly demonstrated. The major goal of this study was to demonstrate the direct anti-seizure effects of KB and to identify an underlying target mechanism.
Methods
We studied the effects of both the KD and KB in spontaneously epileptic Kcna1-null mice using a combination of behavioral, planar multi-electrode, and standard cellular electophysiological techniques. Thresholds for mitochondrial permeability transition (mPT) were determined in acutely isolated brain mitochondria.
Results
KB alone were sufficient to: (1) exert anti-seizure effects in Kcna1-null mice; (2) restore intrinsic impairment of hippocampal long-term potentiation (LTP) and spatial learning-memory defects in Kcna1-null mutants; and (3) raise the threshold for calcium-induced mPT in acutely prepared mitochondria from hippocampi of Kcna1-null animals. Targeted deletion of the cyclophilin D (CypD) subunit of the mPT complex abrogated the effects of KB on mPT, and in vivo pharmacological inhibition and activation of mPT were found to mirror and reverse, respectively, the anti-seizure effects of the KD in Kcna1-null mice.
Interpretation
The present data reveal the first direct link between mPT and seizure control, and provide a potential mechanistic explanation for the KD. Given that mPT is increasingly being implicated in diverse neurological disorders, our results suggest that metabolism-based treatments and/or metabolic substrates might represent a worthy paradigm for therapeutic development.
Keywords: ketones, β-hydroxybutyrate, mitochondrial permeability transition, cyclophilin D, neuroprotection, epilepsy
Recently, interest in metabolism-based treatments for neurological disorders has grown both experimentally and at a clinical level.1–4 The prototypic therapy is the high-fat ketogenic diet (KD) which is effective against medically refractory epilepsy.4 A hallmark feature of the KD is the production of ketone bodies (KB: β-hyroxybutyrate [BHB], acetoacetate [ACA] and acetone) by the liver. Ketone bodies are products of intermediary metabolism and serve as alternative fuels under conditions of fasting or starvation.5–7 Further, they constitute the carbon source for cellular membranes during early development.8
Emerging data indicate that KB may possess neuroprotective activity,1,6,9,10 and hence these substrates may play a broader physiological role than previously understood. As ketosis is observed to varying degrees during caloric restriction – linked to enhanced longevity and improved cognition11,12 – and high-fat diets, the relative importance of KB action is further underscored. And while it is well established that KB can enhance cellular ATP levels13,14 and can attenuate reactive oxygen species (ROS) through several mitochondrial actions,10,15 the precise mechanisms remain unclear.
To determine how KB, and indeed the KD, are functionally neuroprotective, we utilized a clinically relevant rodent model of epilepsy – the Kcna1-null mutant mouse which recapitulates essential features of human temporal lobe epilepsy16,17 – and examined the effects of both the KD and KB on the mitochondrial permeability transition (mPT) pore and hippocampal long-term potentiation (LTP). We chose to examine the effects of KB on mPT since this complex plays a critical role in regulating cell death pathways and is known to regulate ATP and ROS levels much like KB.18,19 Additionally, mPT has been implicated in a growing number of neurological disorders, some of which have overlap with epileptogenic mechanisms.18–20 Our data indicate that KB alone can exert anti-seizure and nootropic (i.e., cognition-enhancing) effects in epileptic brain, but importantly, directly link mPT to the control of epilepsy.
MATERIALS AND METHODS
Animals
Kcna1-null mice (congenic C3HeB/FeJ background) were maintained at the Barrow Neurological Institute (Phoenix, AZ), Creighton University College of Medicine (Omaha, NE), or the University of Calgary (Alberta, Canada). Ppif-null (CypD knockout) mice and control C57bl/6 wild-type mice were bred at the University of Kentucky (Lexington, KY), and were originally obtained as a gift from Dr. J. D. Molkentin (Cincinnati Children’s Research Foundation). All procedures and interventions reported herein were approved by all four Institutional Animal Care and Use Committees (IACUC) and conformed to the National Institute of Health guide on the care of laboratory animals. Genotyping was determined either via PCR on genomic DNA from tail-clips or outsourced to Transnetyx (Cordova, TN, USA). Upon weaning, mice were separated by gender, housed in standard cages, and placed ad libitum on either a standard diet (SD) or ketogenic diet (KD; BioServ F3666, Frenchtown, NJ, USA). Blood BHB and glucose levels were assayed with Precision Xtra Meters (Abbott, Alameda, California) and determined at multiple time-points during KD, SD or KB treatment. All mice were maintained on a 12-h light/dark cycle in a temperature-controlled room.
Behavioral Testing
Kcna1-null mice (P28–30) were anesthetized with isoflurane. Burr holes were made 3 mm anterior and 3 mm lateral to the bregma. Leads from the transmitter were cut to the appropriate length for cranial EEG, placed in the burr holes, and fixed with integrity dental caulk (Dentsply, Milford, DE, USA). Wireless transmitters from Data Sciences International (St. Paul, MN, USA) were implanted subcutaneously and EEG signals were recorded using the time-locked Harmonie video-EEG monitoring system (Stellate Systems, Montreal, Canada). All data were reviewed manually by at least two independent scorers; automated detection software was not utilized. Assessment of spatial learning/memory involved the use of a custom-made circular Barnes maze and an EthoVision tracking system (Noldus, Leesburg, VA, USA). While the water maze is a more widely-accepted behavioral test to measure spatial learning, a violent temperature-sensitive neuromuscular reaction and provocation of seizures specific to Kcna1-null mice precluded any behavioral testing involving water. Like the water maze, the Barnes maze tests an animal’s ability to use spatial cues to find a desired escape hole hidden beneath the table when exposed to an open brightly-lit space.
Osmotic Mini-Pump Studies
Mice (P22–23) were anesthetized using 5% isoflurane for induction and maintained with 2% isoflurane. The nape of the neck was shaved and a 1 cm incision made. Using a hemostat, a small subcutaneous pocket was made along the flank of the animal. Each Alzet (Cupertino, CA, USA) osmotic mini-pump was filled with 10M β-hydroxybutyrate (BHB; pH 7.2) and inserted in the subcutaneous pocket. The incision was sutured quickly and the animal was allowed access to standard rodent chow and water ad libitum.
