

Abstract
The mechanistic target of rapamycin (mTOR) is a ubiquitous serine/threonine kinase that plays pivotal roles in integrating growth signals on a cellular level. To support proliferation and survival under stress, two interacting complexes that harbor mTOR, mTORC1 and mTORC2, promote the transcription of genes involved in carbohydrate metabolism and lipogenesis, enhance protein translation, and inhibit autophagy. While rapamycin was originally developed as an inhibitor of T cell proliferation for preventing organ transplant rejection, its molecular target, mTOR, has been subsequently identified as a central regulator of metabolic cues that drive lineage specification in the immune system. Owing to oxidative stress, the activation of mTORC1 has emerged as a central pathway for the pathogenesis of systemic lupus erythematosus and other autoimmune diseases. Paradoxically, mTORC1 has been also identified as a mediator of the Warburg effect that allows cell survival under hypoxia. Rapamycin and new classes of mTOR inhibitors are being developed to block not only transplant rejection and autoimmunity but also to treat obesity and various forms of cancer. Through preventing these diseases, personalized mTOR blockade holds promise to extend life span.
Keywords: metabolism, autoimmunity, inflammation, T cell activation, systemic lupus erythematosus, oxidative stress, hypoxia, mitochondria, glutathione, mTOR, autophagy, pathogenesis, keloid disease, glycolysis, pentose phosphate pathway, kynurenine, biomarker, treatment, rapamycin, sirolimus
Introduction
The mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that is ubiquitous to eukaryotic cells. mTOR is named after rapamycin, an antifungal macrolide antibiotic. Rapamycin is produced by the bacterium Streptomyces hygroscopicus, which was isolated from a soil sample originating from Rapa Nui, commonly known as Easter Island.1 Rapamycin is a potent inhibitor of antigen-induced proliferation of T cells and antibody production.2 Such blockade of T cell activation has been found to be effective for inhibiting the development of a systemic autoimmune inflammatory disease, lupus, both in animal models3 and in patients with systemic lupus erythematosus (SLE).4 Demonstration of the robust immunosuppressive activity of rapamycin in animal models of organ transplantation led to clinical trials and subsequent approval by regulatory authorities for prophylaxis of renal graft rejection, under the pharmaceutical designation of sirolimus (Rapamune).5,6 Interest in sirolimus as an immunosuppressive therapy in organ transplantation derives from its unique mechanism of action, its unique side effect profile, and its ability to synergize with other immunosuppressive agents.7 The molecular mechanism underlying the antifungal, antiproliferative, and immunosuppressive activities of rapamycin involves the formation of a high-affinity complex with a 12-kD intracellular protein that binds FK506 and was thus named FKBP12.8 This complex of rapamycin and FKBP12 blocks the activation of mTOR in systems ranging from yeast9 to mammalian cells.10 mTOR is present in two distinct complexes. mTOR complex 1 (mTORC1) is composed of mTOR, regulatory-associated protein of TOR (Raptor), mammalian lethal with sec-13 protein 8 (mLST8), DEP domain–containing mTOR-interacting protein (Deptor), and proline-rich Akt substrate of 40 kD (PRAS40).11 mTORC1 integrates growth signals reflecting the availability of nutrients and energy to promote either proliferation when conditions are favorable or autophagy when conditions are unfavorable. mTORC1 activation occurs on the surface of the lysosomal membrane in response to amino acid sufficiency (Fig. 1).12 Alternatively, low ATP levels lead to the AMPK-dependent activation of TSC2 and phosphorylation of Raptor to reduce mTORC1 signaling (Fig. 2).13 mTORC1 has a number of downstream substrates that control the translation of mRNA via the phosphorylation of downstream targets (4E-BP1 and p70 S6 Kinase), suppression of autophagy (Atg13, ULK1), ribosome biogenesis, and activation of transcription.14,15 mTOR complex 2 (mTORC2) comprises mTOR, rapamycin insensitive companion of mTOR (Rictor), stress-activated protein kinase interacting protein 1(mSIN1), Protor, GβL, Deptor, and mLST8 (Fig. 2). mTORC2 promotes cellular survival by activating Akt.16 Although mLST8 is present in both complexes, its absence primarily compromises mTORC2 activity.17 Given that mTOR plays a pivotal role in integrating growth signals on a cellular level, it is not surprising that both complexes have been targeted for reversing increased proliferation associated with carcinogenesis.18,19 In SLE patients, mTORC1 is activated,20,21 while mTORC2 is reduced.22 The activation of mTORC1 causes the proinflammatory increased production of IL-4 and necrotic death of CD4−CD8− double-negative (DN) T cells and depletion of CD4+CD25+FoxP3+ regulatory T cells in SLE.23 Accordingly, the therapeutically effective blockade of mTORC1 is accompanied by correction of these T cell abnormalities in SLE patients.23 Increased mTORC1 was also noted in T lymphocytes of patients with another autoimmune disease, multiple sclerosis (MS), which was underlying the contraction of regulatory T (Treg) cells in these patients.24 Rapamycin has shown therapeutic benefits in mice with experimental autoimmune encephalomyelitis (EAE),25 an animal model of multiple sclerosis (MS). Along these lines, genetic inactivation of mTORC1 prevents the development of EAE in mice.26 Obviously, rapamycin is the drug of choice for treatment of genetic causes of mTORC1 activation, such as lymphangioleiomyomatosis27 and tuberous sclerosis.28. mTORC1 activation has been associated with epilepsy,29–31 learning, and memory.32,33 Given such broad implications of mTOR pathway activation and its involvement in a wide spectrum of common diseases, overall lifespan, and social adaptation, this chapter will critically review its relevance for health and personalized medicine.
