

Summary
Transcription factors of the TCF family are key mediators of the Wnt/β-catenin pathway. TCF usually activates transcription on cis-regulatory elements containing TCF binding sites when the pathway is active and represses transcription when the pathway is inactive. However, some direct targets display an opposite regulation (activated by TCF in the absence of Wnt) but the mechanism behind this atypical regulation remains poorly characterized. Here we use the cis-regulatory region of an opposite target gene, ttx-3, to dissect the mechanism of this atypical regulation. Using a combination of genetic, molecular and biochemical experiments we establish that, in the absence of Wnt pathway activation, TCF activates ttx-3 expression via a Zic binding site by forming a complex with a Zic transcription factor. This mechanism is later reinforced by specific bHLH factors. This study reveals an atypical mode of action for TCF that may apply to other binary decisions mediated by Wnt signaling.
Graphical abstract
Introduction
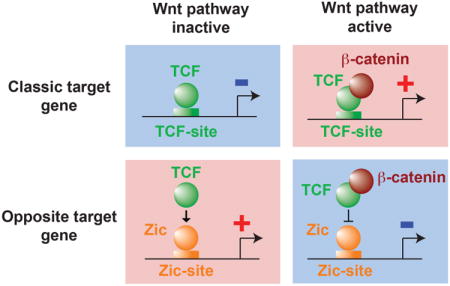
The Wnt/β-catenin pathway is a key signaling pathway that plays important roles at several steps during animal development, cell proliferation and differentiation. Studies in model organisms have established that the key transcriptional mediators of this pathway are transcription factors of the TCF family (MacDonald et al., 2009). Following activation of the Wnt pathway the coactivator β-catenin is stabilized, enters the nucleus and associates with TCF. The TCF/β-catenin complex then directly activates the expression of target genes via TCF binding sites present in their cis-regulatory regions. On the contrary, when the Wnt pathway is inactive, β-catenin is degraded, TCF is not associated with β-catenin and it represses the expression of Wnt target genes via TCF binding sites (Figure 1A).
Figure 1. Activation of ttx-3 expression by POP-1 and repression by SYS-1.
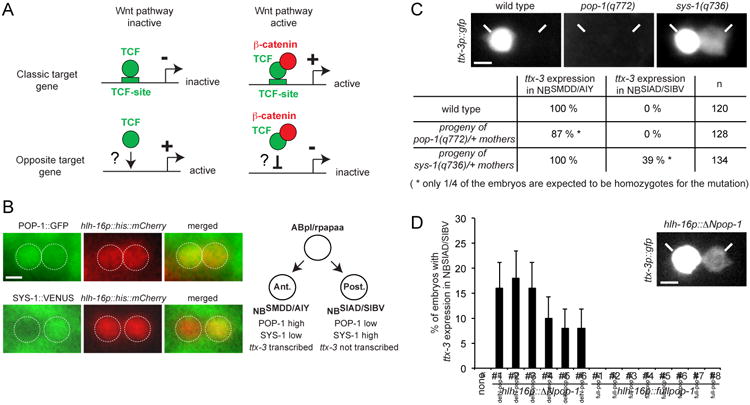
(A) Classic target genes are activated by TCF associated with β-catenin when the Wnt pathway is active and repressed by TCF when the Wnt pathway is inactive. Opposite target genes are activated by TCF when the Wnt pathway is inactive and repressed by the TCF:β-catenin complex when the Wnt pathway is active.
(B) Expression of the POP-1∷GFP (qIs74) and SYS-1∷VENUS (qIs95) fusion proteins in the NBSMDD/AIY (Ant.) and NBSIAD/SIBV (Post.) neuroblasts identified with the hlh-16p∷his∷mCherry transgene (promoter of the hlh-16 gene driving an histone fused to mCherry, stIs10546) at epidermal enclosure.
(C) Effect of pop-1 and sys-1 loss of function mutants on the initiation of ttx-3 expression (ttx-3p∷gfp, otIs173) at epidermal enclosure. As pop-1(q772) and sys-1(q736) homozygote mutants are lethal (at later embryonic stages), we scored the progeny of heterozygote mothers. Around 1/4 of the progeny displays a phenotype as expected from mendelian segregation (n = number of lineages analyzed). Note that in sys-1 mutants the ectopic expression in NBSIAD/SIBV is always weaker than the endogenous expression in NBSMDD/AIY (see picture). This may be due to the fact that sys-1 loss of function is only partial (due to maternal contribution) and/or to the fact that NBSIAD/SIBV has a lower nuclear concentration of POP-1 than NBSMDD/AIY. See also Figure S1.
(D) Expression of a POP-1 version lacking the SYS-1 interaction domain (hlh-16p∷ΔNpop-1) but not a full length version (hlh-16p∷fullpop-1) ectopically activates ttx-3 expression (ttx-3p∷gfp, mgIs18) in the NBSIAD/SIBV neuroblast. The hlh-16 promoter which drives expression in NBSMDD/AIY and NBSIAD/SIBV is used as a driver. 6 independent lines were analyzed for hlh-16p∷ΔNpop-1 and 8 for hlh-16p∷fullpop-1 (none: control without any pop-1 transgene). The percentage of embryos showing ectopic expression in each of the hlh-16p∷ΔNpop-1 lines is low but statistically significant (p<0.01, Fisher's exact test; n=50; error bars show standard error of proportion). Note that the ectopic expression in NBSIAD/SIBV is always weaker than the endogenous expression in NBSMDD/AIY (see picture). Scale bar = 2 μm.
