

Abstract
Background and Purpose
Acute communicating hydrocephalus and cerebral edema are common and serious complications of subarachnoid hemorrhage (SAH), whose etiologies are poorly understood. Using a mouse model of SAH, we determined if soluble epoxide hydrolase (sEH) gene deletion protects against SAH-induced hydrocephalus and edema by increasing levels of vasoprotective eicosanoids and suppressing vascular inflammation.
Methods
SAH was induced via endovascular puncture in WT and soluble epoxide hydrolase knockout (sEHKO) mice. Hydrocephalus and tissue edema were assessed by T2-weighted magnetic resonance imaging (MRI). Endothelial activation was assessed in vivo using T2*-weighted MRI after intravenous administration of iron oxide particles linked to anti-vascular cell adhesion molecule-1 (VCAM-1) antibody 24h after SAH. Behavioral outcome was assessed at 96h after SAH with the open field and accelerated rotarod tests.
Results
SAH induced an acute sustained communicating hydrocephalus within 6h of endovascular puncture in both WT and sEHKO mice. This was followed by tissue edema, which peaked at 24h after SAH and was limited to white matter fiber tracts. sEHKO mice had reduced edema, less VCAM-1 uptake and improved outcome compared to WT mice.
Conclusions
Genetic deletion of sEH reduces vascular inflammation and edema, and improves outcome after SAH. sEH inhibition may serve as a novel therapy for SAH.
Keywords: Subarachnoid Hemorrhage, Acute Communicating Hydrocephalus, Soluble Epoxide Hydrolase, VCAM-1, Edema, EETs
Introduction
Acute communicating hydrocephalus and global cerebral edema are common life-threatening complications of subarachnoid hemorrhage (SAH), which occur in 20% of patients1-3 and are independent risk factors for poor outcome3,4. While both represent a dysfunction in water handling within the cranium5, their etiologies are likely different and possibly unrelated. Current treatments for hydrocephalus and cerebral edema are largely supportive and do not target the underlying pathologies, especially for hydrocephalus, which leaves some patients requiring permanent ventricular shunts due to unremitting disease6,7. A better understanding of the mechanisms underlying these complications is needed to identify viable therapeutic targets.
Mouse models of SAH have been employed to study mechanisms of cerebral edema8,9 but do so without acknowledging the potential contribution of hydrocephalus to brain water content10. To date, there are no studies describing hydrocephalus in mouse models of SAH. In the current study, we employ high field magnetic resonance imaging (MRI) to study the timing, severity and localization of acute communicating hydrocephalus as well as cerebral edema occurring simultaneously in the mouse endovascular puncture model of SAH.
Vasogenic edema is caused by extravasation of ions and proteins through a disrupted blood-brain barrier and is often preceded by activation of the vascular inflammatory cascade11. Within endothelial cells, nuclear translocation of NF-κB is an essential step in the expression of endothelial pro-inflammatory adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1)12. Epoxyeicosatrienoic acids (EETs) are eicosanoids formed by cytochrome P450 enzymes in brain glia and endothelium13 which oppose VCAM-1 expression by blocking NF-κB translocation14. We have previously demonstrated that mice with elevated levels of EETs due to genetic deletion of their metabolizing enzyme soluble epoxide hydrolase (sEH knockout, sEHKO mice) are protected from experimental cerebral ischemia15 and delayed microvascular dysfunction16 after experimental SAH. Further, we have shown that patients with genetic polymorphisms that reduce sEH activity have improved outcomes after SAH17. We hypothesized that the beneficial effects of EETs also modulate acute inflammation and edema formation after SAH.
Methods
An extended version of methods can be found in the online supplementary material. Please see http://stroke.ahajournals.org.
Animals
All experiments were approved by the institutional animal care and use committee of Oregon Health & Science University. Adult (8-12 week) male wild-type (WT) C57BL/6J mice obtained from Jackson Laboratories and homozygous sEHKO mice in the C57BL/6J background were used15
Endovascular Puncture
SAH was induced in mice using the endovascular perforation technique as previously described 18. Briefly, a nylon suture was introduced into the internal carotid artery and advanced into the Circle of Willis to induce a hemorrhage. In sham operated animals the suture was advanced without arterial perforation.
Physiological Monitoring
In a subset of non-survival surgeries, animals were monitored invasively for intracranial pressure (ICP), mean arterial pressure (MAP) and cerebral blood flow (CBF) with laser doppler (LDF) for 30 minutes following SAH
Vascular Cell Adhesion Molecule-1 (VCAM-1)-bound micro particles of iron oxide (MPIO)
We conjugated monoclonal rat anti-mouse CD106 (VCAM-1; 1510-01 Southern Biotech) or mouse IgG1 (0102-01 Southern Biotech) to Dynabeads MyOne Tosylactivated MPIOs (Invitrogen) per manufacturer’s instructions. MPIOs were used at a final concentration of 5mg MPIO per ml in PBS plus 0.1% BSA and 0.05% Tween 20 at 37 °C.