Figure 1.

Schematic diagram of mTOR-mediated T cell activation and lineage specification. mTOR is a sensor of metabolic stress and integrator of environmental cues. Activation of mTOR complex 1 (mTORC1) is triggered by oxidative stress,82 sufficiency of amino acids, and growth factors, which, in turn, promote proinflammatory skewing of T cell development. Mitochondrial oxidative stress and activation of mTORC1 inhibit the expression of FoxP3 and contract Treg cells.21,23 Downstream of mTORC1,135,136 HIF1α-dependent activation of glycolytic enzymes drives the proinflammatory metabolic changes,137 stimulating the expression of RORγt and the expansion of TH17 cells.22,26,137 This is consistent on the reliance of TH17 and Treg cells on glycolysis and mitochondrial metabolism, respectively.137
Figure 2.
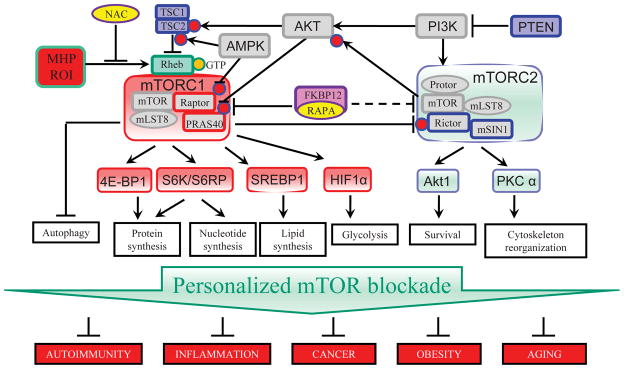
Pharmacologically targetable regulatory checkpoints of the mTOR pathway. While rapamycin and rapalogs selectively block mTORC1, ATP-competitive mTOR kinase domain inhibitors abrogate both mTORC1 and mTORC2 activities. Dual inhibitors of the ATP-binding kinase domains of PI3K and mTOR provide the most overwhelming blockade of cell growth and proliferation. A personalized approach to mTOR pathway blockade holds promise to control autoimmune disease, cancer, obesity, and aging.
The causes and consequences of mTOR pathway activation
mTORC1 is activated by nutrients and the availability of cellular energy, such as amino acids and ATP11 (Fig. 1). In turn, growth factors (e.g., insulin) stimulate mTORC1 via the tuberous sclerosis complex (TSC), comprising TSC1 and TSC2. Further upstream, phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) belongs to a family of enzymes that phosphorylate the 3 position hydroxyl group of the inositol ring of phosphatidylinositol and thus transmit growth signals and block apoptosis via activating another kinase, Akt (Fig. 2).34 As an example, insulin stimulates the PI3K–AKT kinase complex, which phosphorylates TSC2 (also known as tuberin). While TSC1 serves as a stabilizer of the TSC complex, TSC2 acts as a GTPase-activating protein (GAP) to promote the activity of the small GTPase RAS homologue enriched in brain (Rheb). Amino acid sufficiency is transduced via the RAS-related GTP-binding protein (RAG) family of small GTPases which mediate the translocation of mTORC1 from the cytoplasm to the surface of the lysosome (Fig. 1), where mTORC1 is activated by Rheb. Redox-dependent activation of mTORC1 involves cysteine oxidation and interaction with Rheb35 and Raptor.35,36 These findings are consistent with earlier observations that mitochondrial oxidative stress is a trigger of mTORC1 activation.37
Upon activation, mTORC1 controls protein synthesis by inducing ribosome biogenesis38 and mRNA translation.39,40 mTORC1 phosphorylates S6 ribosomal kinase (S6K) to induce protein synthesis and 4E binding protein 1 (4E-BP1) to promote translation.41 Specifically, mTORC1 phosphorylates S6K on Thr389, which activates S6K to phosphorylate ribosomal protein S6 (S6RP), a component of the 40S ribosomal subunit. mTORC1 also phosphorylates the translation inhibitor 4E-BP1, causing the liberation of a key initiation factor of eukaryotic translation initiation, factor eIF4E. However, these fundamental checkpoints do not exert complete control of de novo protein synthesis in skeletal muscle and liver tissue.42 Mice lacking S6K and S6RP activate a compensatory mechanism through inhibition of 4E-BP.38 These findings indicate significant cross talk between the ribosome biogenesis and protein translation pathways, which are separately controlled by mTORC1 via S6K and 4E-BP1, respectively. mTORC1 promotes the transcription of genes involved in glycolysis, the pentose phosphate pathway (PPP), and de novo lipogenesis.43 Upregulation of glycolysis is mediated via the transcription factor hypoxia-inducible factor 1 α (HIF1α)44,45 (Fig. 2). As revealed by a recent metabolomic study, most of the mTORC1-regulated metabolites belong to the PPP.46 A signature substrate of mTORC1, S6K, directly phosphorylates serine 1859 of the enzyme CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase), which catalyzes the first three steps of de novo nucleotide synthesis46 (Fig. 2).