Most of the direct target genes of the Wnt/β-catenin pathway display this classic type of regulation (“classic target genes”, Figure 1A). However in Drosophila and vertebrates a few direct target genes have been observed to display the opposite regulation. These “opposite target genes” are repressed by TCF in presence of Wnt and/or activated by TCF in absence of Wnt (Blauwkamp et al., 2008; Cadigan, 2012; Jamora et al., 2003) (Figure 1A). The mechanism by which TCF mediates this intriguing opposite regulation remains poorly characterized (Cadigan, 2012). In this study we used the asymmetric divisions of C. elegans neuronal precursors as an experimental model to analyze this mechanism.
In both vertebrates and invertebrates, postmitotic neurons are often generated by asymmetric divisions of neuronal progenitors (Sawa, 2010). In C. elegans most neurons are generated during neurulation (called epidermal enclosure) by asymmetric divisions oriented along the antero-posterior axis (Sulston et al., 1983). These terminal divisions are regulated by a particular Wnt pathway called the Wnt/β-catenin asymmetry pathway (Bertrand and Hobert, 2009a, b). This pathway also regulates multiple other asymmetric divisions during C. elegans embryonic and larval development (Bertrand and Hobert, 2010; Mizumoto and Sawa, 2007; Phillips and Kimble, 2009). Following asymmetric division this pathway is active in the posterior daughter where the TCF transcription factor POP-1 associates with its coactivator, a β-catenin called SYS-1, to directly activate the transcription of genes specific of the posterior daughter via TCF binding sites. In the anterior daughter SYS-1 is low or absent and POP-1 represses the target genes specific of the posterior daughter following a “classic target gene” logic. On the contrary the target genes expressed in the anterior daughter display an “opposite target gene” logic: they are activated in the absence of SYS-1 (in the anterior daughter) and repressed in the presence of SYS-1 (in the posterior daughter). While the mechanism by which classic target genes are activated in posterior daughters has been well characterized (Phillips and Kimble, 2009), how opposite target genes are activated in anterior daughters remains poorly understood (Bertrand and Hobert, 2010).
In this study we used the opposite target gene ttx-3, which encodes a LIM homeodomain transcription factor, as an entry point to analyze how gene expression is activated in anterior daughter cells. ttx-3 expression starts at the end of gastrulation in a neuroblast (ABpl/rpapaaa) that generates the SMDD and AIY neurons (Bertrand and Hobert, 2009a). We refer to this neuroblast cell from here on as NBSMDD/AIY. The NBSMDD/AIY neuroblast is produced by an asymmetric division which generates the NBSMDD/AIY neuroblast (anterior daughter) and the NBSIAD/SIBV neuroblast (posterior daughter) (Figure 1B). The NBSMDD/AIY neuroblast divides afterwards to generate the SMDD motoneuron and the AIY interneuron, while the NBSIAD/SIBV neuroblast divides to generate the cholinergic motoneurons SIAD and SIBV. In wild type embryos ttx-3 is only expressed in the NBSMDD/AIY neuroblast but not in its sister NBSIAD/SIBV and this expression is regulated by the Wnt/β-catenin asymmetry pathway (Bertrand and Hobert, 2009a). We have previously shown that when the Wnt/β-catenin asymmetry pathway is inactivated just before this division using temperature-sensitive alleles of the upstream kinases lit-1 and mom-4, the ttx-3 gene becomes ectopically expressed in the NBSIAD/SIBV neuroblast (Bertrand and Hobert, 2009a). Vice versa, when the pathway is ectopically activated in the NBSMDD/AIY neuroblast by overexpression of the Wnt receptor mom-5, expression of ttx-3 is lost (Bertrand and Hobert, 2009a). Therefore ttx-3 expression is activated in the anterior daughter where the Wnt/β-catenin asymmetry pathway is inactive and is repressed in the posterior daughter where the Wnt/β-catenin asymmetry pathway is active. Following division of the NBSMDD/AIY ttx-3 is expressed in both the SMDD and AIY postmitotic neurons in the embryo but then gradually disappears from the SMDD neuron to be restricted to the AIY neuron at larval and adult stages where it regulates AIY terminal differentiation. There are two AIY lineages, one on either side of the embryo.
Here we use a combination of genetic, molecular and biochemical analysis to dissect how the TCF transcription factor POP-1 activates ttx-3 expression in the NBSMDD/AIY neuroblast. We show that ttx-3 activation by POP-1 in this anterior daughter is primarily mediated by the Zic transcription factor REF-2, which forms a complex with POP-1 and activates transcription via a Zic binding site present in the ttx-3 cis-regulatory region. The asymmetric activation of ttx-3 expression is later reinforced via bHLH binding sites by the bHLH factor HLH-3 whose expression becomes gradually asymmetric and is regulated by the Wnt pathway. This study reveals the mechanism of transcriptional activation in an anterior daughter and suggests an atypical mode of action for a TCF factor, which may apply to opposite target genes in other cellular and organismal contexts.