Cellular Electrophysiology
Multi-electrode recordings in hippocampal slice cultures were conducted as previously described.17 Briefly, 400-μm thick hippocampal slices from P5–7 Kcna1-null mice were collected in cold preparation buffer, placed on moist membrane inserts in 6-well plates filled with 1 ml culture medium (50% minimal essential medium, 25% Hank’s balanced salt solution, 20% inactivated horse serum, 30 mM HEPES, 30 mM glucose, 3 mM glutamine, 0.5 mM ascorbic acid, 1 mg/ml insulin, 5 mM NaHCO3, pH 7.3) and incubated in humidified, CO2-enriched atmosphere at 36°C. Pairs of adjacent slices were compared for control and experimental conditions. Beginning on the third day in vitro (DIV), a fresh ketone body cocktail (5 mM BHB and 1 mM ACA) was added daily to the culture medium. On the seventh DIV, cultures were monitored for spontaneous network activity, including seizure-like events (SLEs) using a 64-channel multi-microelectrode array (MED64 probe; electrode size: 50×50 μm; electrode separation: 150 μm; Alpha Med Systems, Osaka, Japan). Spontaneous and evoked extracellular potentials were recorded with Conductor v3 (Alpha Med Systems) and analyzed with Spike2 (v6) software (Cambridge Electronic Design, Cambridge, England). Spontaneous SLEs were identified as having high-frequency spiking (tonic features) with or without evolution of slower, large amplitude DC shifts (i.e., “clonic” features). SLE incidence, duration and intensity were quantified, and SLE intensity was derived from coastline burst index (CBI) analysis. Briefly, SLE waveforms and baseline waveforms of similar durations were linearized and total lengths quantified. CBI was calculated with the following equation: CBI = (SLE length-baseline length)/Δtime.
For LTP experiments, transverse hippocampal slices (400 μm thickness) were prepared acutely from brains of mice exposed to various treatments. Following decapitation, the whole brain was rapidly isolated and submerged in ice-cold oxygenated physiological saline (composition in mM: 124 NaCl, 1.8 MgSO4, 4 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, and 10 D-glucose; pH: 7.4). Slices were cut with a standard vibratome, and then transferred to an incubation chamber containing physiological saline bubbled with 95% O2/5% CO2 at 35 °C for 1 hr. Each slice was transferred to a submersion-type recording chamber attached to an Axioskop FS2 microscope (Zeiss Instruments, Thornwood, NY, USA) and superfused with warm (31 ± 1°C) physiological saline at a rate of 2–3 ml/min before recording. Upon electrical stimulation of Schaffer collaterals, excitatory post-synaptic potentials (EPSPs) were recorded at a control test frequency of 0.05 Hz (0.1 ms, 20–100 μA) in the stratum radiatum of CA1 hippocampus. Using a standard input-output curve (stimulus intensity vs. EPSP amplitude), baseline EPSP amplitude (over 1 mV) was set to 30–40 % of the maximum responses. LTP was expressed as the percent of mean baseline EPSP amplitude. Recorded data were filtered at 3 kHz, sampled at 10 kHz using pClamp, and analyzed with Clampfit (Molecular Devices, Sunnyvale, CA, USA).
Mitochondrial Studies
Mitochondria were isolated using Ficoll density gradient centrifugation, and CaG5N (excitation, 506 nm; emission, 532 nm) was used to monitor Ca2+ uptake and determine the mitochondrial permeability transition threshold as previously described.21,22 The time-point at which the CaG5N signal was 150% above the average baseline was considered to be the threshold for mitochondrial permeability transition. Mitochondrial swelling was assessed under de-energized conditions as a decrease in light absorbance (light scattering) using a Synergy HT 96-well plate reader. Mitochondrial swelling rate was measured as the decrease in absorbance (540 nm) over 20 min following addition of calcium ions.22 Briefly, 400μg mitochondria were suspended in 1 ml of isotonic buffer (150mM KCl, 20mM MOPS, 10mM Tris, 1μM rotenone, 1 μM antimycin and 2 μM ionomycin at pH 7.2. After a 5-min pre-incubation at 37 °C and baseline measurement, CaCl2 (200 μM) was added into wells and mitochondrial swelling rate was measured as the decrease in absorbance (540 nm) over 20 min following addition of calcium ions.
Chemicals
Sodium pyruvate, malate, succinate and adenosine di-phosphate were purchased from Sigma-Aldrich (St. Louis, MO, USA); 2,4-dinitrophenol carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) was purchased from Biomol (Plymouth Meeting, PA, USA); calcium green-5N, hexapotassium salt (CaG5N) was purchased from Invitrogen Molecular Probes (Grand Island, NY, USA). NIM811 was obtained from Novartis Pharma Ltd. (Basel, Switzerland). All other chemicals were purchased from Sigma-Aldrich.
Statistics
Significance was determined by either an unpaired t-test or ANOVA followed by Bonferroni post-hoc test when appropriate using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA, USA). The day-five Barnes maze data was compared among groups using the ANOVA for pairwise multiple comparison procedures according to the Holm-Sidak method using SigmaStat 3.5 software (Systat, Chicago, IL, USA). Results are expressed as the group means (± S.E.M.). Statistical significance was established at p<0.05.
RESULTS
KD and KB Both Induce Anti-Seizure Effects
Kcna1-null mice display spontaneous recurrent seizures (SRS) lasting 20–30 sec or longer (on average 6–12 times daily) by the third to fourth postnatal week. These seizures possess many of the characteristics of limbic system seizures in other rodent models (e.g., rearing, forelimb clonus) as they evolve into whole-body tonic-clonic activity, suggesting that the hippocampus may play an important role in their pathogenesis.16,23 Figure 1A illustrates typical EEG findings in wild-type mice, as well as interictal and ictal EEG changes seen in Kcna1-null mutants. Consistent with our earlier observations,23 we found that a widely employed experimental KD significantly reduced SRS in Kcna1-null mice compared to SD-fed controls during active treatment (Fig 1B), and was accompanied by elevations in plasma BHB and decreases in blood glucose levels (Fig 2A, 2B). We next asked whether in vivo KB administration alone would block SRS. Given technical constraints in administering ACA on a long-term basis due to its high volatility and rapid breakdown, we exposed Kcna1-null mice solely to BHB using subcutaneously implanted osmotic mini-pumps. We found that BHB was sufficient to produce an anti-seizure effect identical to the full KD itself in animals fed an ad lib SD (Fig 1C). Glycolytic restriction was not a factor in these experiments as glucose levels were unaffected by exogenous BHB supplementation (Fig 2E).