In addition to responding to growth signals and promoting cell proliferation, mTORC1 is also actively involved in blocking autophagy, a complex lysosomal degradation pathway that allows cell survival during starvation. The initiation of autophagy is inhibited by mTORC1 through phosphorylation of autophagy/beclin-1 regulator 1 (AMBRA1).47 Upon separation from mTORC1, unc-51–like kinase 1/autophagy related gene 1 (ULK1/ATG1) phosphorylates beclin-1 and binds to membranes to start autophagosome formation.47
Although mTORC2 regulation is less well understood, it involves its PI3K-dependent association with ribosomes and phosphorylation of Akt (Fig. 2).48 Further downstream, mTORC2 promotes insulin-like growth factor 2 (IGF2) production and ultimately cell proliferation by phosphorylating IGF2 mRNA-binding protein 1 (IMP1).49 Similar to mTORC1, mTORC2 activates SREBP1 transcriptionally and posttranslationally to enhance glycolysis and lipogenesis.50 Via mTORC2, insulin also promotes cell survival via cytoskeleton reorganization51–53 (Fig. 2).
Duration and selectivity of mTORC1 and mTORC2 blockade is critical for control of diabetes and obesity
Increased mTOR signaling has been implicated in metabolic diseases, such as diabetes and obesity.54 mTORC1 and its downstream target S6K are involved in amino acid–induced insulin resistance. Combined hyperaminoacidemia and postprandial hyperinsulinemia increase S6K phosphorylation and inhibitory insulin receptor substrate-1 (IRS-1) phosphorylation at Ser312 and Ser636.55 Activation of mTORC1 is also required for the differentiation of adipocytes in mice56 and humans.57 Accordingly, long-term blockade of mTORC1 by rapamycin reduced high-fat diet–induced obesity in mice.58 However, this beneficial effect of mTORC1 blockade impaired glucose tolerance.59 It appears that short-term blockade of mTORC1, for 2 weeks or so, causes insulin resistance,60,61 which is likely to occur via secondary activation of mTORC2.16 As reinforced by a seminal follow-up study, the duration of treatment with rapamycin is critical. While 2-week treatment has detrimental metabolic effects, 6-week treatment leads to a metabolic transition and 20-week treatment improves metabolic profiles and insulin sensitivity.62
Proinflammatory effects of mTOR pathway activation within the adaptive and innate immune systems
Signaling pathways that control the proliferation, survival, and differentiation of cells in the immune system regulate metabolic pathways to provide nutrients required to support specialized lymphocyte functions.63 Recently, mTOR was identified as a central integrator of metabolic cues that drive lineage specification in the T cell compartment.26 In order to support cell proliferation, mTORC1 promotes the transcription of genes involved in glycolysis, the pentose phosphate pathway (PPP) and de novo lipogenesis.43 In particular, mTORC1 induces glucose 6-phoshate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PDG).43 It has been generally assumed that mTORC1 signaling increases flux through the oxidative PPP to generate NADPH, which is needed for reducing power and for many biosynthetic processes, and ribose 5-phosphate, which is needed for the synthesis of nucleotides.43 Earlier studies suggest that myc- and mTORC1-dependent activation of T cells involves dramatic upregulation of glucose consumption via PPP.64 Selective activation of mTORC1 is required for the development of TH17 cells that mediate the development of EAE, which is induced by myelin oligodendrocyte glycoprotein (MOG) immunization of mice.26 Both mTORC1 and mTORC2 are required for TH1 development, while only mTORC2 is required for TH2 development.26 Inactivation of both mTORC1 and mTORC2 favor the development of Treg cells.26 Inhibition studies with rapamycin suggest that mTORC1 blocks the development of CD8+ memory T cells.65
mTOR pathway activation: a biomarker for diagnosis and target for treatment in SLE
The fundamental role for mTOR pathway activation in T cell lineage specification is consistent with its involvement in transplant rejection6,7 and autoimmunity, as demonstrated by its central role in the pathogenesis of the prototypical systemic autoimmune disease, SLE.20,66,67 The involvement of mTOR activation was initially suggested by successful blockade of T cell activation, autoimmunity, and nephritis in rapamycin-treated lupus-prone mice.3 Rapamycin also blocked T cell activation in patients with SLE20 with remarkable therapeutic efficacy.4,23 The activation of mTORC1 preceded disease flares by 4 months and responded to therapeutic intervention with rapamycin.23 In particular, rapamycin blocked the proinflammatory, necrotic death of CD4−CD8− DN T cells and depletion of CD4+CD25+FoxP3+ Treg cells.23 Diminished Treg frequency may be linked to mTORC1-sensitive methylation of the Foxp3 promoter.68 Diminished expression of FoxP3 in Treg cells may be due to reduced mTORC2 activity in lupus T cells.22,69,70 Rapamycin treatment in vivo reduced the production of IL-4 by DN T cells,23 which accounts for increased production of anti-DNA auto-antibodies by B cells.71,72 The benefit of rapamycin may also be attributed to Treg expansion23 via activation of mTORC2.22