MRI
MR imaging employed a Bruker-Biospin 11.75T small animal MR system with Paravision 4.0. To quantify brain size, ventricle size and white matter edema, a T2-weighted image set was obtained at baseline, 6h, 12h, 24h and 72h after SAH or sham surgery. To quantify nanoparticle uptake 24h after SAH or sham surgery, 3D T2*-weighted images were obtained without injection of MPIOs, then 80 minutes after injection with VCAM-1 MPIO or IgG MPIO at 4.5 mg Fe/kg body weight. All image processing was done using tools from FSL19-21 and Jim (Xinapse Systems).
Histology
Fixed and embedded brains from mice 72h after SAH were cut into 8um sections and stained with hematoxilin & eosin. Images were obtained on a BX40 microscope (Olympus).
Behavioral Assessment
A subset of animals were survived for 96h after surgery and tested for behavioral deficit. To test general locomotor activity, mice were placed in an open field apparatus (Columbus instruments). To test sensorimotor function, mice were placed on the accelerated rotarod (Columbus instruments).
Statistics
Group data are expressed as mean ± sem unless otherwise stated. LDF, ICP, MAP, ventricular volume, brain size, brain edema, and behavioral performance were compared between groups using a two-way ANOVA with repeated measures where appropriate Vacuolization data and VCAM-1 MPIO uptake were compared using one way ANOVA. The Holm-Sidak post-hoc test was used for all pairwise comparisons.
Results
SAH induces immediate and sustained rise in ICP
Endovascular puncture caused blood to fill the basal cisterns and subarachnoid space of the mouse within minutes. T2-weighted MRI scans within 30 minutes showed blood had flowed retrogradely as far as the 4th ventricle (Figure 1a; note the change in T2 signal in CSF spaces from white to black). We monitored ICP, MAP and CBF in a cohort of WT and sEHKO mice for 30 minutes following vessel puncture. At the moment of hemorrhage, ICP spiked as CBF decreased (figure 1b). Arterial pressure also increased. The ICP waveform changed after hemorrhage with increased pressure fluctuations during systole indicating reduced CSF compliance (Figure 1b inset). In both sEHKO and WT mice, ICP rose considerably at the time of hemorrhage (WT 60.2 ± 9.9 mmHg vs sEHKO 69.2 ± 6.0 mmHg) and returned to an elevated set point by 30 minutes (WT 33.0 ± 3.2 mmHg vs sEHKO 30.4 ± 1.1 mmHg). LDF also decreased substantially at the time of SAH (WT 55.2 ± 19.2 % vs sEHKO 37.3 ± 6.8 % of baseline) and returned to a point below baseline by 30 minutes (WT 80.9 ± 28.2 %baseline vs sEHKO 77.9 ± 20.7 %baseline). Baseline MAP appeared lower in sEHKO mice but was not statistically significant (WT 74.3 ± 1.0 mmHg vs sEHKO 60.8 ± 2.3 mmHg) and rose to similar levels after hemorrhage (WT 83.6 ± 8.6 mmHg vs sEHKO (85.4 ± 6.8 mmHg) before returning back towards baseline by 30 minutes (WT 74.2 ± 1.8 mmHg vs sEHKO 70.2 ± 2.1 mmHg). There were no significant differences between WT and sEHKO mice at any time point.
Figure 1.
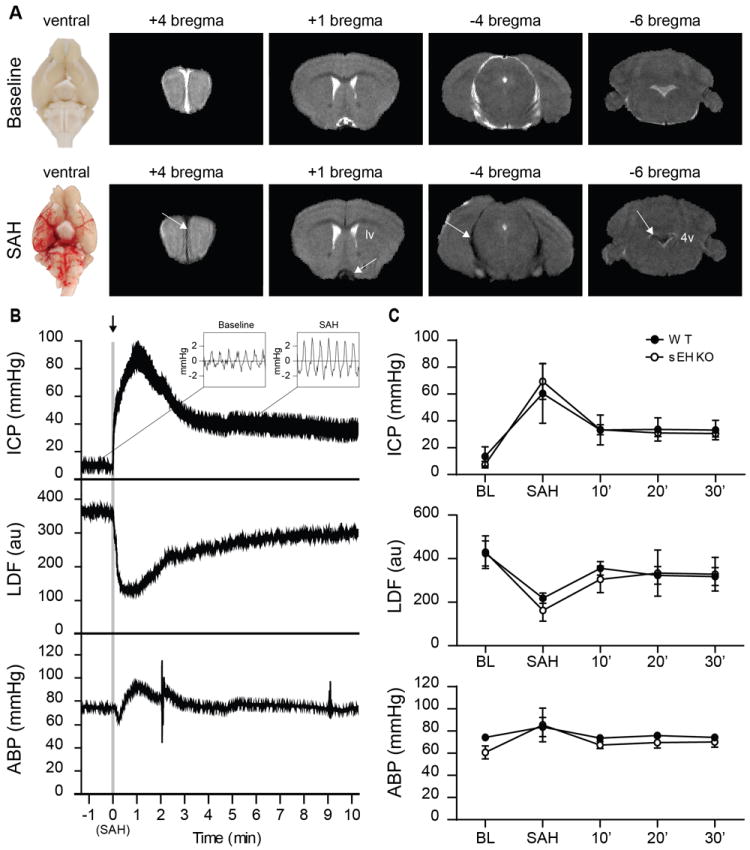
SAH causes similar changes to physiology in both WT and sEHKO mice. A.) Representative gross images (left) and T2-weighted MRI (right) of naïve (top) and SAH (bottom) mice 30 minutes after induction. Blood within CSF space causes the T2-weighted signal to change from white to black (arrows). B.) Representative tracing of ICP with view of waveform (inset), LDF and MAP in a WT SAH mouse during SAH. C.) Average changes in ICP, LDF, and MAP in WT(n=5) and sEHKO(n=5) mice after SAH. There were no significant differences between groups.
