

Abstract
Background and Purpose
Hypoperfusion-induced thrombosis is an important mechanism for post-surgery stroke and cognitive decline, but there are no perioperative neuroprotectants to date. This study investigated whether prophylactic application of Edaravone, a free radical scavenger already used in treating ischemic stroke in Japan, can prevent infarct and cognitive deficits in a murine model of transient cerebral hypoxia-ischemia.
Methods
Adult male C57BL/6 mice were subjected to transient hypoxic-ischemic (tHI) insult that consists of 30-min occlusion of the unilateral common carotid artery and exposure to 7.5% oxygen. Edaravone or saline was prophylactically applied to compare their effects on cortical oxygen saturation, blood flow, coagulation, oxidative stress, metabolites, and learning-memory using methods that include photoacoustic imaging, laser speckle contrast imaging, solid state NMR and Morris water maze. The effects on infarct size by Edaravone application at different time-points after tHI were also compared.
Results
Prophylactic administration of Edaravone (4.5 mg/kg × 2, IP, 1 h before and 1 h after tHI) improved vascular reperfusion, oxygen saturation, and the maintenance of brain metabolites, while reducing oxidative stress, thrombosis, white-matter injury, and learning impairment after tHI insult. Delayed Edaravone treatment after 3 h post-tHI became unable to reduce infarct size.
Conclusions
Acute application of Edaravone may be a useful strategy to prevent post-surgery stroke and cognitive impairment, especially in patients with severe carotid stenosis.
Keywords: Stroke, Radicut, MCI-186, thrombosis, Virchow’s triad, antioxidants, cognitive decline
Introduction
Stroke, a dangerous complication of surgery, occurs in 5–8% of carotid endarterectomy and cardiac procedures and is more common in patients with symptomatic carotid stenosis1–4. A high incidence of silent brain infarct (~25%) and cognitive decline (33–83% in short term and 42% by 5 years) also occurs after coronary artery bypass grafting (CABG)5–6. Studies suggest that transient ischemia triggers a sustained pro-thrombotic tendency in the cerebral vasculature7. The addition of hypoxia to ischemia further accelerates coagulation and leads to infarction8–11. Yet, due to the risk of bleeding, anticoagulants are unsuitable for perioperative neuroprotection. To date, there is no specific drug to prevent post-surgery stroke and cognitive deficits12.
Edaravone is a free radical scavenger that readily crosses blood-brain-barrier (BBB)13,14. It has been used for treating acute ischemic stroke in Japan since 2001 and is in clinical trials in Europe15,16. When combined with tissue-type plasminogen activator (tPA), Edaravone confers synergistic benefits in both animal models (reduction of the mortality and infarct size) and stroke patients (a greater recanalization rate)9,17. Research indicates that Edaravone markedly reduces oxidative stress in multiple components of the neurovascular unit, including neurons, platelets, endothelial cells, and pericytes18–21, to interrupt the Virchow’s triad of thrombosis (the stasis of blood flow, endothelial injury, and hyper-coagulability)11. Hence, prophylactic administration of Edaravone may confer perioperative neuroprotection, but this utility is yet to be tested.
To this end, we examined the effects of prophylactic Edaravone treatment in a transient hypoxia-ischemia (tHI)-induced thrombotic stroke model9. In this model, the unilateral common carotid artery of adult mice is occluded when the animal inhales hypoxic gas (7.5% oxygen) via a face-mask. We have shown that as brief as 30 min tHI induces thrombosis, reperfusion deficits, and cortical infarct11. In the present study, we examined the effects of two doses of Edaravone (1 h before and 1 h after tHI) to mimic pre- and post-operative care for patients, and evaluated the therapeutic window of this treatment.
Our results showed that pre- and acute post-tHI application of Edaravone mitigates brain injury and learning-memory deficits, suggesting that Edaravone is a promising neuroprotectant against post-surgery stroke and cognitive decline. Further translational study of this prophylactic treatment merits consideration.
Material and Methods
Stroke surgery
Male C57BL/6 mice and Thy1-YFP mice at the age of 10 to 13 weeks were subjected to transient cerebral HI, as previously described9 with minor modifications. Animals weighing 22–30 g were anesthetized under 2% isofluorane to perform right common carotid artery occlusion (RCCAo) with two releasable knots of 4.0 silk sutures that were released after the hypoxic stress. After RCCAO, mice were infused with 7.5% O2/92.5% N2 via face-mask for 30 min, while the animal core body temperature was maintained at 37.5 ± 0.5°C by a rectal thermoprobe coupled to a heating lamp. After transient hypoxia-ischemia (tHI), mice were randomized for treatments and investigators analyzing brain infarction were blinded to the treatments. Treatment consisted of intraperitoneal injection of 3 or 4.5 mg/kg Edaravone (a gift of the Mitsubishi Tanabe Pharma Cooperation, Osaka, Japan) at designated time-points in each regimen, which was illustrated in the corresponding figure in the result section. The number of operated animals, the mortality, the outliers (>2 SD), and animals for quantification of infarction were also tabulated. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and conform to the National Institutes of Health Guide for Care and Use of Laboratory Animals, as well as the ARRIVE guidelines (Animals in Research: Reporting In-Vitro Experiments).