Figure 1.
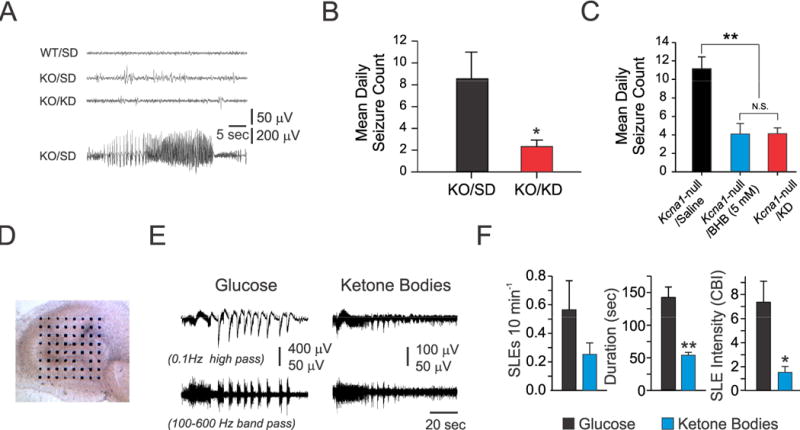
A ketogenic diet (KD) and ketone bodies (KB) provide anti-seizure effects in epileptic Kcna1-null (KO) mice. (A) Interictal (upper three traces) and ictal EEG (bottom trace) recordings from wild-type (WT) and KO mice treated with either standard rodent diet (SD) or a KD. The bottom trace (KO/SD) depicts an electrographic seizure coincident with a behavioral generalized tonic-clonic seizure (N=12 in each group). (B) Mean daily tonic-clonic seizure frequency in KO mice over 3-day recording period between SD-treated vs. KD-fed animals, *, p<0.05. (C) Mean daily seizure frequencies over a 3-day recording period in KO mice after a 10–14 day treatment with either BHB or saline. Chronic osmotic mini-pump administration of BHB resulted in anti-seizure effects. One-way ANOVA followed by Tukey test; **, p<0.01. (D-F) Long-term exposure to KB decreases seizure-like events (SLEs) in organotypic slice cultures of KO hippocampi. (D) Light microscopic view of a hippocampal slice culture (14 days in vitro [DIV]) on a 64-microelectrode array. (E) Representative SLEs from a slice cultured for two weeks with either normal glucose or KB (5 mM BHB and 1 mM ACA). High-frequency oscillations (HFOs) revealed by band-pass filtering were present during SLEs in both culture conditions (lower traces). (F) KB significantly reduced SLE duration (~67%) and intensity (~80%) (n=4 cultures harvested from three P3–5 mice per group, unpaired t-test: *, p<0.05; **, p<0.001). For panels (B, C and F), data points reflect the mean ± SEM.
Figure 2.
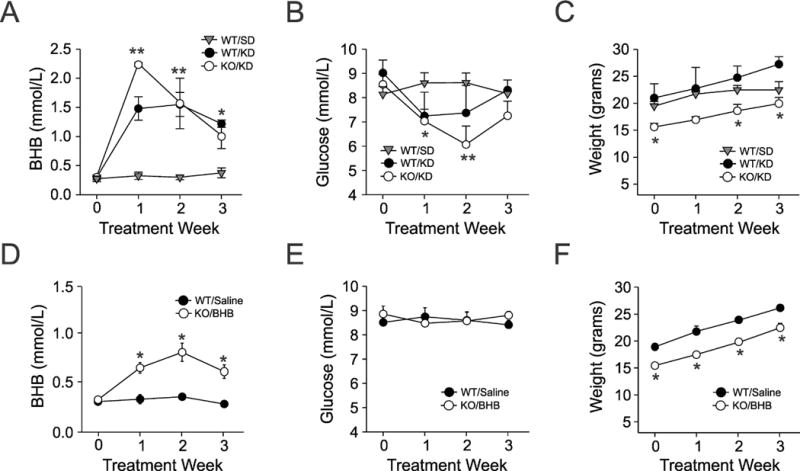
Metabolic parameters determined during the course of treatment. (A, B, C) Plasma β-hydroxybutyrate (BHB), blood glucose levels and weight changes in three cohorts of wild-type (WT) or Kcna1-null (KO) mice treated with either a standard diet (SD) or a ketogenic diet (KD). (D, E, F) Similar measurements made in separate groups of mice administered BHB through osmotic mini-pumps (N=12 mice in each group). Each symbol indicates the mean ± S.E.M; *p<0.05, **p<0.01. One-way ANOVA followed by Tukey test.
KB Block Seizure-Like Events In Vitro
Despite many mechanistic hypotheses,3,7,24 we asked whether KB alone – administered chronically – might be directly responsible for the beneficial actions of the KD against SRS. Using a planar 64-channel multi-electrode recording system, we examined the effects of a cocktail of BHB and ACA at clinically relevant physiological concentrations and ratio25 on organotypic slice cultures prepared from Kcna1-null mutants (Fig 1D). These cultures exhibited spontaneous electrographic seizure-like events (SLEs) which were significantly attenuated by chronic supplementation with KB, as compared to normal glucose-containing media devoid of KB (Fig 1E, 1F). Importantly, glucose concentrations were maintained under both conditions at 10 mM; hence, the effects of glycolytic restriction could not be responsible for the effects observed in our model.26 However, inhibition of glycolysis, such as with 2-deoxyglucose, may play a more important role in other epilepsy models through alternative mechanisms such as trkB/BDNF inhibition.27
Ketogenic Diet and Ketone Bodies Prevent mPT
Having established the beneficial effects of the KD and KB in our epileptic mouse model, we next sought to determine an underlying mechanism. Previously, we reported that KB inhibited mitochondrial permeability transition (mPT) – a biochemical phenomenon linked to apoptotic and necrotic cell death – in acutely isolated mitochondria from normal rat brain.10 We conducted similar experiments in mitochondria isolated from various treatment groups in epileptic Kcna1-null mice, and found that the KD reversed the significantly lowered threshold for calcium-induced mPT transition (Fig 3A, 3B). The mPT threshold for Kcna1-null mice was elevated by KD treatment to a level similar to WT animals fed a SD. And consistently, we demonstrated that KB when applied alone in vitro raised mPT threshold in brain mitochondria from Kcna1-null mutants (Fig 3C).