Acute communicating hydrocephalus forms rapidly and persists after SAH
Before and at several time points after (6h, 12h, 24h, and 72h) inducing SAH, we imaged the animals using T2-weighted MRI. Within 6h of SAH, ventricular volume had increased substantially (Figure 2a). Signs of mass effect in the form of central sulcus effacement were also evident at 6h (figure 2a arrowheads). To ascertain whether the hydrocephalus was obstructive or communicating in nature, we looked for signs of uneven enlargement of the ventricular system or large blood clots within the cerebral aqueduct that could block CSF flow. In all scans, the enlargement of the ventricular system was uniform from the anterior horns of the lateral ventricles to the cisterna magna. Additionally, we found enlargement of the CSF spaces outside of the ventricular system including the intrathecal space around the spinal cord and the subarachnoid space separating the cortex from midbrain structures (figure 2a arrows). WT and sEHKO mice had a similar increase in ventricular volume at each time points beginning as early as 6h (WT 181.3 ± 4.9 %baseline vs sEHKO 185.4 ± 17.5 %baseline) and persisting the length of the study at 72h (WT 206.2 ± 13.2 %baseline vs sEHKO 208.2 ± 23.9 %baseline). Total brain volume also increased in both groups beginning at 6h (WT 0.67 ± 0.5 % vs sEHKO 1.7 ± 0.8 % change from baseline) persisting to 72h (WT 3.0 ± 0.5 vs sEHKO 2.2 ± 0.6 change from baseline). Brain tissue volume (total brain volume – ventricular volume) did not increase significantly during the study (supplemental figure 1). There were no differences between WT and sEHKO mice at any time point.
Figure 2.
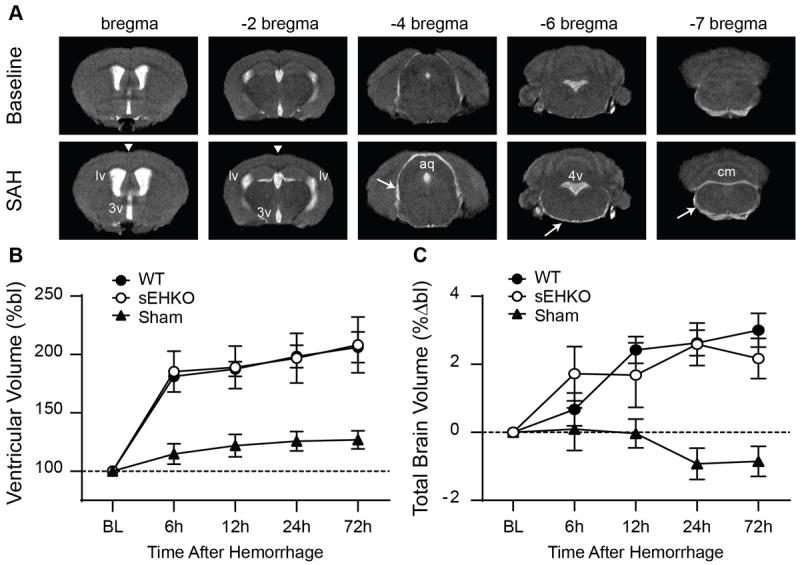
SAH induces acute communicating hydrocephalus in both WT and sEHKO mice. A.) representative T2-weighted MRI images at baseline (top) and 6h after SAH (bottom). Expansion of the CSF space occurs at all levels including lateral ventricles (lv), third ventricle (3v), cerebral aqueduct (aq), fourth ventricle (4v), cisterna magna (cm) and the subarachnoid space (arrows). Effacement of the central sulcus is also apparent at 6h after SAH (arrowheads). B.) Ventricular volume changes WT (n=10), sEHKO (n=7) SAH animals and sham(n=5). There is no significant difference between WT and sEHKO SAH mice. C.) Total brain volume change in WT (n=10, sEHKO (n=10) SAH animals and sham (n=5). There is no significant difference between WT and sEHKO SAH mice.