Post-surgery monitoring and measurement of brain infarction
Detection of infarction was performed by in-vivo TTC staining at 24 h after tHI by a lab member unaware of the treatment. Brains were sectioned into 0.7 mm thickness slices (8 per brain) and analyzed using the ImageJ 1.4 software (NIH, Bethesda, MD). The infarct size was quantified as the ratio of the infarcted area to the total area of the uninjured, contralateral hemisphere9.
Photoacoustic and ultrasound imaging
Ultrasound and photoacoustic imaging was performed using the Vevo LAZR photoacoustic micro-ultrasound imaging system (FUJIFILM VisualSonics, Toronto, Canada). All images including B-Mode imaging for high-resolution anatomical images, color Doppler imaging for blood flow in cerebral vessels, and photoacoustic imaging for oxygen saturation (SaO2) were generated with the LZ250 transducer at 21 MHz. Parametric maps of oxygen saturation in coronal sections of the brain co-registered with the B-Mode images were generated using a dual-wavelength approach22. Regions of interest (ROIs) were drawn to encompass the left and right cortical regions of the brain and SaO2 values were plotted over time.
Laser speckle contrast imaging and cortical oxygen saturation measurement
Cortical oxygen saturation (SaO2) was measured using the tissue oxygenation monitoring system (moorVMS-OXY™; Moor Instruments Inc. Delaware, USA). Briefly, anesthetized mice with skull exposed were measured in both cerebral hemispheres (2 mm posterior and 2 mm lateral to Bregma) using optic probe and recorded immediately after the RCCAO surgery, RCCAO plus hypoxia (30 min), and 1h post-tHI. CBF measurment was performed by a two-dimensional laser speckle contrast imaging system9 following the manufacturer’s instruction (MoorFLPI-2; Moor Instruments Inc). Briefly, post-stroke mice at 2 h post-tHI were re-anesthetized and placed in the prone position with the skull exposed yet unopened. CBF were measured in both hemispheres and recorded continuously for at least 7 min. The CBF images are shown with arbitrary units in a 16-color palette by the MoorFLPI software. SaO2 and CBF were quantified as the percentage to the contralateral hemisphere.
Immunohistochemistry and immunoblot
Immunohistochemistry and immunoblotting were performed as previously described9. The following antibodies were used: rabbit anti-fibrinogen (a gift of Dr. J. Degen), Alexa Fluor 488 isolectin GS-IB4 conjugate (#I21411, Invitrogen, Carlsbad, CA), rabbit anti-ERG (#92513, Abcam, Cambridge, MA), and mouse anti- β-actin (Sigma, St. Louis, MO).
Histological assay of vascular perfusion and white-matter injury
Evans blue albumin was used to evaluate vascular perfusion and BBB leakage as previously described9. Vascular perfusion was quantified using the CellSens software (Olympus, Tokyo, Japan) with adjusted threshold and background to obtain the total intensity of vasculature. The value of ipsilateral vasculature intensity was normalized to the contralateral hemisphere. To detect white-matter injury, Thy1-YFP-labeled nerve fiber and the FluoroMyelin Red (Molecular Probes, Invitrogen) in the external capsule were measured using CellSens (Olympus).
Detection of oxidative stress
Lipid peroxidation was measured by quantifying malondialdehyde (MDA) in brain extracts using a commercial kit (OxiSelect™; Cell Biolabs Inc., San Diego, CA), as previously described9. The superoxide production was determined by oxidized hydroethidine detection (HEt) (Invitrogen), as previously described9. Briefly, the HEt solution (1 mg/ml) was injected through the tail vein to the animals at 30 min before sacrifice. Oxidized HEt was detected at an emission of 590 nm and quantified as the ratio to DAPI-positive nuclei in four randomly selected visual fields (100X).
High-resolution magic angle spinning (HRMAS) nuclear magnetic resonance study
For ex vivo NMR analysis, tissue samples were prepared as previously reported23. Each sample was cut from the snap frozen tissue slice and thawed in 99.996 % saline deuterium oxide (D2O, Sigma) before loading to the sample holder/rotor (4 mm ZrO2). 99.996 % D2O containing 0.75 % 3-(Trimethylsilyl) propionic acid (TSP) was added to obtain a frequency-lock signal for NMR, and to serve as an internal reference for chemical shift and concentration measurements.