Figure 3.
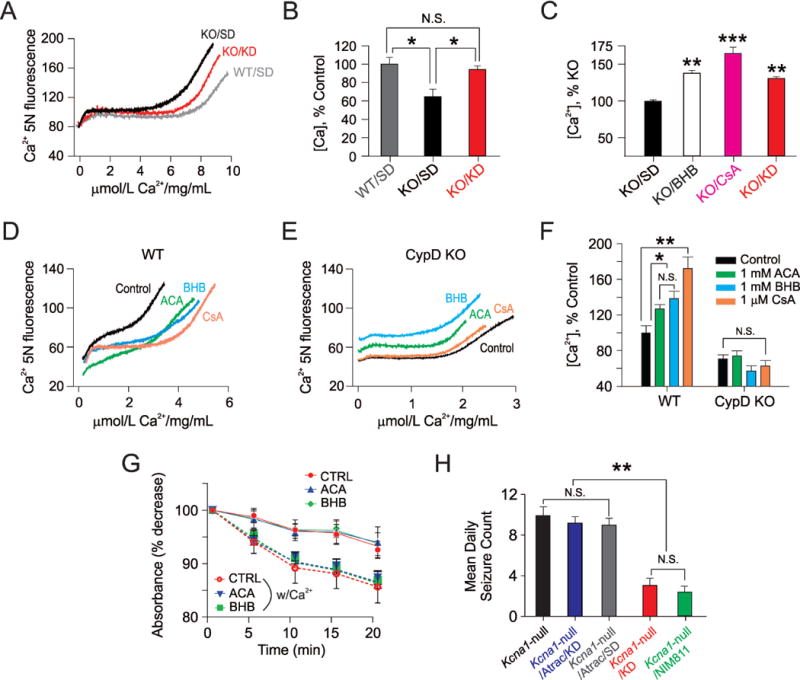
The cyclophilin D (CypD) subunit of the mitochondrial permeability transition (mPT) complex is required for functional neuroprotection by ketone bodies (KB). (A and B) Ketogenic diet (KD) treatment increases mPT threshold in Kcna1-null (KO) mice. Isolated brain mitochondria from wild-type (WT) C3H mice were able to accumulate more Ca2+ than mitochondria prepared from KO mice. (A) Representative spectrofluorometric traces indicating the capacity of mitochondria to sequester Ca2+ administered through bolus injections. Two weeks of KD treatment increased the capacity for Ca2+ uptake and fully restored mPT threshold in KO mitochondria. (B) Summary bar graphs of the different treatment groups. Each sample comprised pooled mitochondria from 3 mice. * p <0.05, n=4 per group. (C) β-hydroxybutyate (BHB) increases mPT threshold in acutely isolated cortical mitochondria from Kcna1-null (KO) mice. Graph indicates the different treatment groups normalized to KO mitochondria. For comparison, the mPT threshold in mitochondria from ketogenic diet (KD)-treated KO mice is shown (black bar). Each bar indicates the mean ± S.E.M, and each mitochondrial sample tested came from a single mouse. ** p<0.01, *** p<0.001, one-way ANOVA with post-hoc Bonferonni correction, n=4–5 samples per group. (D and E) Traces depicting thresholds for Ca2+-induced mPT in acutely isolated hippocampal mitochondria from B6 wild-type mice (D) and age-matched CypD-null mice (E). For these experiments, calcium was administered through micro-infusion. In CypD-null mice, neither BHB nor ACA – each 1 mM – were effective in raising the mPT threshold. Cyclosporin A (1 μM) was also not effective in altering Ca2+-induced mPT. (F) Summary bar graphs showing the mean ± S.E.M. of mPT thresholds for the various treatment groups. Note: the thresholds for calcium-induced mPT differed not only amongst the treatment groups, but also varied as a function of different background strain, and whether mice were wild-type or null mutants. (G) Ca2+ -induced swelling of control mitochondria with or without pre-incubation of 1 mM ACA and 1 mM BHB in control C57Bl6 mice. These experiments were performed under de-energized conditions and with a Ca+2 ionophore in order to equilibrate this cation across the mitochondrial inner membrane. Ca+2 (200 μM)-induced swelling increased in control, as well as ketone-treated samples. ACA and BHB treatment did not inhibit Ca+2-induced mitochondrial swelling as shown. Data represent n=5 animals in each group and samples were measured in two replicates per animal. (H) Atractyloside reverses the anti-seizure effects of the KD. Mean daily seizure frequencies over a 3-day recording period in Kcna1-null mice after various two-week treatments – those administered: (1) a standard diet (SD) only; (2) a KD plus atractyloside (ATRAC) 30 mg/kg IP daily; (3) a SD plus ATRAC; (4) a KD only; and (5) the selective cyclophilin D (CypD) inhibitor NIM811, 10 mg/kg IP daily. NIM811 resulted in an anti-seizure effect similar to what was observed with either the full KD or KB (see Figure 1). One-way ANOVA followed by Tukey test; **, p <0.01. N=8–9 per group. For panels (B, F, G and H), data points reflect the mean ± S.E.M.
To further explore this effect, and having observed the similarity in the effect of KB with cyclosporin A (CsA), an established inhibitor of mPT that binds specifically to the cyclophilin D (CypD) subunit of the mPT complex,28 we next tested the effects of these substrates in acutely isolated hippocampal mitochondria from mice lacking CypD (Ppif-null mutants). We observed that the effect of KB on raising the mPT threshold was fully prevented in CypD-deficient animals (Fig 3D, 3E, 3F).
Finally, to delineate the effects of KB on mPT threshold, we asked whether KB could enhance calcium buffering under de-energized conditions22,29 which eliminate the potential contribution of other species known to modulate the pore.18 With this approach, modulation of mPT is limited to the effects of calcium on components of mPT, and not upon mitochondrial membrane potential, redox status or ROS production.21 We found that the decrease in light absorbance of mitochondria – a reflection of increased swelling – was unaffected by KB (Fig 3G). These data indicate that the KB effect on mitochondrial calcium buffering and mPT threshold are energy-dependent, consistent with our previous findings demonstrating KB effects on mitochondrial function.7,14,30 While a direct interaction of KB with CypD was not confirmed, these results nevertheless indicate that the anti-seizure activity of KB requires this specific subunit of the mPT complex.