Antiphospholipid antibodies (APLA) represent a component of the diagnostic criteria for SLE.73–75 They also contribute to significant pathologies, specifically antiphospholipid syndrome (APS) in patients with or without SLE.76 In a retrospective study of 10 patients with APS nephropathy who required renal transplantation and received treatment with sirolimus (i.e., rapamycin), 7 of 10 patients (70%) had a functioning allograft 144 months after transplantation, versus 3 of 27 patients treated without sirolimus (11%).77 Interestingly, the majority of APS patients in this study also had SLE (16/28 = 57%).77 However, it has not been disclosed how many of the seven patients who actually benefited from sirolimus77 satisfied the diagnosis of SLE.73,74 mTOR activity has not been measured within the immune system itself,77 which is considered to be the principal mediator of autoimmunity both in APS78 and SLE.79 Therefore, it is likely that sirolimus may selectively benefit renal transplant recipients with underlying SLE.
N-acetylcysteine (NAC), which is a precursor of glutathione, reversed the depletion of this natural antioxidant in peripheral blood lymphocytes (PBL) of patients with SLE in a double-blind placebo-controlled phase I/II clinical trial.21 Importantly, this biological effect of NAC was safe and clinically effective over a 3-month intervention. NAC treatment also reversed the prominent activation of mTORC1 in DN T cells, which is consistent with the role of oxidative stress as a regulatory checkpoint. Oxidative stress originates from increased oxygen consumption by complex I of the mitochondrial electron transport chain (ETC) in lupus T cells.80 In turn, inhibition of mitochondrial oxidative stress by blocking ETC complex I with metformin also reduced mTORC1 activity and prevented nephritis in lupus-prone mice.81
The reliance of certain T cell subsets (e.g., TH17 cells) on mTORC1 during mitochondrial dysfunction–driven oxidative stress82 and the compensatory activation of glycolysis via HIF1α83 are likely to underlie the markedly altered T cell development in SLE.84,85 This is consistent with the induction of HIF1α by oxidative stress86 and its dependence on mTORC1.87 Therefore, the reversal of GSH depletion by NAC may be a particularly safe and mechanistically driven therapeutic intervention in SLE.
As recently revealed, oxidative stress is associated with global metabolome changes in SLE that affect 27 of 80 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, with the most prominent impact on the PPP.88 Moreover, cysteine was depleted and cystine and methionine sulfoxide were accumulated, all of which reflect oxidative stress. Area under the receiver operating characteristic curve (AUC) logistic regression approach identified kynurenine (AUC = 0.859), as the top metabolite predictor of SLE. NAC treatment significantly reduced kynurenine levels relative to placebo in vivo (raw P = 2.8 × 10−7, FDR corrected P = 6.6 × 10−5). Kynurenine was also the top predictor of the NAC effect in SLE (AUC = 0.851). The accumulation of kynurenine may be a result of decreased catabolism by kynurenine hydroxylase owing to NADPH dependence of this enzyme.89 Along these lines, NAC dramatically augmented the levels of NADPH, which can occur through sparing of NADPH via the enhancement of de novo GSH synthesis by NAC.90,91 Thus, the marked suppression of kynurenine in NAC-treated patients can be attributed to the NADPH-sparing effect of NAC.90 Kynurenine stimulated mTOR activity in healthy control PBL in vitro. Therefore, the PPP-connected and NAC-responsive accumulation of kynurenine and its stimulation of mTORC1 are identified as a novel biomarker and as metabolic checkpoints in lupus pathogenesis. 88
A fundamental role of mTOR pathway activation is also illustrated by the first clinical case of fulminant lupus in a patient with TSC.92 In accordance with a negative regulatory role for the TSC1/TSC2 complex, robust mTORC1 activation was documented in all lymphocyte subsets of this patient.92
As evidenced by diminished phosphorylation of serine473 in Akt, activity of mTORC2 is reduced in DN T cells of SLE patients.22 Rapamycin treatment reduced mTORC1 and enhanced mTORC2 activities of lupus T cells in vitro.22 This is consistent with an inhibitory role for mTORC1/S6K-mediated phosphorylation of Rictor in mTORC2 and Akt signaling.93 It is presently unknown whether the therapeutic benefit of rapamycin is solely associated with blockade of mTORC1 or whether it also involves the activation of mTORC2. Pursuit of such studies is justified by the notion that TOR inhibitors other than rapamycin may be more effective. As examples, Torin1 or INK128, two ATP-competitive inhibitors of mTOR, inhibit both mTORC1 and mTORC2.94 Such dual inhibitors are currently in clinical trials in patients with cancer. Using flow cytometry, dual monitoring of mTORC1 and mTORC2 via intracellular staining for pS6RP Ser235/236 and pAktSer473 has been clearly delineated and proposed as relevant biomarkers for disease pathogenesis, prediction of flares, and monitoring of treatment efficacy of mTOR blockade in SLE.21–23
mTOR is a metabolic regulator of survival in cancer cells
Inherited mutations in the tuberous sclerosis genes TSC1 and TSC2 cause autosomal-dominant hamartomas95 and lymphangioleiomyomatosis.96 The first clinical case of lupus in a patient with mTORC1 activation due to tuberous sclerosis was recently documented.92 However, most cancers do not carry genetic defects in TSC1 or TSC2, and the activation of mTORC1 is triggered by mutations that inactivate p53.97,98 p53 restrains mTORC1 by transactivating its negative regulators, namely TSC2 and AMPKβ1.99 Downstream, the role of mTOR in carcinogenesis is linked to multiple layers of metabolic regulatory networks that activate glycolysis via increasing the expression of pyruvate kinase.100 This enzyme, isoenzyme M2, has been identified as the mediator of the so-called Warburg effect, which is a long-recognized metabolic feature of cancer cells that rely on glycolysis rather than mitochondrial respiration.101 This allows cancer cell survival under hypoxic conditions through reliance on glycolysis, which involves the transcription factor HIF1α, which orchestrates the coordinate upregulation of glycolytic gene expression.44,45