sEHKO mice have reduced periventricular white matter edema after SAH
Edema, visualized by hyperintensities on T2-weighted images, began to form specifically in the periventricular white matter of SAH mice within 6h then peaked at 24h (figure 3a). The white matter structures affected included the corpus callosum and the dorsal hippocampal commissures (figure 3a arrows). Histological sections of the corpus callosum showed substantial vacuolization of the white matter in WT mice. sEHKO mice had significantly less edema at 24h (WT 3.1 ± 0.5 % brain volume vs sEHKO 1.7 ± 0.4 % brain volume p < 0.05) and 72h (WT 2.7 ± 0.5 % brain volume vs sEHKO 0.7 ± 0.2 % brain volume p < 0.05) compared to WT mice (figure 3b). Vacuolization found in histological sections of the corpus callosum was also significantly reduced in sEHKO mice compared to WT mice (WT 1.6 ± 0.4 a.u. vs sEHKO 1.2 ± 0.09 a.u. p < 0.05). Further histological analysis of the white matter identified subtle evidence of axonal injury within the corpus callosum of SAH mice and variable expression of microglial marker Iba-1 (supplemental figure 2 and 3).
Figure 3.
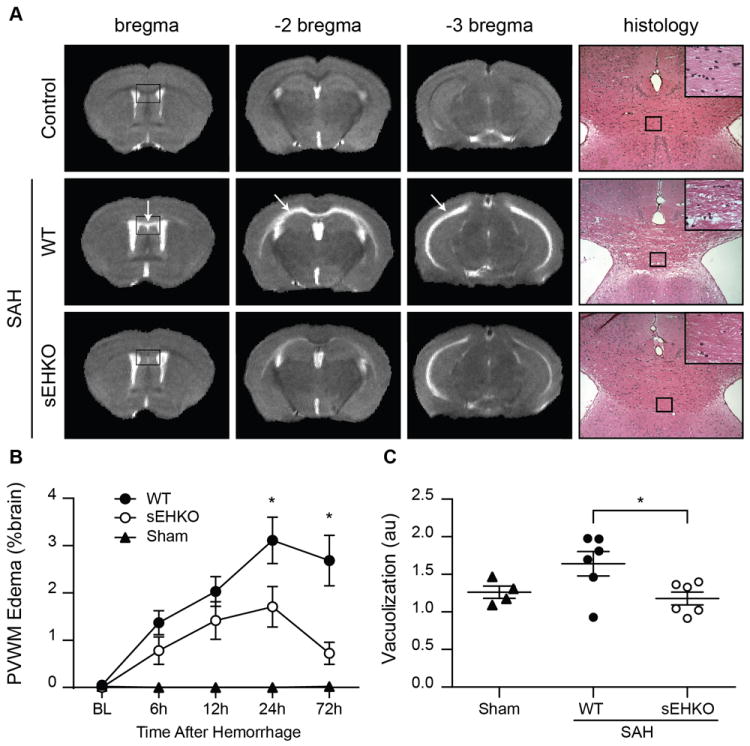
sEHKO mice have less edema than WT mice. A.) representative T2-weighted MRI images in sham (top) WT SAH (middle) and sEHKO SAH (bottom) at 24h after SAH. Edema forms in the specifically within the white matter of the corpus callosum and dorsal hippocampal commissures (arrows). Histological sections (right) of the corpus callosum 72h after SAH show vacuolization within the white matter of SAH mice. B.) Periventricular white matter edema formation as a percent of brain volume in WT (n=10), sEHKO (n=7) and sham (n=5) mice. sEHKO mice have less edema at 24h and 72h after SAH (*=p<0.05). C.) Vacuolization determined by changes in mean pixel intensity of the corpus callosum in WT SAH (n=6), sEHKO SAH (n=6) and sham (n=4) mice 72h after SAH. sEHKO mice have less vacuolization than WT mice (*=p<0.05).
sEHKO mice have reduced expression VCAM-1 following SAH
As a measure of vascular inflammation in vivo, we studied the expression of VCAM-1 on brain endothelium using VCAM-1 MPIOs which have been well validated in previous studies to detect endothelial VCAM-1 expression in vivo22-24. 24h after SAH or sham surgery, we injected the labelled particles into the vasculature and imaged the animals via T2*-weighted MRI. VCAM-1 MPIO uptake showed as hypointensities on the scans and was greatest in the vasculature along the midline and surrounding the midbrain of the mice (figure 4a). Immunolabeling of brain tissue sections from SAH mice show that in areas with high MPIO uptake, VCAM-1 primarily localized in the large caliber cerebral veins with relative sparing of the microvasculature (Supplemental figure 4). We found very little uptake in the IgG-tagged MPIO mice as well as the sham operated mice. After injection (80min), bound VCAM-1 MPIO caused a negative shift in the voxel intensity histogram when compared to pre-injection scans (figure 4b). sEHKO mice had reduced overall VCAM-1 uptake in the whole brain compared to WT mice (WT 9.3 ± 1.9 × 103 voxels vs sEHKO 3.7 ± 1.3 × 103 voxels p < 0.05) (figure 4c).