Activation of mPT Negates Anti-Seizure Effects of the KD
Given these results, we reasoned that if mPT is a critical variable in the neuroprotective activity of the KD and KB, activation of mPT in epileptic Kcna1-null mice should prevent the anti-seizure effects of the KD. Indeed, we found that chronic intraperitoneal (IP) administration of atractyloside (ATRAC) – an mPT activator which binds to the adenine nucleotide translocase (ANT) subunit of the mPT complex,31 resulted in a loss of seizure control afforded by the KD (Fig 3H). Importantly, to more directly demonstrate that inhibition of CypD can result in anti-seizure effects, we found that the selective CypD antagonist N-methyl-4-isoleucine-cyclosporin (NIM811), a non-immunosuppressive CsA analogue which is devoid of effects on calcineurin32 – an inherent confound with CsA (which inhibits both mPT and calcineurin) – induced both anti-seizure and LTP protective effects in Kcna1-null mutants when administered peripherally (Fig 3H).
KD and KB Afford Nootropic Effects in Epileptic Brain
To assess whether the KD and/or KB exert additional functional neuroprotective effects in the epileptic brain, we conducted further behavioral and cellular electrophysiological experiments in Kcna1-null mice. For the former, we employed the Barnes maze test of spatial learning/memory.33 Compared to wild-type mice, SD-fed Kcna1-null animals showed a significant increase in the latencies for finding the hidden escape chamber during testing, and KD treatment fully rescued this memory deficit in Kcna1-null mice (Fig 4A). These findings were corroborated by cellular electrophysiological measures of synaptic plasticity (EPSP). We found that SD-fed Kcna1-null mice exhibited intrinsic impairment of CA1 hippocampal LTP evoked by Schaffer collateral stimulation using a high-frequency protocol (Fig 4B). The EPSP amplitudes of SD-fed WT and SD-treated Kcna1-null mice at 60 min post-high frequency stimulation (HFS) differed significantly, but the KD fully restored LTP maintenance in Kcna1-null mice to a level comparable to SD-fed WT animals (Fig 4B). Moreover, osmotic mini-pump administration of BHB alone resulted in a similar restoration of compromised CA1 hippocampal LTP in these epileptic mice (Figure 4C).
Figure 4.
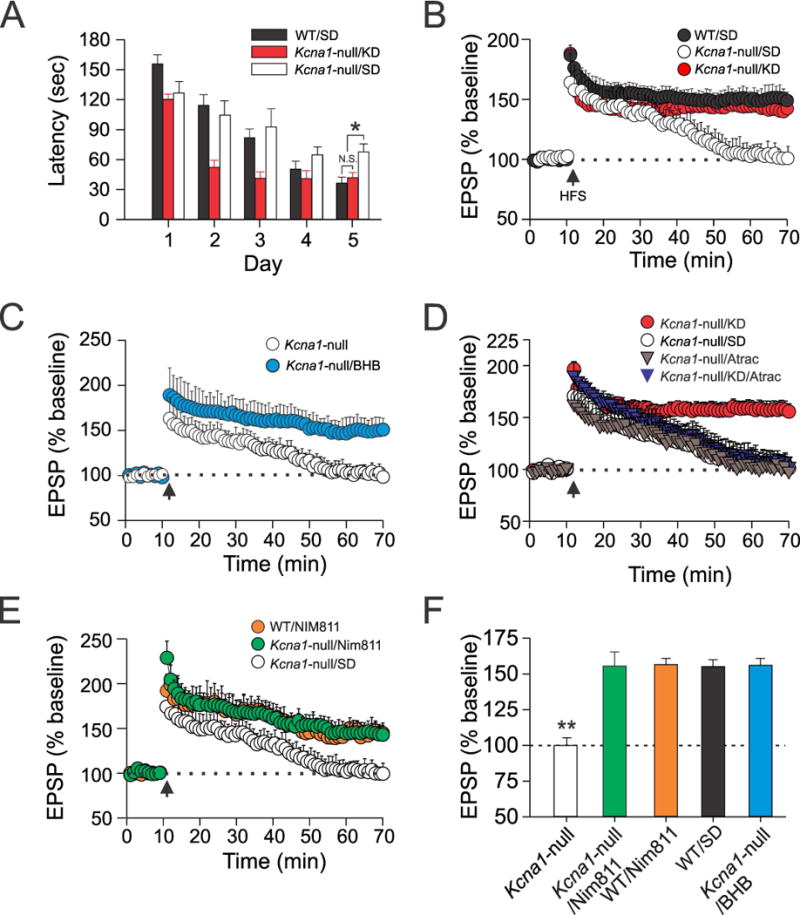
The KD and KB induce nootropic (cognitive-enhancing) effects in epileptic brain. (A) Average time latencies for mice to find the escape chamber in the Barnes maze are summarized for three groups of mice: (1) wild-type (WT) mice fed a standard diet (SD); (2) Kcna1-null mice fed a SD; (3) Kcna1-null mice fed a KD. KD-treated Kcna1-null mice exhibited improved performance in the Barnes maze (as indicated by the shorter latencies) compared to Kcna1-null mice fed only a SD. Interestingly, Kcna1-null mice fed the KD performed as well or better than WT mice fed the SD. N=18–24 per group *, p<0.05. (B) Kcna1-null mice fed a KD exhibit restoration of LTP induction after a two-week treatment period (red circles; N=16 slices from 6 mice). Standard diet (SD)-fed Kcna1-null mice show impairment of LTP induction (open circles; N=13 slices from 6 mice) compared to WT mice fed regular rodent chow (black circles). (C) Chronic in vivo BHB administration for one week via osmotic mini-pumps results in restoration of intrinsically impaired hippocampal LTP in Kcna1-null mice (N=14 slices from 8 mice). (D) ATRAC treatment negates the protective effects of the KD on intrinsic impairment of CA1 hippocampal long-term potentiation (LTP) induced by high-frequency stimulation (HFS; 100 Hz, 1 sec) of Schaffer collaterals. ATRAC alone does not influence the EPSP amplitude after HFS in Kcna1-null mice. N=12–14 slices from 6–8 mice in each group. (E) NIM811 – similar to either the KD or beta-hydroxybutyrate (BHB) alone restores intrinsic impairment of hippocampal LTP (n=10–16 slices from 4–7 mice in each group). (F) Summary data of EPSP amplitudes 1 hour after LTP induction for various treatment groups. For all panels, data points and bars reflect the mean ± SEM. **, p<0.01.