mTORC1 also promotes carcinogenesis by inhibiting physiological protein turnover via autophagy. This occurs at the phase of autophagosome formation, which is inhibited by mTORC1 through phosphorylation of AMBRA1.47 Therefore, mTORC1 promotes tumorigenesis both through reversing glycolysis and promoting autophagy. While mTORC2 is essential for cell survival, its activation is a critical mediator of cancer cells lacking PTEN.102 Meta-analyses indicate that mTOR blockade reduces the incidence of a variety of cancers.103 The efficacy of such treatment may depend on the relative involvement of mTORC1 and mTORC2.
Pharmacological blockade of mTOR beyond rapamycin
Since mTOR forms an integral part of both enzyme complexes, mTORC1 and mTORC2, inhibitors have been developed that compete with ATP for access to the active site of mTOR. For example, Torin1, an ATP-competitive mTOR inhibitor, directly inhibits both mTORC1 and mTORC2.104 Torin1 blocks cell growth and proliferation to a far greater degree than rapamycin.104 These effects are independent of mTORC2 inhibition, and they are thought to be mediated by mTORC1-dependent and relatively rapamycin-resistant phosphorylation of 4E-BP1.104 Therefore, these direct inhibitors of mTORC1 kinase activity may be more successful than rapamycin for cancer treatment105 via inhibiting tumor growth and triggering autophagy that depend on mTORC1.104 ATP-competitive inhibitors that target the active sites of the holoenzymes in both PI3K and mTOR have been also developed.105 These drugs, which also exert greater proapoptotic and antiproliferative effects over rapamycin,106 have entered phase I clinical trials.107 While the overall safety and potency of rapalogs and ATP-competitive mTOR and dual PI3P/mTOR inhibitors are unknown, their large numbers offer tremendous hope for treatment of various forms of cancer.105
Personalized mTOR blockade for extension of life span
mTORC1 blockade by rapamycin is associated with increased life span in mice.108 Such beneficial effect of rapamycin is independent of calorie restriction,109 and it does not compromise mitochondrial homeostasis and muscle endurance.110 However, the blockade of mTORC2 by rapamycin increases insulin resistance, which appears to be independent from extending life span.16 Although this is presently far from certain, blockade of mTORC2 may compromise the extension of life span in subjects with obesity and hyperglycemia. In contrast, blocking of both mTORC1 and mTORC2 favors the development of Treg cells;26 thus, an ATP-competitive mTOR inhibitor may have greater therapeutic benefit and potency for extension of life span over rapamycin in patients with the autoimmune disease SLE. Oral rapamycin increases life span without changes in body weight, even when started late in life.108 Such an effect was reproduced in C57BL/6 mice were treated with rapamycin at 4 mg/kg body weight by intraperitoneal injection every other day for 6 weeks starting at age 22 months.111 However, 1 mg/kg rapamycin given 3 times weekly markedly reduced the body weight of lupus-prone mice, while completely blocking autoimmunity and nephritis.112 Interim analysis of a prospective open-label clinical trial with rapamycin in SLE patients revealed marked improvement of disease activity with blockade of mTORC1.23 Final analysis of rapamycin impact on weight, glucose tolerance, and hyperlipidemia are expected following the completion of this study in 2015. Owing to its potent anti-proliferative effects on fibroblasts, rapamycin has been used to block keloid disease113 and to maintain patency of coronary artery stents.114
However, mTOR blockade is associated with serious side effects. For example, rapamycin treatment induces hyperlipidemia, at least in renal transplant recipients.115 Of the potentially fatal adverse events, the most common were pneumonia (30.8%) and sepsis (38.5%).103 These potential side effects are of concern because infections and cardiovascular disease are the leading causes of increased mortality in SLE.116 Importantly, oxidative stress significantly contributes to cardiovascular disease,117 which is responsive to treatment with NAC in patients with end-stage renal disease.118 NAC was also shown to increase high-density lipoprotein (HDL) cholesterol in patients with hyperlipidemia,119,120 which has long been recognized as a hallmark of enhanced atherosclerosis in SLE.121–129 mTORC2 protects the heart from ischemic damage.130,131 Thus, in line with selectively blocking mTORC1, NAC may also be beneficial for reducing cardiovascular disease and fatigue in SLE. Fatigue is a common side effect noted in clinical trials, even with the newer rapalogs, everolimus and temsirolimus.132 Future studies should be directed at deciphering whether GSH depletion and activation of mTORC1 predict responsiveness to treatment by NAC and mTOR inhibitors, alone or in combination, and whether selective or non-selective mTORC1, mTORC2, or dual mTORC1/mTORC2 inhibitors deliver greater clinical benefit (Table 1).