Solid-state HRMAS NMR experiments were conducted, as previously reported23, using a Bruker AVANCE 600 WB NMR spectrometer (Bruker Instruments, Inc., Billerica, MA) with a dedicated 4 mm HRMAS probe. Sample spinning rates were controlled in the range of 2,800 KHz (±2 Hz), or at the lower spin rate of 800 Hz if the rotor-synchronized delay alternating with nutation for tailored excitation (DANTE) sequence. A rotor-synchronized Carr–Purcell–Meibom–Gill (CPMG) pulse sequence was used to suppress broad signals from macromolecules. The one-dimensional NMR spetra were recorded using the repetition time of 5.0 s, the spectral width was 10 kHz and the number of transients of 256. The presence and the level of selected brain metabolites were determined based on their chemical shifts in the spectra.23
Morris water maze testing (MWM)
To evaluate working (short-term, trial-to-trial) and reference (longer-term, day-to-day) memory MWM procedure was performed as described24. Briefly, the animal position in the maze and latency to reach platform was recorded and analyzed using computer-assisted tracking system (Clever System Inc., Reston, Virginia). Beginning on post-tHI day 10, all mice were tested for acquisition in the MWM. Each animal received two trials per day, separated by a 5-min interval. The mice were placed in the pool and allowed to swim until they reached the platform or until 90 sec had elapsed. When mice were unable to locate the MWM platform within 90 sec, the experimenter guided the mice to the platform and kept for 20 sec. After 5 min, subjects were again released from the adjacent quadrant of the tank and allowed to swim to the platform. Then, before a probe test (on day 8 from the beginning of MWM) to determine in which quadrant they spent the most time was given, there was a two-day break during which the animal swam for 60 s without a platform in the arena.
Statistical analysis
Statistical analysis was performed using repeated measure ANOVA in MWM or one-way ANOVA followed by the post-test of Newman-Keuls or unpaired t-test for two samples. P values less than 0.05 were considered a significant difference. Values were expressed as mean ± SEM.
Results
Prophylactic Edaravone prevents reperfusion and reoxygenation deficits after tHI insult
To mimic cerebral hypoperfusion and hypoxia possibly encountered in surgical procedures, we subjected adult C57BL/6 mice to a tHI insult that consists of unilateral occlusion of the common carotid artery and exposure to 7.5% oxygen for 30 min (Figure 1A). Color flow Doppler imaging and 3D reconstruction showed that the right common carotid artery occlusion (RCCAO) reduced blood flow in the ipsilateral hemisphere and reversed blood flow in the right internal carotid artery (RICA) in the circle of Willis (Figure 1B). Photoacoustic imaging showed that oxygen saturation (SaO2) in the ipsilateral cortex was reduced from ~70% to ~61% upon RCCAO and plummeted to ~20% upon exposure to hypoxic air (Figure 1C). After 30 min tHI, the ipsilateral hemisphere showed poorer recovery of SaO2 than the contralateral cortex (~28% compared with ~39% in normoxia and ~53% versus ~70% under 100% oxygen) (Figure 1D) (n>3 times). This pattern suggests that a transient episode of HI may impair subsequent cortical oxygenation5.
Figure 1.

Transient hypoxia-ischemia (tHI) caused prolonged deficits in cortical oxygenation. A, A schematic diagram of the tHI model. MCA/ICA/ECA, middle, internal, and external carotid artery. RCCA, the right common carotid artery. B, Doppler flow imaging and 3D reconstruction showed reduced perfusion in the ipsilateral hemisphere and the reversal of blood flow in the right ICA (RICA) in the Circle of Willis after RCA occlusion (RCCAO). C, Co-registered anatomical image (B-mode micro-ultrasound) and oxygen saturation (SaO2 in a red-white-blue color-map by photoacoustic imaging) showed bilateral difference of SaO2 between the left (L, contralateral) and right (R, ipsilateral) cerebral cortex of an adult C57BL/6 mouse after RCCAO in normoxic (20.9% sO2) or hypoxic (7.5% O2) condition. D, After 30 min tHI, the ipsilateral cortex showed poorer recovery of SaO2 in either normixa or 100% O2 condition. Shown is the representative response (n>3).
Next we tested the effects of prophylactic Edaravone treatment (4.5 mg/kg × 2, 1 h before and 1 h after tHI). Despite comparable reduction of cortical SaO2 under RCCAO and combined RCCAO-hypoxia insult, Edaravone-treated animals exhibited better recovery of cortical SaO2 at 1 h post-tHI (p=0.012, n=8–9) (Figure 2A). Similarly, laser speckle contrast imaging showed greater recovery of cerebral blood flow (CBF) in Edaravone-treated than saline-treated animals at 2 h post-tHI (85% versus 64% of the contralateral hemisphere, p=0.011, n=8–9) (Figure 2B). Tail-vein injection of Evans blue dye at 2 h post-tHI also showed better vascular perfusion in the ipsilateral cortex of Edaravone-treated than saline-treated mice (83% versus 43%, p=0.006, n=4) (Figure 2C). Immunostaining showed greater fibrin(ogen) precipitation and immuno-reactivity to P-Selectin (a marker for endothelial activation) and GPIIb/CD41 (a platelet surface receptor) in the ipsilateral cortex (n=4, Figure DC and data not shown). Immunoblot analysis also uncovered greater deposition of fibrin(ogen) in the ipsilateral cortex in saline-treated mice at 2 h after tHI (n=3, p=0.04 compared to untouched animals) (Figure 2D). These data suggest that tHI induces thrombosis and reperfusion-reoxygenation deficit, which is attenuated by Edaravone treatment.