Figure 4.
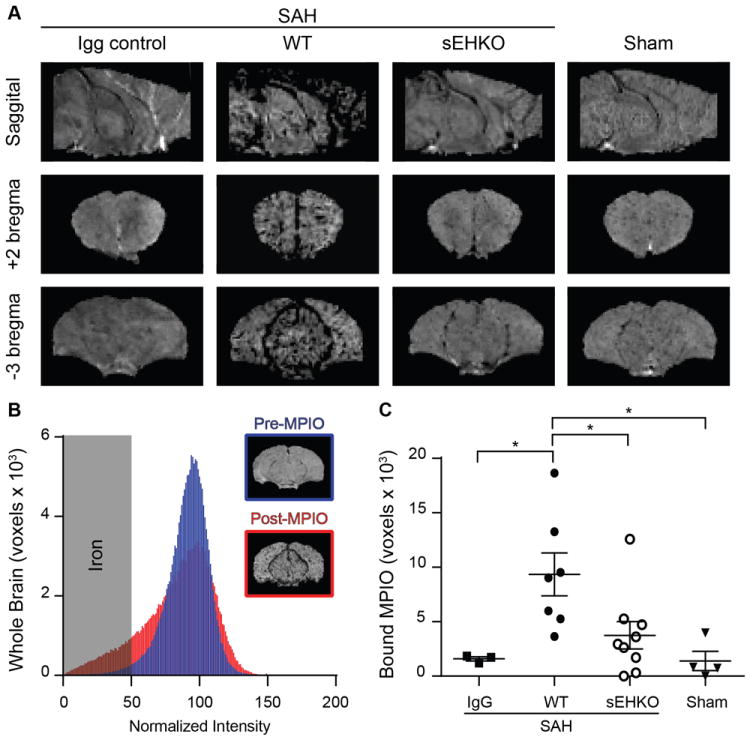
sEHKO mice have less VCAM-1 expression than WT mice after SAH. A.) Representative T2* weighted images in SAH mice (IgG control, WT, and sEHKO) and sham operated mice. Deposition of the VCAM-1 MPIO causes hypointensity on MRI. B.) Representative normalized histogram of voxel intensities within a single WT brain 24h after SAH pre-MPIO (blue) and post-MPIO (red) injection (80min). Voxels below the normalized intensity threshold were used to measure the extent of iron particle deposition. C.) Quantification of MPIO deposition as the increase in iron particles detected 80min after injection in IgG control (n=3) WT SAH (n=7) sEHKO SAH (n=9) and Sham (n=4). sEHKO mice have reduced MPIO deposition compared to WT mice (*=p<0.05).
sEHKO mice have improved outcome after SAH
To assess behavioral outcome after SAH, the open field test and the accelerated rotarod were performed 96h after SAH or sham was induced. Sham operated sEHKO mice performed differently on the open field test than sham operated WT mice (WT 6944.4 ± 433.9 cm vs sEHKO 5675.9 ± 418.2 cm), which did not allow direct comparison between WT and sEHKO mice (figure 5a). Comparing within genotype, we found that SAH caused a reduction in total movement on the open field in WT (sham 6944.4 ± 433.9 cm vs SAH 5106.9 ± 525.1 cm p <0.05) but not sEHKO mice (sham 5675.9 ± 418.2 cm vs SAH 4663.2 ± 589.4 cm). On the accelerated rotarod, WT and sEHKO sham mice remained on the rotating rod for similar amounts of time (WT 42.5 ± 3.0 s vs sEHKO 43.1 ± 3.5 s), allowing a direct comparison between genotypes after SAH (figure 5b). At 96h after SAH, sEHKO mice remained on the accelerating rotarod longer compared to WT mice (WT 36.5 ± 2.7 s vs sEHKO 47.3 ± 2.5 s p <0.05).
Figure 5.
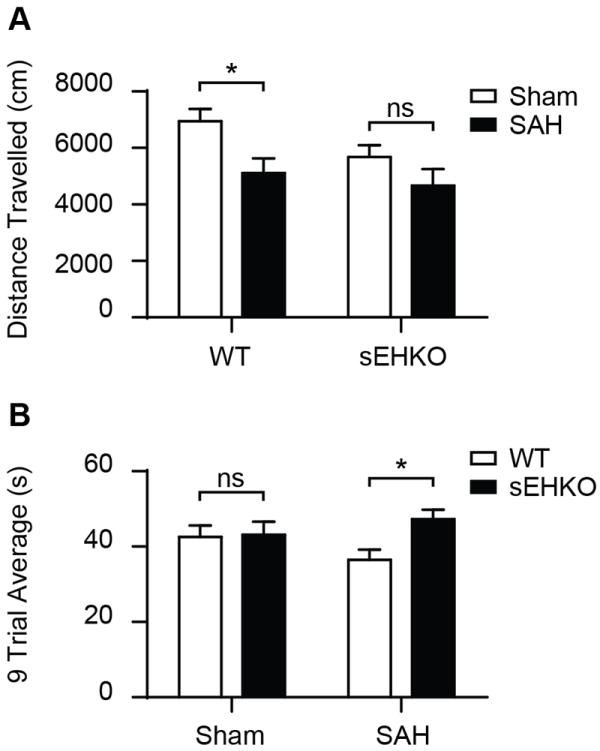
sEHKO mice have improved outcome after SAH. A.) 96h after SAH (WT n=12, sEHKO n=10) or sham (WT n=10, sEHKO n=10), mice were placed in the open field for 10 min. SAH reduces movement on open field for WT mice (*=p<0.05) but not sEHKO mice (ns=no significance) B.) 96h after SAH (WT=12, sEHKO=8) or sham (WT n=10, sEHKO n=10), animals were timed on the accelerated rotarod for three trials per day for three days. The average time to fall over the nine trials was increased in the sEHKO SAH mice compared to the WT SAH mice (*=p< 0.05).