Results
POP-1/TCF activates ttx-3 expression in the absence of SYS-1/β-catenin
To establish how ttx-3 expression is activated in this anterior daughter we first tested whether the classic transcriptional effectors of the Wnt/β-catenin asymmetry pathway, the TCF transcription factor POP-1 and its coactivator the β-catenin SYS-1, are involved in the initiation of ttx-3 expression. The Wnt/β-catenin asymmetry pathway usually regulates the nuclear level of both SYS-1 and POP-1 by modulating the degradation of SYS-1 and the nuclear export of POP-1 (Phillips and Kimble, 2009). Following asymmetric division of their mother cell, the anterior NBSMDD/AIY neuroblast displays a high nuclear concentration of POP-1 and low concentration of SYS-1 while the posterior NBSIAD/SIBV neuroblast has a low nuclear concentration of POP-1 and high concentration of SYS-1 (Figure 1B). This suggests that nuclear POP-1 is mostly free of SYS-1 in the NBSMDD/AIY neuroblast (anterior) and mostly bound to SYS-1 in the NBSIAD/SIBV neuroblast (posterior), as observed in other asymmetric divisions regulated by this pathway (Phillips and Kimble, 2009). In sys-1 loss of function mutants ttx-3 becomes ectopically expressed in the NBSIAD/SIBV neuroblast similar to what we have previously observed when upstream components of the pathway are inactivated (Figure 1C). Interestingly, in pop-1 loss of function mutants ttx-3 expression is lost (Figure 1C, an effect is observed in a bit less than the 25% pop-1(q772) homozygote embryos expected from mendelian segregation; this is possibly due to remains of the very strong maternal contribution of pop-1 observed at earlier stages). Loss of ttx-3 expression in pop-1 mutants is not due to a loss of the NBSMDD/AIY neuroblast itself since the NBSMDD/AIY neuroblast still expresses hlh-16, a very restricted marker that is turned on in the NBSMDD/AIY and NBSIAD/SIBV neuroblasts just before ttx-3 (hlh-16p∷his∷mCherry, stIs10546, (Bertrand et al., 2011)) (Supplementary Figure 1A). The NBSMDD/AIY neuroblast also still undergoes its terminal division at the right time during neurulation (Supplementary Figure 1B). Taken together, our data suggest that POP-1 is required for the activation of ttx-3 expression in the anterior daughter and that this activation is blocked in the posterior daughter by SYS-1.
Zic and bHLH binding sites contribute to the asymmetry of ttx-3 expression
In order to determine how POP-1 activates ttx-3 in the absence of SYS-1 we examined the cis-regulatory regions of ttx-3. We have previously identified a 113 bp cis-regulatory element responsible for the initiation of ttx-3 expression in the NBSMDD/AIY neuroblast (Bertrand and Hobert, 2009a). This element does not contain any POP-1/TCF binding sites but contains binding sites for bHLH and Zic transcription factors that are required for the initiation of ttx-3 expression (Bertrand and Hobert, 2009a) (Figure 2). To determine whether these binding sites could mediate the asymmetric activation in the anterior daughter (NBSMDD/AIY neuroblast) versus the posterior daughter (NBSIAD/SIBV neuroblast) we generated multimers of bHLH or Zic binding sites, placed them upstream of GFP and tested their activity in transgenic embryos. Multimers of 6 or 12 bHLH binding sites drive expression in both the anterior and posterior daughters (Figure 2). Interestingly, while the activity is initially mostly symmetrical, it becomes stronger in the anterior daughter than in the posterior as the neuroblasts approach their terminal division (Figure 2). A multimer of 6 Zic binding sites does not drive any expression on its own but greatly increases the antero-posterior asymmetry of bHLH binding sites both early and late (Figure 2). Zic binding sites increase the expression in the anterior daughter and decrease the expression in the posterior daughter. As expected, the activity of this multimer of Zic and bHLH binding sites is regulated by pop-1 (Supplementary Figure 2). In pop-1 mutants the expression of the multimer is either lost or its asymmetry is reduced. Taken together this suggests that both bHLH and Zic binding sites contribute to the asymmetric activation of ttx-3 expression. Zic binding sites contribute to the asymmetry both early and late, while bHLH binding sites contribute mostly to the late asymmetry.
Figure 2. Role of the Zic and bHLH binding sites in the generation of the asymmetry.

Activity of multimers of Zic or bHLH binding sites placed in front of gfp. On top the structure of the cis-regulatory element responsible for the initiation of ttx-3 expression is shown (“ttx-3 initiator”). The graphs present the percentage of lineages with expression in both NBSMDD/AIY and NBSIAD/SIBV at similar level (black), expression in both NBSMDD/AIY and NBSIAD/SIBV with higher level in NBSMDD/AIY (grey), or expression only in NBSMDD/AIY (white). Expression in the NBSMDD/AIY and NBSIAD/SIBV neuroblasts was scored early (during interphase) or late (when entering mitosis as monitored by chromosome condensation using hlh-16p∷his∷mCherry and by the appearance of the cleavage furrow) (n>30, error bars show standard error of proportion). The intensity of the GFP signal in the NBSMDD/AIY neuroblast in each line is indicated above the graph (+++: strong, ++: medium, +: low). Numbers below graph indicate independent lines. See also Figure S2.