Figure 2.

Edaravone abated tHI-induced cortical hypoxia, thrombosis, and reperfusion deficits. A, Comparison of the cortical SaO2, normalized to the contralateral hemisphere, in saline- (n=8) or Edaravone- (n=9) treated mice under RCCAO, dual RCCAO-hypoxia, and at 1 h post-tHI. The Edaravone treatment led to better recovery of SaO2 after tHI (p=0.012 by t-test). B, Laser speckle contrast imaging showed that mice received Edaravone treatment (n=9) had significantly better CBF recovery at 2 h post-tHI than those received saline treatment (n=8) (p=0.011 by t-test). C, Tail vein injection of Evans blue dye at 2 h post-tHI also indicated greater recovery of CBF in the ipsilateral hemisphere in Edaravone- than saline-treated mice (p=0.006 by t-test, n=4 for each). D, Accordingly, saline-treated mice showed more widespread fibrin(ogen) deposition than Edaravone-treated mice at 2 h after tHI in immunostaining (n=4) and immunoblot analysis (n=3 for each). Scale bar: 250 μm.
Edaravone abates tHI-induced oxidative stress in the brain parenchyma and vascular wall
Tissue ischemia accumulates succinate in the mitochondria, which drives superoxide formation upon reperfusion by reverse electron transport25. To detect superoxide, tHI-injured mice were intravenously injected with hydroethidine, which emit 590 nm fluorescence when it is oxidized (oxHET) by superoxide. This analysis showed far more oxHET-positive cells in the ipsilateral hemisphere in saline-treated mice than Edaravone-treated mice at 2.5 h post-tHI (26.3% versus 5.3% of all DAPI+ nuclei, p=0.002, n=8) (Figure 3A). Double-labeling with ERG (a marker for endothelial cell) showed co-localization with oxHET+ nuclei in the ipsilateral cortex of saline-treated, but not Edaravone-treated animals (arrows in Figure 3A and Figure 3B). Saline-treated mice also exhibited greater increase of MDA, a marker of lipid peroxidation, than Edaravone-treated mice at 24 h after tHI (1.69 versus 1.30-fold, p=0.0004, n=5) (Figure 3C). These results suggest that prophylactic Edaravone treatment ameliorates tHI-induced oxidative stress in both brain parenchyma and the vascular endothelium.
Figure 3.
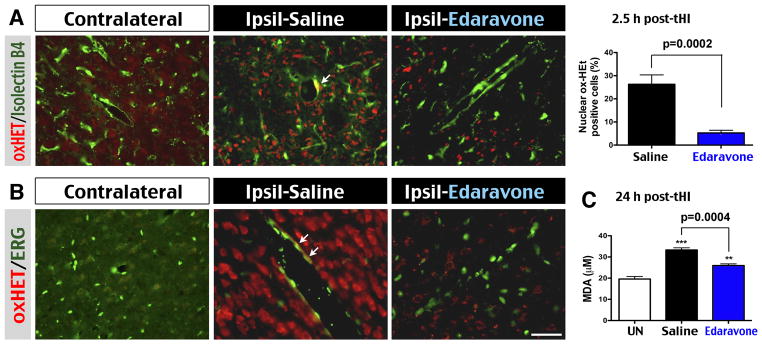
Edaravone abated oxidative stress in the brain parenchyma and vascular endothelium. A, Merged image of oxidized hydroethidine (oxHET) and isolectin B4 (an endothelial marker) labeling revealed more superoxide-positive cells in the ipsilateral hemisphere in saline-treated than Edaravone-treated mice at 2.5 h post-tHI (p=0.0002 by t-test, n=8 for each). B, Higher magnification showed frequent co-localization of oxHET in ERG+ endothelial cells (arrows) in the ipsilateral hemisphere of saline-treated mice. Note the seemingly more numerous oxHET+ nuclei due to brighter fluorescence under higher magnification in B than in A. C, Quantification of malondialdehyde (MDA) suggested significant reduction of lipid peroxidation by the Edaravone treatment at 24 h post-tHI (p=0.0004 by t-test, n=5 for each). **p<0.01, ***p<0.001 compared with UN group by unpaired t-test. Scale bar: 40 μm in A; 20 μm in B.