Discussion
The present study shows that acute communicating hydrocephalus and cerebral edema occur simultaneously in the mouse endovascular puncture model of SAH. The onset of hydrocephalus was rapid and sustained, occurring within 6h of SAH and remaining present for at least 72h. Cerebral edema formed primarily in the white matter fiber tracts and followed a time course that was distinct from that of the hydrocephalus. Specifically, edema formation was gradual, peaked at 24h and began to recede by 72h after SAH, while hydrocephalus persisted. Mice with genetic deletion of sEH, which have elevated basal levels of EETs, had a similar severity of hydrocephalus and brain swelling compared to WT mice, but significantly less edema formation within the white matter. We tested one potential mechanism of this edema formation, vascular inflammation indicated by expression of VCAM-1, which was reduced in sEHKO mice compared to WT. Finally, sEHKO mice have improved behavioral outcome after SAH.
We have identified additional features of the endovascular puncture model that mimics human disease seen clinically. Specifically, a substantial number of SAH patients present with acute hydrocephalus at admission25 and our model replicates this rapid time scale, a feature not found in other rodent models of hydrocephalus10,26,27. While we see the hydrocephalus lasting at least 72h in our model, we do not know the time frame at which hydrocephalus resolves, so whether our model represents the 10-20% of those chronically hydrocephalic patients that require implantation of semi-permanent shunts to manage unremitting disease7,6 remains to be determined. Edema formation in our model shares similarities with the human condition as well. While human brains have a substantially larger percentage of white matter than mice28, the phenomenon that edema forms preferentially within white matter, sparing the grey matter3,29, holds true in this model.
The anti-inflammatory effects of EETs are well documented in mice14,30-32 and inform us about the etiology of hydrocephalus and edema after SAH. First, the rate of formation, severity, and persistence of hydrocephalus are not altered in sEHKO mice which have elevated levels of basal EETs. This supports our hypothesis that hydrocephalus is likely mechanical in nature and not inflammatory. We have previously shown that CSF flow is blocked within minutes of SAH by fibrin deposition within the CSF pathways18 and is likely sufficient to induce acute communicating hydrocephalus after SAH. Additionally, while hydrocephalus is unchanged in the sEHKO mice, edema is significantly reduced in association with the reduction in vascular inflammation as measured by reduced VCAM-1 expression in the cerebrovasculature. VCAM-1 is a cell adhesion molecule expressed by damaged or activated endothelium and important to the trans-migration of infiltrating inflammatory cells33. Elevated levels of VCAM-1 have been detected in the plasma and CSF of SAH patients34,35. Its activation is generally accepted to be a part of the brain inflammatory response33,36 and has been associated with breakdown of the blood-brain barrier in other models of disease22,23,37,38. In this context, we interpret our finding to mean that the inflammatory response to SAH is responsible in part for cerebral edema after SAH and that modulation of sEH to increase levels of EETs is protective.
Important considerations in the interpretation of our data are the multiple complications other than edema and hydrocephalus previously documented in this model, including elevated intracranial pressure (ICP)18, and acute18 and delayed cortical hypoperfusion16. Therefore, the improvement in behavior may result from improvement in any of these parameters, independent of hydrocephalus. Furthermore, one must consider the wide range of beneficial effects of EETs in the brain, which include their anti-inflammatory14, anti-thrombotic39, cytoprotective40 and vasodilator properties13. Thus it is likely that the benefits of sEH deletion are multifactorial. Overall, these findings complement our previous work demonstrating improved outcomes after SAH in both humans17 and mice16 when sEH activity is altered, and support the hypothesis that sEH inhibition may be a viable therapeutic strategy in SAH.
In conclusion, the endovascular puncture model in the mouse is useful to study the etiology of acute communicating hydrocephalus and cerebral edema after SAH. Genetic deletion of sEH reduces vascular inflammation and edema formation in this model as well as improving outcome. This finding adds to the body of literature supporting further investigation of sEH inhibition as a therapeutic target in SAH.
Supplementary Material
Acknowledgments
Funding Sources:
DS-NHLBI F30 HL108624, Oregon Brain Institute.