Table 1.
Pharmacological blockade of mTOR pathway activation
| Drug | Molecular Target | Mechanism action | Pathway blockade | Reference |
|---|---|---|---|---|
| Rapamycin | FKBP12 | Allosteric | mTORC1 | 1, 2, 13) |
| Everolimus | FKBP12 | Allosteric | mTORC1 | 132, 139 |
| Temsirolimus | FKBP12 | Allosteric | mTORC1 | 132, 139 |
|
| ||||
| Torin1 | mTOR kinase domain | ATP competitive | mTORC1/mTORC2 | 104 |
| AZD8055 | mTOR kinase domain | ATP competitive | mTORC1/mTORC2 | 140 |
| INK128 | mTOR kinase domain | ATP competitive | mTORC1/mTORC2 | 94 |
|
| ||||
| BGT226 | PI3K/mTOR kinase | ATP competitive | PI3K/mTORC1/mTORC2 | 107 |
|
| ||||
| BAL | Disulfide bonds | H donor | Raptor/mTORC1 | 36 |
|
| ||||
| NAC | GSH | Antioxidant | mTORC1 | 21 |
|
| ||||
| Metformin | ETC complex I | Antioxidant | mTORC1 | 81 |
Constitutive mutations in TSC1/TSC2 lead to hamartomas95 and lymphangioleiomyomatosis via activation of mTORC1.96 In turn, cell type–specific inactivation of p53 triggers uncontrolled cell proliferation in lung and thyroid cancer133 and other solid tumors via upregulation of mTORC1 as well.97–99 Therefore, the preferential blockade of mTORC1 by rapamycin and rapalogs may be the optimal treatment for these types of neoplasia (Table 1). In contrast, the selective blockade of mTORC2 is preferred in another subset of cancers that exhibit the loss of PTEN, such as prostate cancer102 and breast cancer.134 Nevertheless, the importance of preserving mTORC2 function is supported by the notion that no spontaneous mutations have been found in essential components of this complex. Along these lines, inactivation of mTORC2 via disruption of Rictor causes embryonic lethality.17 mTORC2 is required for the expression of FoxP3.69,70 Diminished mTORC2 activity may underlie the reduced expression of FoxP3 and loss of Treg cells in patients with SLE.22 Therefore, the selective blockade of mTORC1 is desirable for SLE23 and prevention of autoimmunity in general.70 Moreover, owing to the involvement of mTORC1 in lipogenesis, the inhibition of this complex is likely to be advantageous in most clinical cases of obesity. Taken together, the personalized blockade of mTORC1—in time and space—appears to be a promising approach to extend life span via preventing obesity and the development of some cancers, as well as autoimmune inflammatory and connective tissue diseases.
Acknowledgments
This work was supported in part by Grants AI 048079 and AI 072648 from the National Institutes of Health and the Central New York Community Foundation, and Investigator-Initiated Research Grant P0468X1-4470/WS1234172 from Pfizer.