Edaravone prevents tHI-induced alteration of brain metabolites and learning deficits
Depletion of N-acetyl-aspartate (NAA) is a hallmark of brain damage after cerebral ischemia26. Using HRMAS NMR analysis of tHI-injured tissue samples, we found that 30 min tHI produced progressive reduction of NAA in saline-treated mice from 6 to 24 h recovery (n=4 each, p=0.034 compared to untouched mice). In contrast, the level of NAA was suppressed at 6 h, but recovered at 24 h in Edaraovne-treated mice (n=3, p=0.032 compared to saline-treated mice) (Figure 4A). We also subjected Thy1-YFP mice to tHI insult, and found attenuated white matter injury in the ipsilateral hemisphere after prophylactic Edaravone treatment (p=0.024, n=4) (Figure 4B). Using FluoroMyelin dye, we also found reduction and diffusion of myelin in the external capsule (EC) of ipsilateral hemisphere in saline-treated mice at 24 h post-tHI (n=4) (Figure 4B).
Figure 4.

Edaravone prevented tHI-induced alteration of brain metabolites and learning deficits. A, Representative HRMAS NMR spectra of the cortical tissue from brain slices of untouched, tHI-injured and saline- versus Edaravone-treated mice at 24 h recovery. The brain NAA (N-acetyl-aspartate) level was reduced to a similar degree in saline- and Edaravone- treated mice at 6 h post-tHI (n=4 each), but more diminished in saline-treated mice at 24 h recovery (n=3 for untouched and each treatment). Shown are p-values determined t-test. Labeled are the peaks for Lac (lactate), GABA (γ-aminobutyric acid), Cr/PCr (creatine and phosphocreatine), PC (P-choline), GPC (glucocerphosphocholine), Glu (glutamate), and myo-I (myo-inisitol). B, Thy1-YFP mice received Edaravone treatment showed less white-matter damage than those received saline (n=6 for each). Shown is the quantification of Thy1-YFP fluorescence in the ipsilateral external capsule (p=0.024 by t-test, n=4 each). Fluoromyelin stain showed attenuated and diffused signals in the ipsilateral external capsule (EC) in mice received the saline treatment (n=4 each). C, Effects of Edaravone on learning-memory preservation were assessed by the latency to location in run 1 (reference memory) and run 2 (working memory) in Morris water maze in 5 consecutive day, and the probe test on day 8. Saline-treated mice showed longer latency than sham and Edaravone-treated mice in day 4 and 5 (*p<0.05 by post-hoc LSD multiple comparison test, n=12 for each group), and less time spent in the probe quadrant than sham-treated mice on day 8 (p=0.047 by t-test). Values are mean ± standard error. Scale bar: 100 μm.
Next we compared the learning capacity in Morris water maze (MWM) between the sham and tHI-injured mice that received prophylactic saline or Edaravone treatment after 10 d recovery (n=12 each). In 5 consecutive days, sham and Edaravone-treated mice showed progressive reduction in the latency to find the platform, while saline-treated mice showed a slower decline in latency that became significant at day 4–5 (p=0.032 in run 1 [reference memory]; p=0.012 in run 2 [working memory] by ANOVA) (Figure 4C). On probe test, the saline-treated mice spent significantly less time in the former platform quadrant than sham animals (p=0.047 by t-test). Edaravone-treated mice spent more time in the platform quadrant than saline-treated mice on probe test, but the difference was not statistically significant. These results suggest that prophylactic Edaravone treatment attenuates tHI-induced cognitive impairment.
Prophylactic, but not delayed application of Edaravone, reduces tHI-induced infarction
Last, we compared the effects of saline (S) versus three regimens of Edaravone treatment in tHI-induced mortality and infarct size. In the first protocol (E1), 4.5 mg/kg Edaravone was applied at 1 h before and 1 h after tHI. In the second protocol (E2), three doses of 3 mg/kg Edaravone were injected at -1, 1, and 2 h after tHI to maintain the same total dosage. In the third protocol (E3), two doses of 4.5 mg/kg Edaravone were administered at 3 and 4 h after tHI to simulate delayed treatment. We recorded the mortality rate and infarct size at 24 h recovery.
This experiment showed that both E1 and E2 protocols conferred protection against tHI-induced mortality and cerebral infarct (Figure 5A). Specifically, the 24 h mortality rate in saline-treated mice was 27.6% (n=29), but dropped to 11.8% by the E1 protocol (n=17) and 16.7% by the E2 regimen (n=18) (Figure 5B). In saline-treated animals (n=19), the mean infarct size was 30.5% of the contralateral hemisphere, which was reduced to 18.8% by the E2 protocol (n=14, p=0.002) and to 11.1% by the E1 protocol (n=15, p<0.0001) (Figure 5C). In contrast, delayed Edaravone treatment in the E3 protocol led to a trend of higher mortality rate (38.5%, n=26) and a larger, more variable infarct size (33.8%, n=16) (Figure 5B and 5C).
Figure 5.