JC-Veteran’s Affairs Merit Award
MP-NIH/NCI grant 1R21CA167302–0, Nishijima Hospital
NA-NINDS R01 NS070837
Footnotes
Disclosures: None
References
- 1.Hasan D, Vermeulen M, Wijdicks EF, Hijdra A, van Gijn J. Management problems in acute hydrocephalus after subarachnoid hemorrhage. Stroke. 1989;20:747–753. doi: 10.1161/01.str.20.6.747. [DOI] [PubMed] [Google Scholar]
- 2.Milhorat TH. Acute hydrocephalus after aneurysmal subarachnoid hemorrhage. Neurosurgery. 1987;20:15–20. doi: 10.1227/00006123-198701000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: Frequency, predictors, and impact on outcome. Stroke. 2002;33:1225–1232. doi: 10.1161/01.str.0000015624.29071.1f. [DOI] [PubMed] [Google Scholar]
- 4.Dupont S, Rabinstein AA. Extent of acute hydrocephalus after subarachnoid hemorrhage as a risk factor for poor functional outcome. Neurol Res. 2013;35:107–110. doi: 10.1179/1743132812Y.0000000122. [DOI] [PubMed] [Google Scholar]
- 5.Ko S-B, Choi HA, Parikh G, Schmidt JM, Lee K, Badjatia N, et al. Real time estimation of brain water content in comatose patients. Ann Neurol. 2012;72:344–350. doi: 10.1002/ana.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoh BL, Kleinhenz DT, Chi YY, Mocco J, Barker FG., 2nd Incidence of ventricular shunt placement for hydrocephalus with clipping versus coiling for ruptured and unruptured cerebral aneurysms in the nationwide inpatient sample database: 2002 to 2007. World Neurosurg. 2011;76:548–554. doi: 10.1016/j.wneu.2011.05.054. [DOI] [PubMed] [Google Scholar]
- 7.Vale FL, Bradley EL, Fisher WS. The relationship of subarachnoid hemorrhage and the need for postoperative shunting. J Neurosurg. 1997;86:462–466. doi: 10.3171/jns.1997.86.3.0462. [DOI] [PubMed] [Google Scholar]
- 8.Li T, Wang H, Ding Y, Zhou M, Zhou X, Zhang X, et al. Genetic elimination of nrf2 aggravates secondary complications except for vasospasm after experimental subarachnoid hemorrhage in mice. Brain Res. 2014;1558:90–99. doi: 10.1016/j.brainres.2014.02.036. [DOI] [PubMed] [Google Scholar]
- 9.Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1β in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40:2519–2525. doi: 10.1161/STROKEAHA.109.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloch O, Auguste KI, Manley GT, Verkman AS. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J Cereb Blood Flow Metab. 2006;26:1527–1537. doi: 10.1038/sj.jcbfm.9600306. [DOI] [PubMed] [Google Scholar]
- 11.Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. American Journal of Physiology - Heart and Circulatory Physiology. 2014;306:H317–H325. doi: 10.1152/ajpheart.00182.2013. [DOI] [PubMed] [Google Scholar]
- 12.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: Nf-kappa b and cytokine-inducible enhancers. The FASEB Journal. 1995;9:899–909. [PubMed] [Google Scholar]
- 13.Iliff JJ, Jia J, Nelson J, Goyagi T, Klaus J, Alkayed NJ. Epoxyeicosanoid signaling in cns function and disease. Prostaglandins Other Lipid Mediat. 2010;91:68–84. doi: 10.1016/j.prostaglandins.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. Anti-inflammatory properties of cytochrome p450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, et al. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–2078. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siler DA, Martini RP, Ward JP, Nelson JW, Borkar RN, Zuloaga KL, et al. Protective role of p450 epoxyeicosanoids in subarachnoid hemorrhage. Neurocrit Care. 2015;22:306–319. doi: 10.1007/s12028-014-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martini RP, Ward J, Siler DA, Eastman JM, Nelson JW, Borkar RN, et al. Genetic variation in soluble epoxide hydrolase: Association with outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2014;121:1359–1366. doi: 10.3171/2014.7.JNS131990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siler DA, Gonzalez JA, Wang RK, Cetas JS, Alkayed NJ. Intracisternal administration of tissue plasminogen activator improves cerebrospinal fluid flow and cortical perfusion after subarachnoid hemorrhage in mice. Transl Stroke Res. 2014;5:227–237. doi: 10.1007/s12975-014-0329-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TEJ, Johansen-Berg H, et al. Advances in functional and structural mr image analysis and implementation as fsl. NeuroImage. 2004;23(Supplement 1):S208–S219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 20.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. NeuroImage. 2002;17:825–841. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- 21.Jenkinson M, Smith S. A global optimisation method for robust affine registration of brain images. Medical Image Analysis. 2001;5:143–156. doi: 10.1016/s1361-8415(01)00036-6. [DOI] [PubMed] [Google Scholar]