To demonstrate that modulation of mPT can mirror the effects of the KD, we observed that IP administration of ATRAC prevented the protective effects of the KD against LTP impairment in Kcna1-null mice (Fig 4D). NIM811 was comparably effective as the KD and KB (Fig. 4B, 4C, 4E). Summary data for the various treatments are provided in Fig. 4F.
Phenobarbital Blocks SRS But Impairs Hippocampal LTP
Finally, to provide evidence that the effects on LTP are independent of the anti-seizure action activity of these metabolic treatments, we compared the effects of phenobarbital (PB), a broad-spectrum anti-seizure drug, to those induced by the KD and KB. PB blocked SRS in Kcna1-null mice to a degree similar to these metabolic treatments (Fig 5A), but in contrast to the KD and KB, PB was unable to restore compromised CA1 hippocampal LTP (Fig 5B). Based on this inability to render beneficial effects on LTP, we hypothesized that PB would not positively affect mPT. Indeed, we found that PB, at a clinically significant concentration (100 μM), was unable to alter the threshold for calcium-induced mPT in acutely isolated mitochondria from normal mice (Fig 5C, 5D).
Figure 5.
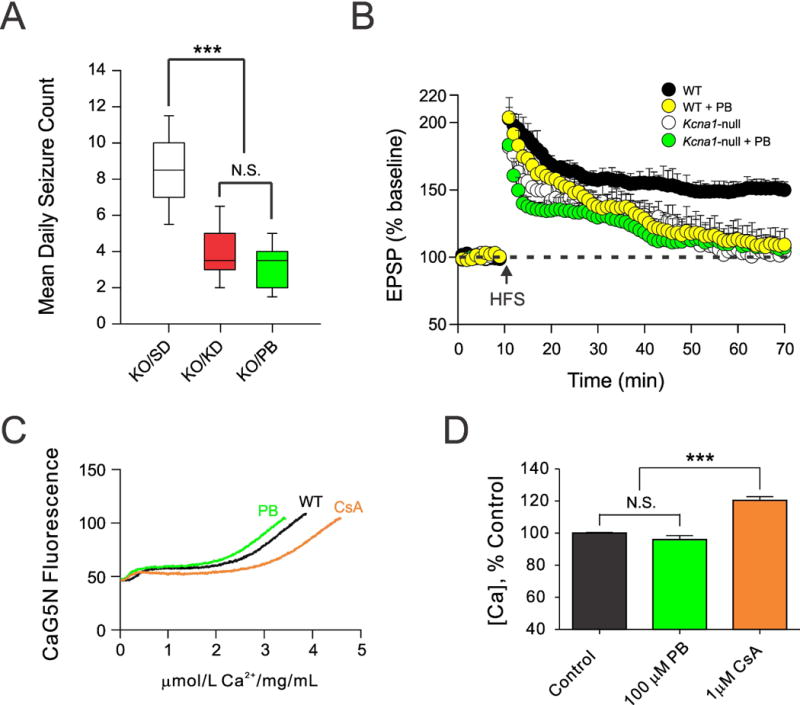
Comparison between the ketogenic diet (KD) and phenobarbital (PB) on spontaneous recurrent seizures, hippocampal long-term potentiation (LTP) and mPT thresholds. (A) Seizure frequency in Kcna1-null (KO) mice over a 3-day recording period after anti-seizure treatments. Both the KD and PB (30 mg/kg/d IP daily for 10 days) resulted in a significant decrease in seizure frequency compared to standard diet (SD) treatment, and were similarly effective (N=9 in each group). (B) PB worsened LTP maintenance in WT mice, and did not confer protective effects on the EPSP amplitude in Kcna1-null animals. PB (30 mg/kg daily) was administered IP over 10 days. Each set of traces was derived using 16 hippocampal slices from 9 mice. (C) Representative traces demonstrating that PB (100 μM) did not significantly alter the threshold for Ca+2-induced mPT in acutely isolated mitochondria from C57/Bl6 mice, compared to the control compound 1 μM CsA. (D) Summary bar graph reflecting the means ± S.E.M. of mPT thresholds in the different treatment groups. N=10–13 separate runs in each group from mitochondria prepared from 9 mice. *, p<0.05, ***, p<0.001; N.S., not significant. One-way ANOVA followed by Tukey test.
DISCUSSION
There are several major findings from the current study. First, we present evidence that the functional neuroprotective effects of the anti-seizure KD in a clinically relevant developmental mouse model of epilepsy can be explained by KB alone, independent of glycolytic restriction. This addresses a fundamental question of how the KD might exert its broad-spectrum clinical effects, and clarifies the protective role of BHB relative to ACA and acetone.3 Second, we show that KB effects require the CypD subunit of the mPT complex, a still controversial entity linked to regulation of mitochondrial respiratory function, calcium and free radical homeostasis, among other actions.18,34 Third, and most importantly, while it has been well understood that mitochondrial bioenergetic impairment can occur as a consequence of prolonged or repetitive seizure activity,35,36 either dietary or pharmacological inhibition of mPT results in consistent anti-seizure effects. Collectively, our results provide the first direct evidence that mPT may be pivotal to seizure control.
Earlier, Kovac and colleagues37 demonstrated that prolonged in vitro “seizure-like” activity, induced with low-Mg2+ in rat glio-neuronal co-cultures, resulted in depolarization of the mitochondrial membrane potential and mPT opening with subsequent cell death. These effects were reversed by CsA (a known blocker of CypD), but this clinically utilized drug is also known to inhibit calcineurin (or protein phosphatase 3) which can regulate N-methyl-D-aspartate and γ-aminobutyric acid type A receptors.38,39 Nevertheless, despite recent data that demonstrate anti-seizure and neuroprotective properties of CsA,37,40 neither clinical nor direct experimental evidence implicating mPT as a pharmacological target for epilepsy has been forthcoming.