Prophylactic, but not delayed application of Edaravone abated tHI-induced infarction. A, Scheme of the saline (S) versus Edaravone treatment (E1, E2, and E3) against tHI injury and the representative TTC-stained brain slices at 24 h recovery for each group. B, Tabulation of the number of operated mice, death within 24 h, outliers (beyond mean ± 2 SD within group), mice included for infarct size comparison as the percentage to the contralateral hemisphere. C, (Left) Mean and the infarct size of individual animals in each treatment group. (Right) Statistical analysis of the infarct size among groups. Shown is the mean ± standard error. The p-values are determined by t-test.
These results suggest that acute prophylactic administration of Edaravone within a <3 h therapeutic window reduces tHI-induced death and infarction, but delayed Edaravone treatment may be harmful.
Discussion
Stroke is a dreaded complication of cardiovascular surgery and a particular threat to individuals with symptomatic carotid artery stenosis1,3,4. A high rate of silent cortical infarct also occurs after the cardiopulmonary bypass procedure. A recent study showed that 27.5% of CABG patients developed new brain infarct on diffusion-weighted imaging (DWI). Among them, 3.1% showed overt symptoms while the other 24.1% were clinically silent5. This high rate of silent infarct may relate to the phenomenon of post-surgery decline of cognitive functions6. In terms of the etiology, showers of micro-emboli during cardiovascular operations have been reported, and hypoperfusion is a critical mechanism linked to prolonged cardiopulmonary bypass, low cardiac output, and infarct in the watershed area2. Conceivably, a long duration of cortical hypoperfusion may exacerbate the stasis of blood flow and oxidative stress in the neurovascular unit, leading to hyper-coagulability and a greater pro-thrombotic tendency10,11.
Despite understanding of the etiology, there are no specific drugs to prevent post-surgery stroke or cognitive deficits to date12. This is because anticoagulants are unsuitable prophylactic agents due to the risk of hemorrhage. Past studies of pharmacological neuroprotectants suggested the benefits of reacemide (a NMDA receptor antagonist) and fluvastatin (a HMG-CoA reductase inhibitor) with pre- and post- surgery treatment for several days, while intraoperative infusion of magnesium (a multi-functional neuroprotectant) was ineffective (see Supplemental Table I)27–29. A recent study suggests the improvement of cognitive functions following experimental tibial fracture by peri-operative stimulation of the nicotinic receptor, but whether this treatment is able to prevent post-surgery stroke is uncertain30.
In the present study, we used a model of transient cerebral hypoxia-ischemia to assess the effect of Edaravone as a perioperative protectant against stroke and cognitive decline. Edaravone is a lipophilic free radical scavenger that crosses BBB and reduces post-stroke oxidative stress in multiple components of the neurovascular unit18–21. Edaravone also possesses anti-apoptosis and anti-inflammation functions in preclinical stroke models13,14. Clinically, Edaravone is used in Japan to treat acute ischemic stroke with a two-week, twice-a-day regimen15. A recent clinical trial in Europe shows that a shorter course of higher-dose Edaravone is also safe, tolerated, and improving the neurological outcome in ischemic stroke patients (although this study was not designed to determine efficacy)16. Finally, the combinational tPA-Edaravone treatment provides synergistic benefits of reduced infarct size in animal models and an increased recanalization rate for patients9,17. Given its clinical efficacy and safety in ischemia stroke therapy, we hypothesize that Edaravone may be an useful prophylaxis against post-surgery stroke and cognitive decline.
The tHI model used in this study has unique features tailored for testing this hypothesis. First, unlike focal ischemia models that block terminal branches of a cerebral artery, occlusion of RCCA in the tHI model only reduces CBF to half in the ipsilateral hemisphere 9, making it closer to the extent of hypoperfusion during surgery. Second, the addition of hypoxia to hypoperfusion wields endogenous components into in-situ thrombosis, making the tHI model ideal to study the cerebral vascular bed-specific hemostasis-coagulation31.
Our experiments showed that the dual hypoxia-hypoperfusion insult triggers long-lasting reduction of CBF and oxygen saturation after the release of vascular obstruction and returning to a normoxic environment (Figure 1 and 2). These results highlight the importance of maintaining adequate blood flow and brain oxygenation for patients during surgical operations. Interestingly, when Edaravone was administered in a regimen to mimic pre- and post-operative patient care, it significantly improved cerebral reperfusion and oxygenation, while abating in-situ thrombosis (Figure 2). These physiological effects by Edaravone are associated with marked reduction of superoxide in the brain parenchyma and the vascular wall following tHI (Figure 3). Our finding is consistent with the reports of Edaravone mitigating oxidative stress in the neurovascular unit18–21, which may attenuate thrombosis and improve CBF recovery. Prophylactic administration of Edaravone also attenuated tHI-induced brain damage and learning deficits based on a variety of assays (Figure 4). Finally, pre- and acute post-tHI administration of Edaravone in a 2 h window reduces the mortality rate and infarct size. However, delayed Edaravone treatment after 3 h is either ineffective or harmful (Figure 5). These findings suggest that prophylactic Edaravone is an effective neuroprotectant against post-surgery stroke and cognitive decline, but future studies are needed to ensure its therapeutic window in clinical settings.