- 22.McAteer MA, Sibson NR, von zur Muhlen C, Schneider JE, Lowe AS, Warrick N, et al. In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide. Nat Med. 2007;13:1253–1258. doi: 10.1038/nm1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoyte LC, Brooks KJ, Nagel S, Akhtar A, Chen R, Mardiguian S, et al. Molecular magnetic resonance imaging of acute vascular cell adhesion molecule-1 expression in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1178–1187. doi: 10.1038/jcbfm.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gauberti M, Montagne A, Marcos-Contreras OA, Le Béhot A, Maubert E, Vivien D. Ultrasensitive molecular mri of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke. 2013;44:1988–1996. doi: 10.1161/STROKEAHA.111.000544. [DOI] [PubMed] [Google Scholar]
- 25.van Gijn J, Hijdra A, Wijdicks EF, Vermeulen M, van Crevel H. Acute hydrocephalus after aneurysmal subarachnoid hemorrhage. J Neurosurg. 1985;63:355–362. doi: 10.3171/jns.1985.63.3.0355. [DOI] [PubMed] [Google Scholar]
- 26.Braun KP, Dijkhuizen RM, de Graaf RA, Nicolay K, Vandertop WP, Gooskens RH, et al. Cerebral ischemia and white matter edema in experimental hydrocephalus: A combined in vivo mri and mrs study. Brain Res. 1997;757:295–298. doi: 10.1016/s0006-8993(97)00345-4. [DOI] [PubMed] [Google Scholar]
- 27.Tada T, Kanaji M, Shigeaki K. Induction of communicating hydrocephalus in mice by intrathecal injection of human recombinant transforming growth factor-β1. Journal of neuroimmunology. 1994;50:153–158. doi: 10.1016/0165-5728(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 28.Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proceedings of the National Academy of Sciences. 2000;97:5621–5626. doi: 10.1073/pnas.090504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nag S, Manias JL, Stewart DJ. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009;118:197–217. doi: 10.1007/s00401-009-0541-0. [DOI] [PubMed] [Google Scholar]
- 30.Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, et al. Anti-inflammatory effects of [omega]-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-ii-dependent hypertension. Journal of Cardiovascular Pharmacology. 2013;62:285–297. doi: 10.1097/FJC.1090b1013e318298e318460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, et al. Endothelial cyp epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. The FASEB Journal. 2011;25:703–713. doi: 10.1096/fj.10-171488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmelzer KR, Kubala L, Newman JW, Kim I-H, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, del Zoppo GJ, Doolittle ND, et al. Immunologic privilege in the central nervous system and the blood-brain barrier. J Cereb Blood Flow Metab. 2013;33:13–21. doi: 10.1038/jcbfm.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaynar MY, Tanriverdi T, Kafadar AM, Kacira T, Uzun H, Aydin S, et al. Detection of soluble intercellular adhesion molecule—1 and vascular cell adhesion molecule—1 in both cerebrospinal fluid and serum of patients after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;101:1030–1036. doi: 10.3171/jns.2004.101.6.1030. [DOI] [PubMed] [Google Scholar]
- 35.Frijns CJM, Fijnheer R, Algra A, van Mourik JA, van Gijn J, Rinkel GJE. Early circulating levels of endothelial cell activation markers in aneurysmal subarachnoid haemorrhage: Associations with cerebral ischaemic events and outcome. Journal of Neurology, Neurosurgery & Psychiatry. 2006;77:77–83. doi: 10.1136/jnnp.2005.064956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenwood J, Etienne-Manneville S, Adamson P, Couraud P-O. Lymphocyte migration into the central nervous system: Implication of icam-1 signalling at the blood–brain barrier. Vascular Pharmacology. 2002;38:315–322. doi: 10.1016/s1537-1891(02)00199-4. [DOI] [PubMed] [Google Scholar]
- 37.Serres S, Mardiguian S, Campbell SJ, McAteer MA, Akhtar A, Krapitchev A, et al. Vcam-1-targeted magnetic resonance imaging reveals subclinical disease in a mouse model of multiple sclerosis. The FASEB Journal. 2011;25:4415–4422. doi: 10.1096/fj.11-183772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Q, Chu M, Wang H, Lu S, Gao H, Li P, et al. Sevoflurane preconditioning protects blood-brain-barrier against brain ischemia. Frontiers in Bioscience. 2011;E3:10. doi: 10.2741/e303. [DOI] [PubMed] [Google Scholar]
- 39.Heizer ML, McKinney JS, Ellis EF. 14,15-epoxyeicosatrienoic acid inhibits platelet aggregation in mouse cerebral arterioles. Stroke. 1991;22:1389–1393. doi: 10.1161/01.str.22.11.1389. [DOI] [PubMed] [Google Scholar]
- 40.Koerner IP, Jacks R, DeBarber AE, Koop D, Mao P, Grant DF, et al. Polymorphisms in the human soluble epoxide hydrolase gene ephx2 linked to neuronal survival after ischemic injury. The Journal of Neuroscience. 2007;27:4642–4649. doi: 10.1523/JNEUROSCI.0056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.