REF-2/Zic forms an activator complex with POP-1/TCF
REF-2 is the only transcription factor of the Zic family in C. elegans. In ref-2 mutants the initiation of ttx-3 expression in the NBSMDD/AIY neuroblast is lost (Figure 3A, (Bertrand and Hobert, 2009a)). By Electrophoretic Mobility Shift Assay (EMSA) we observed that in vitro produced REF-2 is able to bind directly to the Zic binding site present in the ttx-3 promoter while in vitro produced POP-1 is unable to directly bind to this Zic binding site (Figure 3B, Supplementary Figure 4A). REF-2 may therefore mediate the asymmetric activity of POP-1 on Zic binding sites. The REF-2 protein is expressed at a similar level in the NBSMDD/AIY and NBSIAD/SIBV neuroblasts (Figure 3D, assessed with a REF-2∷VENUS translational fusion that rescues the ref-2 mutant phenotype (Bertrand and Hobert, 2009a)) suggesting that POP-1 does not regulate REF-2 expression but rather regulates its activity at a post-translational level. Consistent with this idea, REF-2 expression is not affected in pop-1 mutants (Supplementary Figure 1C). Interestingly a genome wide yeast two-hybrid screen has revealed that POP-1 and REF-2 can directly interact (Simonis et al., 2009). Using co-immunoprecipitation we confirmed that POP-1 and REF-2 form a complex in vivo in the C. elegans embryo (Figure 3C). This suggests that POP-1 directly regulates the activity of the REF-2 protein by binding to it.
Figure 3. Regulation of the initiation of ttx-3 expression by REF-2, HLH-2, HLH-3 and HLH-16.
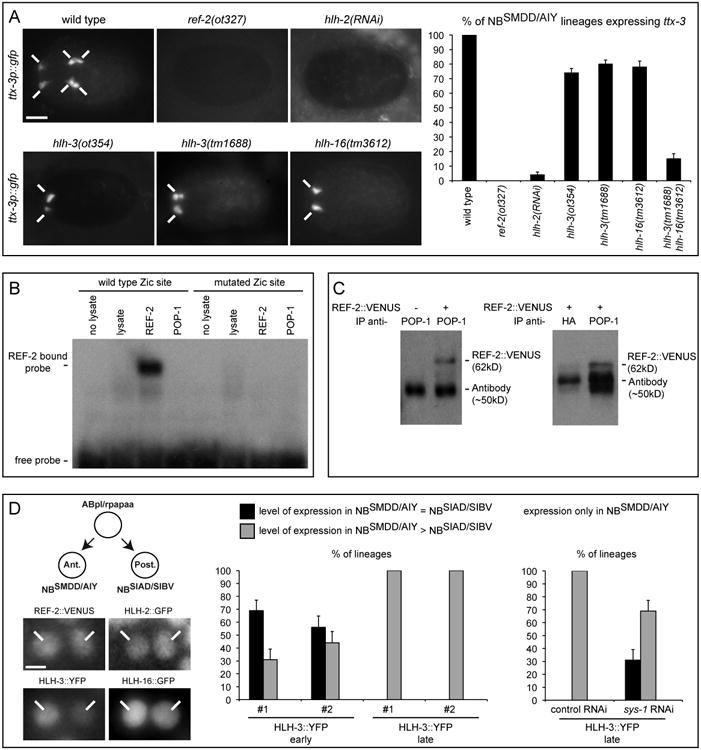
(A) Left: expression of ttx-3 (ttx-3p∷gfp) in embryos at epidermal enclosure just after division of the NBSMDD/AIY neuroblast (ventral view, anterior is left, scale bar = 10 μm, note that the expression of ttx-3 in the AIN lineage (AINm, an unrelated neuronal lineage that also expresses ttx-3) is affected by loss of ref-2 and hlh-2 function but not by loss of hlh-3 and hlh-16 function). Right: percentage of NBSMDD/AIY lineages that display expression of ttx-3 (ttx-3p∷gfp) at epidermal enclosure (n>100, error bars show standard error of proportion). See also Figure S3.
(B) EMSA using in vitro (reticulocyte lysate) produced REF-2 or POP-1 proteins on probes containing the wild type or mutated Zic site from the ttx-3 promoter. See also Figure S4.
(C) Coimmunoprecipitation from C. elegans embryos expressing the REF-2∷VENUS (otEx3091) translational fusion (+) or not (-), using an anti-POP-1 antibody or control anti-HA antibody for immunoprecipitation and an anti-VENUS antibody for western blot. (Note that in addition to the REF-2∷VENUS band (62kD) an additional band corresponding to the antibody used for immunoprecipitation (∼50kD) is also detected. The same quantity of anti-POP-1 and anti-HA antibodies were used in the immunoprecipitations, differences of intensity of the antibody band between anti- POP-1 and anti-HA reflects difference of affinity of these antibodies with the western blot antibodies).
(D) Left: expression of the translational fusions REF-2∷VENUS (otEx3091), HLH-2∷GFP (nIs47), HLH-3∷YFP (otEx4140), HLH-16∷GFP (otEx4503) in the NBSMDD/AIY (Ant.) and NBSIAD/SIBV (Post.) neuroblasts at epidermal enclosure (scale bar = 2 μm). Right: percentage of lineages expressing the translational fusion HLH-3∷YFP in both NBSMDD/AIY and NBSIAD/SIBV at similar level (black), in both NBSMDD/AIY and NBSIAD/SIBV with higher level in NBSMDD/AIY (grey), or only in NBSMDD/AIY (white). Two independent HLH-3∷YFP lines (#1 otEx4140, #2 otEx4142) were analyzed. Expression in the NBSMDD/AIY and NBSIAD/SIBV neuroblasts was scored early (during interphase) or late (when entering mitosis as monitored by chromosome condensation using hlh-16p∷his∷mCherry and by the appearance of the cleavage furrow). The effects of control and sys-1 RNAi were tested on otEx4140. (n>30, error bars show standard error of proportion).