A number of drugs, Edaravone included32, have been shown to reduce infarct size if they are applied before or during experimental focal cerebral ischemia, which could signify potential to prevent post-surgery stroke. Yet, Edaravone has the unique attribute of demonstrated utility in acute ischemic stroke therapy and a favorable safety and pharmacokinetics profile. Furthermore, the use of scavengers to counter free radical-induced thrombotic tendency in the neurovascular unit carries a smaller risk of bleeding, when compared to anticoagulants. Hence, to prioritize the candidates for perioperative protection, we suggest that Edaravone merits further evaluation for potential clinical trials.
Supplementary Material
Acknowledgments
We thank the Mitsubishi Tanabe Pharma Co. for the generous gift of Edaravone, and Mr. Tim Brown (Moor Instruments Inc.) for lending the moorVMS-OXYTM machine for experiments.
Sources of Funding:
This study was supported by the NIH grant NS084744 and a Children’s Healthcare of Atlanta Research Center Pilot Project grant (to C.-Y. K.).
Footnotes
Disclosure:
Dr. Abe received honoraria for speaking engagements with the Mitsubishi Tanabe Pharma Co. The other authors report no conflicts.
References
- 1.Selim M. Perioperative stroke. New England journal of medicine. 2007;356:706–713. doi: 10.1056/NEJMra062668. [DOI] [PubMed] [Google Scholar]
- 2.Likosky DS, Caplan LR, Weintraub RM, Hartman GS, Malenka DJ, Ross CS, et al. Intraoperative and postoperative variables associated with strokes following cardiac surgery. The heart surgery forum. 2004;7:E271–276. doi: 10.1532/HSF98.20041035. [DOI] [PubMed] [Google Scholar]
- 3.Gerraty RP, Gates PC, Doyle JC. Carotid stenosis and perioperative stroke risk in symptomatic and asymptomatic patients undergoing vascular or coronary surgery. Stroke. 1993;24:1115–1118. doi: 10.1161/01.str.24.8.1115. [DOI] [PubMed] [Google Scholar]
- 4.Naylor AR, Mehta Z, Rothwell PM, Bell PR. Carotid artery disease and stroke during coronary artery bypass: a critical review of the literature. European journal of vascular and endovascular surgery. 2002;23:283–294. doi: 10.1053/ejvs.2002.1609. [DOI] [PubMed] [Google Scholar]
- 5.Nah HW, Lee JW, Chung CH, Choo SJ, Kwon SU, Kim JS, et al. New brain infarcts on magnetic resonance imaging after coronary artery bypass graft surgery: lesion patterns, mechanism, and predictors. Annals neurology. 2014;76:347–355. doi: 10.1002/ana.24238. [DOI] [PubMed] [Google Scholar]
- 6.Selnes OA, McKhann GM. Neurocognitive complications after coronary artery bypass surgery. Annals neurology. 2005;57:615–621. doi: 10.1002/ana.20481. [DOI] [PubMed] [Google Scholar]
- 7.Tang YH, Vital S, Russell J, Seifert H, Senchenkova E, Granger DN. Transient ischemia elicits a sustained enhancement of thrombus development in the cerebral microvasculature: Effects of anti-thrombotic therapy. Experimental neurology. 2014;261:417–423. doi: 10.1016/j.expneurol.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am journal of pathology. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun YY, Morozov YM, Yang D, Li Y, Dunn RS, Rakic P, et al. Synergy of combined tPA-edaravone therapy in experimental thrombotic stroke. PloS One. 2014;9:e98807. doi: 10.1371/journal.pone.0098807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Hinsbergh VW. Endothelium--role in regulation of coagulation and inflammation. Seminars in immunopathology. 2012;34:93–106. doi: 10.1007/s00281-011-0285-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson SP. Arterial thrombosis--insidious, unpredictable and deadly. Nature medicine. 2011;17:1423–1436. doi: 10.1038/nm.2515. [DOI] [PubMed] [Google Scholar]
- 12.Bilotta F, Gelb AW, Stazi E, Titi L, Paoloni FP, Rosa G. Pharmacological perioperative brain neuroprotection: a qualitative review of randomized clinical trials. British journal of anaesthesia. 2013;110(Suppl 1):i113–120. doi: 10.1093/bja/aet059. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe T, Tahara M, Todo S. The novel antioxidant edaravone: from bench to bedside. Cardiovascular therapeutics. 2008;26:101–114. doi: 10.1111/j.1527-3466.2008.00041.x. [DOI] [PubMed] [Google Scholar]
- 14.Lapchak PA. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert opinion on pharmacotherapy. 2010;11:1753–1763. doi: 10.1517/14656566.2010.493558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edaravone Acute Infarction Study Group Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovascular diseases. 2003;15:222–229. doi: 10.1159/000069318. [DOI] [PubMed] [Google Scholar]