Cyclophilin D is a key component of the mPT complex, and has been implicated in a number of acute brain and neurodegenerative disorders.18,41 There are a growing number of studies supporting the notion that pharmacological or genetic knockdown of CypD elevates the threshold for mPT opening through modulation of intracellular calcium homeostasis.18,41 For example, CypD deficiency has been found to restore deficits in synaptic plasticity due to Aβ toxicity.42,43 Other reports have indicated that CypD deficiency suppresses mPT activation, and that the subsequent stabilization of calcium levels within mitochondria can enhance normal metabolic functions within this organelle, such as ATP generation, reduction in ROS and prevention of release of pro-apoptotic factors – all of which have been cited as key pathophysiological events leading to axonal degeneration, cerebral ischemia and traumatic brain injury.44–46
Disruptions in energy supply with subsequent mPT opening and calcium influx are known to cause neuronal injury and death.19,37 Thus, in the context of seizure-induced impairment of mitochondrial bioenergetics, it is noteworthy to consider both pharmacological and metabolism-based approaches that are neuroprotective. Earlier, we showed that KB enhance mitochondrial ATP production through increased oxidation of NADH,1,14 raise the threshold for calcium-induced mPT,10 and can counter the effects of mitochondrial respiratory chain inhibitors such as rotenone and 3-nitroproprionic acid.14 The current study expands on these earlier findings, and provides direct evidence that mPT may be an important regulatory target for epilepsy therapeutics. Specifically, our results indicate that inhibition of mPT via KB likely occurs indirectly through decreased ROS levels, as well as enhanced mitochondrial respiration, NADH oxidation, and ATP production.1,10,14
Due to the striking ketosis associated with the KD, and the ease with which BHB can be measured in blood, it is no surprise that historical attention has focused on the role of KB as mediators of the KD’s clinical effects. Currently, KB are generally understood to correlate inconsistently with seizure control, but definitive evidence one way or another remains lacking.47 A major impediment has been the fact that peripheral measurements (whether in urine, blood or via exhalation) may not represent true surrogates of brain levels. Until accurate brain ketone levels can be measured during KD treatment, the controversy surrounding the mechanistic role of KB may persist.
Recently, KB has been shown to produce a multiplicity of other effects, including potential activation of ATP-sensitive potassium channels,24,48 blockade of vesicular glutamate release,49 inhibition of class I histone deacetylases (HDACs),50 and attenuation of the NLRP3 inflammasome51 – all of which could induce neuroprotective and homeostatic effects on cellular function. While these distinct non-overlapping mechanisms could work in concert to preserve biological function, especially in the face of disease or insult, it is possible that inhibition of mPT and subsequent mitochondrial changes may in part secondarily induce beneficial pleiotropic effects. In this regard, while such a hypothesis remains purely speculative at present, there is recent evidence linking mPT to histone-mediated apoptosis.52
In summary, while we have demonstrated that KB may be a fundamental mediator of the anti-seizure effects of the KD through modulation of mPT, our findings have been detailed in only a single animal model of epilepsy. Whether KB inhibition of mPT is central to KD effects in other relevant epilepsy models remains unknown, and will need to be confirmed in future studies. Nevertheless, it is reasonable to hypothesize that this mechanism may also be relevant to KB action both during development and in other neurological disease states,18–20 especially since KB are prominently involved in brain maturation and appear to have broad neuroprotective properties.1,6,9,10 Additionally, mPT has been increasingly implicated in clinical conditions such as hypoxia-ischemia, traumatic brain injury, neuroinflammation and neurodegenerative disorders (Alzheimer’s, Parkinson’s, and Huntington’s diseases as well as amyotrophic lateral sclerosis).18 Importantly, the present study is the first to directly identify the mPT complex as a therapeutic target in epilepsy, and our results support the rationale for further development of mPT inhibitors for the treatment of epilepsy.
Acknowledgments
The authors are grateful for the technical assistance of Heather C. Milligan, Mohamed Abdelwahab, Kaeli Samson, Andrew Sachs, and Thomas Bautista. This work was supported by NIH grants RO1 NS070261 (JMR/DYK), RO1 NS48191 (PGS), RO1 NS062993 (JWG/PGS), RO1 NS072179 (KAS), and P30 NS051220; Kentucky Spinal Cord and Head Injury Research Trust (JWG/PGS); Nebraska LB692 Grant (KAS); Health Future Foundation Award (TAS); the Barrow Neurological Foundation, Phoenix, Arizona (JMR/DYK); the Canadian Institutes of Health Research and the Alberta Children’s Hospital Research Institute for Child and Maternal Health (JMR).
Footnotes
Potential Conflicts of Interest
D.Y.K.: None. K.A.S.: None. T.A.S.: None. J.D.P.: None. J.C.W.: None. Y.A.: None. J.W.G.: None. P.G.S.: None. J.M.R.: Scientific Advisory Boards, Charlie Foundation and Accera Pharma.