We then analyzed whether POP-1 and REF-2 are sufficient to activate transcription via Zic binding sites by using a heterologous mammalian cell culture system. Mammalian COS-7 cells were transfected with a multimer of Zic binding sites placed upstream of a luciferase reporter and cotransfected with various combinations of REF-2, POP-1 and SYS-1. We observed that POP-1 increases the ability of REF-2 to activate the multimer (Figure 4A) confirming that the REF-2:POP-1 complex acts as a transcriptional activator on Zic binding sites. In addition we also observed that REF-2 is able to activate a reporter of POP-1 activity (multimer of TCF binding sites placed upstream of mCherry) in transgenic C. elegans (Supplementary Figure 4B) suggesting that the REF-2:POP-1 complex may also act as a transcriptional activator on TCF binding sites.
Figure 4. Action of REF-2, POP-1, SYS-1 on Zic binding sites and model.

(A) Analysis of the activity in COS-7 cells of a multimer of Zic binding sites placed in front of a luciferase reporter. The multimer is cotransfected with vectors expressing REF-2, POP-1, SYS-1 or an empty vector (none). (*: p<0.05, **: p<0.01, ns: not significant, Student's t-test; n=12, error bars show standard error of the mean).
(B) Activation of the opposite target gene ttx-3 by the TCF transcription factor POP-1. “HLH” represents the 3 bHLH factors HLH-2, HLH-3 and HLH-16. The smaller circles in the NBSIAD/SIBV neuroblast represent lower levels of POP-1 and HLH-3.
(C) General model for the activation of opposite target genes by TCF. X = REF-2 in the case of ttx-3.
In C. elegans embryos SYS-1 represses the activation of ttx-3 expression in the posterior NBSIAD/SIBV neuroblasts (Figure 1C). In addition, in our heterologous mammalian cell culture system addition of SYS-1 blocks the ability of REF-2 and POP-1 to activate transcription on Zic binding sites (Figure 4A). This suggests that SYS-1 blocks the activity of the REF-2:POP-1 complex. Interestingly, a recent study in vertebrates (Pourebrahim et al., 2011) has shown that Zic2, TCF4 and β-catenin can form a Zic:TCF:β-catenin trimolecular complex (β-catenin binding to the N-terminal domain of TCF and Zic binding to the HMG domain of TCF), and that this trimolecular complex is not able to activate transcription on TCF binding sites. This suggests that in C. elegans SYS-1/β-catenin could block the activity of the REF-2:POP-1 complex in the posterior daughter by binding to the POP-1 N-terminal domain. To test this model, we induced, at the time when the NBSMDD/AIY and NBSIAD/SIBV neuroblasts are generated from their mother cell, the expression of a version of POP-1 that lacks its N-terminal SYS-1 interaction domain (Kidd et al., 2005) (ΔNPOP-1, deletion of the amino acids 1 to 43, this deletion does not affect POP-1 nuclear asymmetry (Lo et al., 2004; Maduro et al., 2002)). This construct may form an activator complex with REF-2 that would be resistant to blockage by SYS-1. Indeed, when driven by the weak hlh-16 promoter, ΔNPOP-1, but not wild type (full) POP-1, ectopically activates ttx-3 expression in the NBSIAD/SIBV neuroblast where SYS-1 is present (Figure 1D). When driven by a strong heat shock promoter both ΔNPOP-1 and full POP-1 are able to ectopically activate ttx-3 expression in the NBSIAD/SIBV neuroblast probably because the level of full POP-1 driven by the heat shock promoter is sufficiently high for large fractions of full POP-1 to escape SYS-1 binding (Supplementary Figure 1D).
Taken together, these data suggest that ttx-3 expression is activated in the NBSMDD/AIY by a REF-2:POP-1 complex via a Zic binding site and that this activation is blocked in the NBSIAD/SIBV by SYS-1.
HLH-3/Achaete-scute protein level becomes gradually asymmetric
Contrary to the effects of Zic binding sites, bHLH binding sites do not show an asymmetric activity early but the activity becomes gradually asymmetric as the neuroblasts progress in their cell cycle. We have previously identified two bHLH factors involved in the regulation of ttx-3 expression: the E/Daughterless ortholog HLH-2 and the Beta3/Olig ortholog HLH-16 (Bertrand et al., 2011; Bertrand and Hobert, 2009a). In hlh-2(RNAi) treated embryos ttx-3 expression is lost in nearly all the NBSMDD/AIY lineages while in hlh-16 mutants ttx-3 expression is lost in around 20% of the NBSMDD/AIY lineages (Figure 3A). As general E/Daughterless proteins, like HLH-2, often act as heterodimers with other more tissue specific bHLH factors like Beta3/Olig or Achaete-Scute factors (Bertrand et al., 2002) it is likely that another tissue specific bHLH factor acts in parallel to HLH-16 in the initiation of ttx-3 expression. During a genetic screen for regulators of ttx-3 expression (Bertrand and Hobert, 2009a; Sarin et al., 2010) we have identified a mutant, ot354, that displays a loss of ttx-3 expression in around 20% of the AIY neurons at larval and adult stage (Supplementary Figure 3A, B, C). In ot354 mutant larvae we still observe a neuron at the position normally occupied by AIY and this neuron still expresses the pan-neuronal marker rgef-1 (Supplementary Figure 3D) suggesting that the neuron is correctly generated but its subtype-specific identity (marked by ttx-3) is not correctly specified.