- 16.Kaste M, Murayama S, Ford GA, Dippel DW, Walters MR, Tatlisumak T, et al. Safety, tolerability and pharmacokinetics of MCI-186 in patients with acute ischemic stroke: new formulation and dosing regimen. Cerebrovascular diseases. 2013;36:196–204. doi: 10.1159/000353680. [DOI] [PubMed] [Google Scholar]
- 17.Kimura K, Aoki J, Sakamoto Y, Kobayashi K, Sakai K, Inoue T, et al. Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients--a preliminary study. Journal of the neurological sciences. 2012;313:132–6. doi: 10.1016/j.jns.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Higashi Y. Edaravone for the treatment of acute cerebral infarction: role of endothelium-derived nitric oxide and oxidative stress. Expert opinion on pharmacotherapy. 2009;10:323–331. doi: 10.1517/14656560802636888. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe T, Morita I, Nishi H, Murota S. Preventive effect of MCI-186 on 15-HPETE induced vascular endothelial cell injury in vitro. Prostaglandins, leukotrienes, and essential fatty acids. 1998;33:81–87. doi: 10.1016/0952-3278(88)90127-5. [DOI] [PubMed] [Google Scholar]
- 20.Lee BJ, Egi Y, van Leyen K, Lo EH, Arai K. Edaravone, a free radical scavenger, protects components of the neurovascular unit against oxidative stress in vitro. Brain research. 2010;1307:22–27. doi: 10.1016/j.brainres.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deguchi K, Liu N, Liu W, Omote Y, Kono S, Yunoki T, et al. Pericyte protection by edaravone after tissue plasminogen activator treatment in rat cerebral ischemia. Journal of neuroscience research. 2014;92:1509–1519. doi: 10.1002/jnr.23420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Needles A, Heinmiller A, Sun J, Theodoropoulos C, Bates D, Hirson D, et al. Development and initial application of a fully integrated photoacoustic micro-ultrasound system. IEEE transactions on ultrasonics, ferroelectrics, and frequency control. 2013;60:888–897. doi: 10.1109/TUFFC.2013.2646. [DOI] [PubMed] [Google Scholar]
- 23.Mao H, Toufexis D, Wang X, Lacreuse A, Wu S. Changes of metabolite profile in kainic acid induced hippocampal injury in rats measured by HRMAS NMR. Experimental brain research. 2007;183:477–485. doi: 10.1007/s00221-007-1061-6. [DOI] [PubMed] [Google Scholar]
- 24.Wali B, Ishrat T, Won S, Stein DG, Sayeed I. Progesterone in experimental permanent stroke: a dose-response and therapeutic time-window study. Brain. 2014;137:486–502. doi: 10.1093/brain/awt319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gyngell ML, Busch E, Schmitz B, Kohno K, Back T, Hoehn-Berlage M, et al. Evolution of acute focal cerebral ischaemia in rats observed by localized 1H MRS, diffusion-weighted MRI, and electrophysiological monitoring. NMR in biomedicine. 1995;8:206–214. doi: 10.1002/nbm.1940080505. [DOI] [PubMed] [Google Scholar]
- 27.Arrowsmith JE, Harrison MJ, Newman SP, Stygall J, Timberlake N, Pugsley WB. Neuroprotection of the brain during cardiopulmonary bypass: a randomized trial of remacemide during coronary artery bypass in 171 patients. Stroke. 1998;29:2357–2362. doi: 10.1161/01.str.29.11.2357. [DOI] [PubMed] [Google Scholar]
- 28.Schouten O, Boersma E, Hoeks SE, Benner R, van Urk H, van Sambeek MR, et al. Fluvastatin and perioperative events in patients undergoing vascular surgery. New England journal of medicine. 2009;361:980–989. doi: 10.1056/NEJMoa0808207. [DOI] [PubMed] [Google Scholar]
- 29.Mathew JP, White WD, Schinderle DB, Podgoreanu MV, Berger M, Milano CA, et al. Intraoperative magnesium administration does not improve neurocognitive function after cardiac surgery. Stroke. 2013;44:3407–3413. doi: 10.1161/STROKEAHA.113.002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terrando N, Eriksson LI, Ryu JK, Yang T, Monaco C, Feldmann M, et al. Resolving postoperative neuroinflammation and cognitive decline. Annals neurology. 2011;70:986–995. doi: 10.1002/ana.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenberg RD, Aird WC. Vascular-bed--specific hemostasis and hypercoagulable states. The New England journal of medicine. New England journal of medicine. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Sato K, Hayashi T, Omori N, Nagano I, Kato S, et al. Extension of ischemic therapeutic time window by a free radical scavenger, Edaravone, reperfused with tPA in rat brain. Neurol Res. 2004;26:342–348. doi: 10.1179/016164104225014058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.