References
- 1.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY 22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot. 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 2.Sehgal SN, Bansback CC. Rapamycin: In vitro profile of a new immunosuppressive macrolide. Ann NY Acad Sci. 1993;685:58–67. doi: 10.1111/j.1749-6632.1993.tb35852.x. [DOI] [PubMed] [Google Scholar]
- 3.Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arth Rheum. 1994;37:289–297. doi: 10.1002/art.1780370219. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T-cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54:2983–2988. doi: 10.1002/art.22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collier DSJ, Calne R, Thiru S, Lim S, Pollard SG, Barron P, Da Costa M, White DJG. Rapamycin in experimental renal allografts in dogs and pigs. Transplant Proc. 1990;22:1674–1675. [PubMed] [Google Scholar]
- 6.Calne RY, Lim S, Samaan A, Collier DS, Pollard SG, White DJG, Thiru S. RAPAMYCIN FOR IMMUNOSUPPRESSION IN ORGAN ALLOGRAFTING. Lancet. 1989;334:227. doi: 10.1016/s0140-6736(89)90417-0. [DOI] [PubMed] [Google Scholar]
- 7.Sindhi R, LaVia MF, Paulling E, McMichael J, Burckart G, Shaw S, Sindhi LA, Livingston R, Sehgal S, Jaffe J. Stimulated response of peripheral lymphocytes may distinguish cyclosporine effect in renal transplant recipients receiving a cyclosporine+rapamycin regimen. Transplantation. 2000;69:432–436. doi: 10.1097/00007890-200002150-00022. [DOI] [PubMed] [Google Scholar]
- 8.Aghdasi B, Ye K, Resnick A, Huang A, Ha HC, Guo X, Dawson TM, Dawson VL, Snyder SH. FKBP12, the 12-kDa FK506-binding protein, is a physiologic regulator of the cell cycle. Proc Natl Acad Sci USA. 2001;98:2425–2430. doi: 10.1073/pnas.041614198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dolinski K, Muir S, Cardenas M, Heitman J. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997;94:13093–13098. doi: 10.1073/pnas.94.24.13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 11.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126:1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E-BP1 phosphorylation: A novel two step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fonseca BD, Diering GH, Bidinosti MA, Dalal K, Alain T, Balgi AD, Forestieri R, Nodwell M, Rajadurai CV, Gunaratnam C, Tee AR, Duong F, Andersen RJ, Orlowski J, Numata M, Sonenberg N, Roberge M. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2012;287:17530–17545. doi: 10.1074/jbc.M112.359638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKC[alpha], but Not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Xu K, Liu P, Wei W. MTOR signaling in tumorigenesis. Biochim Biophys Acta. 2014;1846:638–654. doi: 10.1016/j.bbcan.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, Liu D, Wan L, Zhai B, Yu Y, Yuan M, Kim BM, Shaik S, Menon S, Gygi SP, Lee TH, Asara JM, Manning BD, Blenis J, Su B, Wei W. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15:1340–1350. doi: 10.1038/ncb2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, Phillips PE, Crow MK, Oess S, Muller-Esterl W, Perl A. Activation of mTOR controls the loss of TCR in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182:2063–2073. doi: 10.4049/jimmunol.0803600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai Z-W, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, Miklossy G, Jimah J, Doherty E, Tily H, Francis L, Garcia R, Dawood M, Yu J, Ramos I, Coman I, Faraone SV, Phillips PE, Perl A. N-acetylcysteine reduces disease activity by blocking mTOR in T cells of lupus patients. Arthritis Rheum. 2012;64:2937–2946. doi: 10.1002/art.34502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato H, Perl A. MTORC1 expands Th17 and IL-4+ DN T cells and contracts Tregs in SLE. J Immunol. 2014;192:4134–4144. doi: 10.4049/jimmunol.1301859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai Z-W, Borsuk R, Shadakshari A, Yu J, Dawood M, Garcia R, Francis L, Tily H, Bartos A, Faraone SV, Phillips PE, Perl A. mTOR activation triggers IL-4 production and necrotic death of double-negative T cells in patients with systemic lupus eryhthematosus. J Immunol. 2013;191:2236–2246. doi: 10.4049/jimmunol.1301005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carbone F, De Rosa V, Carrieri PB, Montella S, Bruzzese D, Porcellini A, Procaccini C, La Cava A, Matarese G. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat Med. 2014;20:69–74. doi: 10.1038/nm.3411. [DOI] [PubMed] [Google Scholar]
- 25.Esposito M, Ruffini F, Bellone M, Gagliani N, Battaglia M, Martino G, Furlan R. Rapamycin inhibits relapsing experimental autoimmune encephalomyelitis by both effector and regulatory T cells modulation. J Neuroimmunol. 2010;220:52–63. doi: 10.1016/j.jneuroim.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–304. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP, III, Goldberg HJ, Young LR, Kinder BW, Downey GP, Sullivan EJ, Colby TV, McKay RT, Cohen MM, Korbee L, Taveira-DaSilva AM, Lee HS, Krischer JP, Trapnell BC. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–1606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Julich K, Sahin M. Mechanism-based treatment in tuberous sclerosis complex. Ped Neurol. 2014;50:290–296. doi: 10.1016/j.pediatrneurol.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brewster AL, Lugo JN, Patil VV, Lee WL, Qian Y, Vanegas F, Anderson AE. Rapamycin Reverses Status Epilepticus-Induced Memory Deficits and Dendritic Damage. PLoS ONE. 2013;8:e57808. doi: 10.1371/journal.pone.0057808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waltereit R, Welzl H, Dichgans J, Lipp HP, Schmidt WJ, Weller M. Enhanced episodic-like memory and kindling epilepsy in a rat model of tuberous sclerosis. J Neurochem. 2006;96:407–413. doi: 10.1111/j.1471-4159.2005.03538.x. [DOI] [PubMed] [Google Scholar]