We cloned ot354 using whole genome sequencing of the mutant strain and confirmed its identity with another allele and by transformation rescue experiments (Supplementary Figure 3A, B, C). ot354 animals contain an early stop in the bHLH factor HLH-3 (Supplementary Figure 3A). HLH-3 belongs to the Achaete-Scute family and can heterodimerize with HLH-2 (Krause et al., 1997). As in hlh-16 mutants, ttx-3 expression is lost in around 20 % of the AIY lineages in hlh-3 mutant embryos and larvae (Figure 3A, Supplementary Figure 3C). In hlh-3; hlh-16 double mutants ttx-3 expression is lost in nearly all AIY lineages similarly to what is observed in hlh-2 RNAi treated animals (Figure 3A) suggesting that ttx-3 expression may be regulated by two partly redundant bHLH complexes HLH-2:HLH-3 and HLH-2:HLH-16.
We analyzed the expression of HLH-2, HLH-3 and HLH-16 by using rescuing translational fusions (HLH-2∷GFP (Nakano et al., 2010); HLH-3∷YFP generated by fosmid recombineering, see Experimental Procedures; and HLH-16∷GFP (Bertrand et al., 2011)). While the level of HLH-2 or HLH-16 protein is similar between the NBSMDD/AIY neuroblast and its posterior sister the NBSIAD/SIBV neuroblast (Figure 3D), the HLH-3 protein displays some asymmetry. The level of HLH-3 protein is initially symmetric just after the generation of the NBSMDD/AIY and NBSIAD/SIBV neuroblasts but becomes gradually asymmetric as the cell cycle progresses (Figure 3D) which mirrors the asymmetry in the activity of the bHLH binding sites (Figure 2). This late asymmetry of HLH-3 protein levels is also dependent on the Wnt pathway as it is reduced by a RNAi knockdown of sys-1 (Figure 3D). This suggests that the early asymmetry generated by the REF-2:POP-1 complex is later reinforced by an asymmetry in the level of HLH-3 protein.
Discussion
Mechanism of transcriptional activation by POP-1/TCF in the absence of SYS-1/β-catenin
In this paper we have analyzed how the Wnt/β-catenin asymmetry pathway regulates the expression of a target gene ttx-3 in an anterior daughter following asymmetric division of neuronal precursors. The genetic, cis-regulatory and biochemical analyses we have performed suggest an interesting atypical mechanism (Figure 4B). Following asymmetric division the level of POP-1/TCF is high and the level of SYS-1/β-catenin is low in the anterior daughter nucleus. POP-1 associates with the Zic transcription factor REF-2 and the REF-2:POP-1 complex activates ttx-3 expression via a Zic binding site present in its cis-regulatory region. On the contrary, in the posterior daughter nucleus the level of POP-1/TCF is low, the level of SYS-1/β-catenin is high and SYS-1 blocks the activation of ttx-3 expression by the REF-2:POP-1 complex. The precise biochemical mechanism by which POP-1 activates REF-2 transcriptional activity remains to be determined. There is clear evidence that the POP-1 and REF-2 proteins physically interact (co-immunoprecipitation we have performed in C. elegans embryos and yeast two-hybrid). One could imagine that POP-1 and REF-2 form a long term complex on Zic sites and that this complex is able to interact with the basal transcription machinery in a manner different from REF-2 alone. Another possibility is that the direct interaction between the POP-1 and REF-2 proteins is transient and induces a long lasting conformational change in the REF-2 protein modifying its activity. The activation of REF-2 by POP-1 is blocked in the posterior daughter by SYS-1. While we have not analyzed in detail the mechanism of this blockage by SYS-1, a study in vertebrates has shown that Zic2 can directly bind a TCF4:β-catenin complex on TCF binding sites blocking transcriptional activation by TCF4:β-catenin (Pourebrahim et al., 2011). One possible mechanism is therefore that in the NBSIAD/SIBV neuroblast SYS-1 blocks the activation of ttx-3 expression by binding to the REF-2:POP-1 complex blocking its ability to activate transcription.
The REF-2:POP-1 mechanism seems to be the main driver of the asymmetric transcriptional activation. However this process is later reinforced by a partial asymmetry in bHLH activity. While initially symmetric, HLH-3 protein levels and transcriptional activity mediated by bHLH binding sites become gradually asymmetric, decreasing in the posterior daughter. The gradual disappearance of the HLH-3 protein in the posterior daughter is regulated by the Wnt pathway and could be due to an increased degradation or a decreased production in the posterior daughter. Interestingly it has been recently reported that another bHLH factor, the Atonal ortholog LIN-32, displays a similar pattern (gradual disappearance from the posterior daughter) during asymmetric divisions in male specific neuronal lineages of the C. elegans larva and this pattern is also regulated by the Wnt/β-catenin asymmetry pathway (Miller and Portman, 2011). The regulation of asymmetries in the level of proneural bHLH between daughter cells by the Wnt/β-catenin asymmetry pathway may therefore be a common theme of the specification of neuronal lineages in C. elegans.
Intriguingly, the activation of target genes in the anterior daughter of another embryonic blastomere, the endomesoderm precursor EMS, seems to follow a different logic (Broitman-Maduro et al., 2009; Owraghi et al., 2010). In this context the POP-1∷SYS-1 complex seems to activate the expression of the GATA transcription factors END-1 and END-3 in the posterior daughter, which subsequently restrict the expression of the anterior transcription factor ceh-51 to the anterior daughter by repressing it in the posterior daughter. Therefore transcriptional activation in anterior daughters can involve either a direct activation by POP-1 in the absence of SYS-1 in the anterior daughter (ttx-3 in the NBSMDD/AIY neuroblast) or an indirect repression by the POP-1:SYS-1 complex in the posterior daughter (ceh-51 in the EMS lineage). We currently do not know whether both mechanisms (direct activation of an anterior target by POP-1 in the absence of SYS-1 and direct activation of a posterior target by the POP-1:SYS-1 complex) can operate together in the same asymmetric cell division.