References
- 1.Maalouf M, Rho JM, Mattson MP. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res Rev. 2009;59:293–315. doi: 10.1016/j.brainresrev.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol. 2012;3:59. doi: 10.3389/fphar.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masino SA, Rho JM. Mechanisms of Ketogenic Diet Action. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies [Internet] 4. Bethesda (MD): National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- 4.Neal EG, Chaffe H, Schwartz RH, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7:500–506. doi: 10.1016/S1474-4422(08)70092-9. [DOI] [PubMed] [Google Scholar]
- 5.Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005;28:109–121. doi: 10.1007/s10545-005-5518-0. [DOI] [PubMed] [Google Scholar]
- 6.Davis LM, Pauly JR, Readnower RD, et al. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008;86:1812–1822. doi: 10.1002/jnr.21628. [DOI] [PubMed] [Google Scholar]
- 7.Kim do Y, Rho JM. The ketogenic diet and epilepsy. Curr Opin Clin Nutr Metab Care. 2008;11:113–120. doi: 10.1097/MCO.0b013e3282f44c06. [DOI] [PubMed] [Google Scholar]
- 8.Edmond J, Auestad N, Robbins RA, et al. Ketone body metabolism in the neonate: development and the effect of diet. Fed Proc. 1985;44:2359–2364. [PubMed] [Google Scholar]
- 9.Kashiwaya Y, Takeshima T, Mori N, et al. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc Natl Acad Sci USA. 2000;97:5440–5444. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim do Y, Davis LM, Sullivan PG, et al. Ketone bodies are protective against oxidative stress in neocortical neurons. J Neurochem. 2007;101:1316–1326. doi: 10.1111/j.1471-4159.2007.04483.x. [DOI] [PubMed] [Google Scholar]
- 11.Chen D, Steele AD, Lindquist S, et al. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- 12.Halagappa VK, Guo Z, Pearson M, et al. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 13.Malaisse WJ, Lebrun P, Rasschaert J, et al. Ketone bodies and islet function: 86Rb handling and metabolic data. Am J Physiol. 1990;259:E123–130. doi: 10.1152/ajpendo.1990.259.1.E123. [DOI] [PubMed] [Google Scholar]
- 14.Kim do Y, Vallejo J, Rho JM. Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J Neurochem. 2010;114(1):130–141. doi: 10.1111/j.1471-4159.2010.06728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srivastava S, Kashiwaya Y, King MT, et al. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 2012;26:2351–2362. doi: 10.1096/fj.11-200410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wenzel HJ, Vacher H, Clark E, et al. Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia. 2007;48:2023–2046. doi: 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simeone TA, Simeone KA, Samson KK, et al. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol Dis. 2013;54:68–81. doi: 10.1016/j.nbd.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 19.Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464(1):111–21. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uchino H, Hatakeyama K, Morota S, et al. Cyclophilin-D inhibition in neuroprotection: dawn of a new era of mitochondrial medicine. Acta Neurochir Suppl. 2013;118:311–315. doi: 10.1007/978-3-7091-1434-6_61. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan PG, Rabchevsky AG, Waldmeier PC, et al. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79(1–2):231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- 22.Brown MR, Geddes JW, Sullivan PG. Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J Bioenerg Biomembr. 2004;36(4):401–406. doi: 10.1023/B:JOBB.0000041775.10388.23. [DOI] [PubMed] [Google Scholar]
- 23.Fenoglio-Simeone KA, Wilke JC, Milligan HL, et al. Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia. 2009;50:2027–2034. doi: 10.1111/j.1528-1167.2009.02163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36:32–40. doi: 10.1016/j.tins.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasselbalch SG, Knudsen GM, Jakobsen J, et al. Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am J Physiol. 1995;268:E1161–1166. doi: 10.1152/ajpendo.1995.268.6.E1161. [DOI] [PubMed] [Google Scholar]
- 26.Kawamura M, Jr, Ruskin DN, Masino SA. Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci. 2010;30:3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garriga-Canut M, Schoenike B, Qazi R, et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9(11):1382–7. doi: 10.1038/nn1791. [DOI] [PubMed] [Google Scholar]
- 28.Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266:3376–3379. [PubMed] [Google Scholar]
- 29.Jiang D, Sullivan PG, Sensi SL, et al. Zn2+ induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem. 2001;276(50):47524–47529. doi: 10.1074/jbc.M108834200. [DOI] [PubMed] [Google Scholar]
- 30.Maalouf M, Sullivan PG, Davis L, et al. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145(1):256–64. doi: 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowser DN, Petrou S, Panchal RG, et al. Release of mitochondrial Ca2+ via the permeability transition activates endoplasmic reticulum Ca2+ uptake. FASEB J. 2002;16:1105–1107. doi: 10.1096/fj.01-0828fje. [DOI] [PubMed] [Google Scholar]
- 32.Waldmeier PC, Feldtrauer JJ, Qian T, et al. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 33.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 34.Giorgio V, von Stockum S, Antoniel M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA. 2013;110(15):5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cock HR, Tong X, Hargreaves IP, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002;48(3):157–68. doi: 10.1016/s0920-1211(01)00334-5. [DOI] [PubMed] [Google Scholar]
- 36.Waldbaum S, Liang LP, Patel M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J Neurochem. 2010;115(5):1172–82. doi: 10.1111/j.1471-4159.2010.07013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovac S, Domijan AM, Walker MC, et al. Prolonged seizure activity impairs mitochondrial bioenergetics and induces cell death. J Cell Sci. 2012;125(Pt 7):1796–806. doi: 10.1242/jcs.099176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swope SL, Moss SJ, Raymond LA, et al. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/s1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- 39.Bannai H, Levi S, Schweizer C, et al. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron. 2009;62(5):670–682. doi: 10.1016/j.neuron.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 40.Jung S, Yang H, Kim BS, Chu K, et al. The immunosuppressant cyclosporin A inhibits recurrent seizures in an experimental model of temporal lobe epilepsy. Neurosci Lett. 2012;529(2):133–8. doi: 10.1016/j.neulet.2012.08.087. [DOI] [PubMed] [Google Scholar]
- 41.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77(5):1111–1122. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14(10):1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010;1802(1):198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrientos SA, Martinez NW, Yoo S, et al. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31(3):966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korde AS, Pettigrew LC, Craddock SD, Pocernich CB, et al. Protective effects of NIM811 in transient focal cerebral ischemia suggest involvement of the mitochondrial permeability transition. J Neurotrauma. 2007;24(5):895–908. doi: 10.1089/neu.2006.0122. [DOI] [PubMed] [Google Scholar]
- 46.Readnower RD, Pandya JD, McEwen ML, et al. Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. J Neurotrauma. 2011;28(9):1845–1853. doi: 10.1089/neu.2011.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kossoff EH, Rho JM. Ketogenic diets: evidence for short- and long-term efficacy. Neurotherapeutics. 2009;6(2):406–414. doi: 10.1016/j.nurt.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Juge N, Gray JA, Omote H, et al. Metabolic control of vesicular glutamate transport and release. Neuron. 2010;68:99–112. doi: 10.1016/j.neuron.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015 Feb 16; doi: 10.1038/nm.3804. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu ZG, Ni SY, Chen GM, et al. Histones-mediated lymphocyte apoptosis during sepsis is dependent on p38 phosphorylation and mitochondrial permeability transition. PLoS One. 2013;8(10):e77131. doi: 10.1371/journal.pone.0077131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]