Regulation of the expression of opposite target genes by TCF
Can we place the atypical mechanism of transcriptional activation by POP-1 described here in a more general framework of transcriptional activation by TCF transcription factors? In both vertebrates and invertebrates it has been observed that a few direct target genes can display an “opposite” type of regulation: repressed by the TCF:β-catenin complex when the Wnt pathway is active and/or activated by TCF when the Wnt pathway is inactive. The mechanism behind this intriguing opposite regulation remains poorly characterized (Cadigan, 2012). In the case of the E-cadherin and p16 genes in mice or of the stripe gene in Drosophila it has been observed that direct repression by the TCF:β-catenin complex is mediated by classic TCF binding sites. However, it is unclear why TCF:β-catenin bound to these sites represses rather than activates transcription (Delmas et al., 2007; Jamora et al., 2003; Piepenburg et al., 2000). The analysis of the Ugt36Bc gene in Drosophila suggests a second mechanism (Blauwkamp et al., 2008; Zhang et al., 2014). In this case TCF binds to a non canonical binding site and binding to this site is suggested to induce an allosteric change in TCF that reverts the transcriptional output. In the case of the ttx-3 gene of C. elegans we propose a third mechanism (Figure 4C). Here, TCF forms a complex with another transcription factor X, regulating transcription via an X binding site (for ttx-3, X = the Zic transcription factor REF-2). When Wnt signaling is inactive the X:TCF complex activates transcription, while when Wnt signaling is active β-catenin blocks this activation. It will be interesting to see whether this atypical mechanism also applies to other opposite target genes in C. elegans or other organisms.
Experimental Procedures
Genetics
ot354 was isolated from an automated genetic screen using EMS as a mutagen and a worm sorter to identify mutants in which ttx-3p∷gfp (otIs173) expression is defective (Bertrand and Hobert, 2009a; Sarin et al., 2010). Identification of variants in the ot354 mutant strain by whole genome sequencing was performed as described (Sarin et al., 2010) using MAQGene (Bigelow et al., 2009) as data analysis tool. The strain was backcrossed four times before phenotypic characterization. RNAi treatment was performed as described (Bertrand and Hobert, 2009a).
Expression constructs and transgenic strains
The making of constructs and transgenic strains is described in detail in the Supplemental Data.
Cell identifications
In the embryo the NBSMDD/AIY and NBSIAD/SIBV neuroblasts and their daughter cells (the SMDD, AIY, SIAD and SIBV neurons) were identified by using the hlh-16p∷his∷mCherry transgene (otIs10546) which labels only a small subset of neuroblasts and neurons at epidermal enclosure (Bertrand et al., 2011). At larval stages the AIY neuron was identified using DIC optics based on its stereotypical location (Sulston et al., 1983).
Luciferase assay
COS-7 cells (105) were transfected using Lipofectamine 2000 (Invitrogen) with the Zic site firefly luciferase reporter construct (200 ng) with or without expression vectors for REF-2 (500 ng), POP-1 (500 ng) and SYS-1 (2 μg). After an incubation of 48h, Luciferase assay was performed according to manufacturer's guideline (Dual-Luciferase Reporter Assay, Promega). Measurements were normalized to a Renilla luciferase co-transfected control and performed on 12 biological replicates per conditions.
Biochemistry
The conditions used for co-immunoprecipitation and Electrophoretic Mobility Shift Assay are described in detail in the Supplemental Data.
Supplementary Material
Acknowledgments
We thank Q. Chen and C. Couillault for expert injection assistance, A. Boyanov for assistance with whole genome sequencing, the Caenorhabditis Genetics Center, the C. elegans knockout facility at Tokyo Women's Medical University School of Medicine, S. Alper, R. Lin, H. Horvitz, M. Herman and J. Kimble for strains and reagents; the groups of Flavio Maina and Yacine Graba for help with the biochemistry; Harold Cremer, Cédric Maurange and members of our labs for comments on the manuscript. This work was funded by an ATIP/Avenir startup grant from CNRS/INSERM (V.B.), grants from Sanofi-Aventis (V.B.) and the Fédération pour la Recherche sur le Cerveau (V.B.), by the National Institutes of Health (R01NS039996-05; R01NS050266-03) (O.H.) and the Howard Hughes Medical Institute (O.H.). This work was performed using France-BioImaging infrastructures supported by the Agence Nationale de la Recherche (ANR-10-INSB-04-01, call “Investissements d'Avenir”).
Footnotes
Author Contributions: V.B. and O.H. conceived the project. S.M., W.K., U.R., M.I., P.M. and V.B. performed the experiments. S.M., W.K., U.R. and V.B. analyzed the data. V.B., O.H., S.M., U.R. and W.K. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Bertrand V, Bisso P, Poole RJ, Hobert O. Notch-dependent induction of left/right asymmetry in C. elegans interneurons and motoneurons. Curr Biol. 2011;21:1225–1231. doi: 10.1016/j.cub.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand V, Hobert O. Linking asymmetric cell division to the terminal differentiation program of postmitotic neurons in C. elegans. Dev Cell. 2009a;16:563–575. doi: 10.1016/j.devcel.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand V, Hobert O. Wnt asymmetry and the terminal division of neuronal progenitors. Cell Cycle. 2009b;8:1973–1974. doi: 10.4161/cc.8.13.9024. [DOI] [PubMed] [Google Scholar]
- Bertrand V, Hobert O. Lineage programming: navigating through transient regulatory states via binary decisions. Curr Opin Genet Dev. 2010;20:362–368. doi: 10.1016/j.gde.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow H, Doitsidou M, Sarin S, Hobert O. MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods. 2009;6:549. doi: 10.1038/nmeth.f.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwkamp TA, Chang MV, Cadigan KM. Novel TCF-binding sites specify transcriptional repression by Wnt signalling. EMBO J. 2008;27:1436–1446. doi: 10.1038/emboj.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broitman-Maduro G, Owraghi M, Hung WW, Kuntz S, Sternberg PW, Maduro MF. The NK-2 class homeodomain factor CEH-51 and the T-box factor TBX-35 have overlapping function in C. elegans mesoderm development. Development. 2009;136:2735–2746. doi: 10.1242/dev.038307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadigan KM. TCFs and Wnt/beta-catenin signaling: more than one way to throw the switch. Curr Top Dev Biol. 2012;98:1–34. doi: 10.1016/B978-0-12-386499-4.00001-X. [DOI] [PubMed] [Google Scholar]
- Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, Denat L, Goodall J, Luciani F, Viros A, et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev. 2007;21:2923–2935. doi: 10.1101/gad.450107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422:317–322. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd AR, 3rd, Miskowski JA, Siegfried KR, Sawa H, Kimble J. A beta-catenin identified by functional rather than sequence criteria and its role in Wnt/MAPK signaling. Cell. 2005;121:761–772. doi: 10.1016/j.cell.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Krause M, Park M, Zhang JM, Yuan J, Harfe B, Xu SQ, Greenwald I, Cole M, Paterson B, Fire A. A C. elegans E/Daughterless bHLH protein marks neuronal but not striated muscle development. Development. 1997;124:2179–2189. doi: 10.1242/dev.124.11.2179. [DOI] [PubMed] [Google Scholar]
- Lo MC, Gay F, Odom R, Shi Y, Lin R. Phosphorylation by the beta-catenin/MAPK complex promotes 14-3-3-mediated nuclear export of TCF/POP-1 in signal-responsive cells in C. elegans. Cell. 2004;117:95–106. doi: 10.1016/s0092-8674(04)00203-x. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro MF, Lin R, Rothman JH. Dynamics of a developmental switch: recursive intracellular and intranuclear redistribution of Caenorhabditis elegans POP-1 parallels Wnt-inhibited transcriptional repression. Dev Biol. 2002;248:128–142. doi: 10.1006/dbio.2002.0721. [DOI] [PubMed] [Google Scholar]
- Miller RM, Portman DS. The Wnt/beta-catenin asymmetry pathway patterns the atonal ortholog lin-32 to diversify cell fate in a Caenorhabditis elegans sensory lineage. J Neurosci. 2011;31:13281–13291. doi: 10.1523/JNEUROSCI.6504-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumoto K, Sawa H. Two betas or not two betas: regulation of asymmetric division by beta-catenin. Trends Cell Biol. 2007;17:465–473. doi: 10.1016/j.tcb.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Nakano S, Ellis RE, Horvitz HR. Otx-dependent expression of proneural bHLH genes establishes a neuronal bilateral asymmetry in C. elegans. Development. 2010;137:4017–4027. doi: 10.1242/dev.058834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owraghi M, Broitman-Maduro G, Luu T, Roberson H, Maduro MF. Roles of the Wnt effector POP-1/TCF in the C. elegans endomesoderm specification gene network. Dev Biol. 2010;340:209–221. doi: 10.1016/j.ydbio.2009.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BT, Kimble J. A new look at TCF and beta-catenin through the lens of a divergent C. elegans Wnt pathway. Dev Cell. 2009;17:27–34. doi: 10.1016/j.devcel.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepenburg O, Vorbruggen G, Jackle H. Drosophila segment borders result from unilateral repression of hedgehog activity by wingless signaling. Mol Cell. 2000;6:203–209. [PubMed] [Google Scholar]
- Pourebrahim R, Houtmeyers R, Ghogomu S, Janssens S, Thelie A, Tran HT, Langenberg T, Vleminckx K, Bellefroid E, Cassiman JJ, et al. Transcription factor Zic2 inhibits Wnt/beta-catenin protein signaling. J Biol Chem. 2011;286:37732–37740. doi: 10.1074/jbc.M111.242826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Bertrand V, Bigelow H, Boyanov A, Doitsidou M, Poole RJ, Narula S, Hobert O. Analysis of multiple ethyl methanesulfonate-mutagenized Caenorhabditis elegans strains by whole-genome sequencing. Genetics. 2010;185:417–430. doi: 10.1534/genetics.110.116319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa H. Specification of neurons through asymmetric cell divisions. Curr Opin Neurobiol. 2010;20:44–49. doi: 10.1016/j.conb.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Simonis N, Rual JF, Carvunis AR, Tasan M, Lemmens I, Hirozane-Kishikawa T, Hao T, Sahalie JM, Venkatesan K, Gebreab F, et al. Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat Methods. 2009;6:47–54. doi: 10.1038/nmeth.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Zhang CU, Blauwkamp TA, Burby PE, Cadigan KM. Wnt-mediated repression via bipartite DNA recognition by TCF in the Drosophila hematopoietic system. PLoS genetics. 2014;10:e1004509. doi: 10.1371/journal.pgen.1004509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.