- 32.Huynh TN, Santini E, Klann E. Requirement of mammalian target of rapamycin complex 1 downstream effectors in cued fear memory reconsolidation and its persistence. J Neurosci. 2014;34:9034–9039. doi: 10.1523/JNEUROSCI.0878-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gkikas I, Petratou D, Tavernarakis N. Longevity pathways and memory aging. Front Genet. 2014;5:155. doi: 10.3389/fgene.2014.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Franke TF, Kaplan DR, Cantley LC. PI3K: Downstream AKTion Blocks Apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, Okada M, Guan KL, Inoki K. Redox Regulates Mammalian Target of Rapamycin Complex 1 (mTORC1) Activity by Modulating the TSC1/TSC2-Rheb GTPase Pathway. J Biol Chem. 2011;286:32651–32660. doi: 10.1074/jbc.M111.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarbassov dD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005;280:39505–39509. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- 37.Desai BN, Myers BR, Schreiber SL. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc Natl Acad Sci USA. 2002;99:4319–4324. doi: 10.1073/pnas.261702698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau-Aveilla C, Dreazen A, Cagnard N, Carpentier W, Kiss T, Meyuhas O, Pende M. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene. 2014;33:474–483. doi: 10.1038/onc.2012.606. [DOI] [PubMed] [Google Scholar]
- 39.Jung H, Gkogkas CG, Sonenberg N, Holt CE. Remote control of gene function by local translation. Cell. 2014;157:26–40. doi: 10.1016/j.cell.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dowling RJO, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804:433–439. doi: 10.1016/j.bbapap.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Lekmine F, Uddin S, Sassano A, Parmar S, Brachmann SM, Majchrzak B, Sonenberg N, Hay N, Fish EN, Platanias LC. Activation of the p70 S6 Kinase and Phosphorylation of the 4E-BP1 Repressor of mRNA Translation by Type I Interferons. J Biol Chem. 2003;278:27772–27780. doi: 10.1074/jbc.M301364200. [DOI] [PubMed] [Google Scholar]
- 42.Mieulet V, Roceri M, Espeillac C, Sotiropoulos A, Ohanna M, Oorschot V, Klumperman J, Sandri M, Pende M. S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am J Physiol Cell Physiol. 2007;293:C712–C722. doi: 10.1152/ajpcell.00499.2006. [DOI] [PubMed] [Google Scholar]
- 43.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, Liu H, Lu Y, Liao J, Chen X, Chu Y. SIRT1 Limits the Function and Fate of Myeloid-Derived Suppressor Cells in Tumors by Orchestrating HIF-1-α Dependent Glycolysis. Cancer Res. 2014;74:727–737. doi: 10.1158/0008-5472.CAN-13-2584. [DOI] [PubMed] [Google Scholar]
- 46.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F. MTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 48.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 49.Dai N, Christiansen J, Nielsen FC, Avruch J. mTOR complex 2 phosphorylates IMP1 cotranslationally to promote IGF2 production and the proliferation of mouse embryonic fibroblasts. Genes Dev. 2013;27:301–312. doi: 10.1101/gad.209130.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Rüegg MA, Hall MN. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 51.Zhang C, Cooper DE, Grevengoed TJ, Li LO, Klett EL, Eaton JM, Harris TE, Coleman RA. Glycerol-3-phosphate acyltransferase-4-deficient mice are protected from diet-induced insulin resistance by the enhanced association of mTOR and rictor. Am J Physiol Endocrin Metab. 2014;307:E305–E315. doi: 10.1152/ajpendo.00034.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramakrishnan G, Davaakhuu G, Kaplun L, Chung WC, Rana A, Atfi A, Miele L, Tzivion G. Sirt2 deacetylase is a novel AKT Binding partner critical for AKT activation by insulin. J Biol Chem. 2014;289:6054–6066. doi: 10.1074/jbc.M113.537266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sato M, Dehvari N, Öberg AI, Dallner OS, Sandström AL, Olsen JM, Csikasz RI, Summers RJ, Hutchinson DS, Bengtsson T. Improving type 2 diabetes through a distinct adrenergic signaling pathway involving mTORC2 that mediates glucose uptake in skeletal muscle. Diabetes. 2014;63:4115–4129. doi: 10.2337/db13-1860. [DOI] [PubMed] [Google Scholar]
- 54.Khamzina L, Veilleux A, Bergeron S, Marette A. Increased Activation of the Mammalian Target of Rapamycin Pathway in Liver and Skeletal Muscle of Obese Rats: Possible Involvement in Obesity-Linked Insulin Resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- 55.Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Fürnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M. The Mammalian Target of Rapamycin Pathway Regulates Nutrient-Sensitive Glucose Uptake in Man. Diabetes. 2007;56:1600–1607. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- 56.Yeh WC, Bierer BE, McKnight SL. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc Natl Acad Sci USA. 1995;92:11086–11090. doi: 10.1073/pnas.92.24.11086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bell A, Grunder L, Sorisky A. Rapamycin inhibits human adipocyte differentiation in primary culture. Obesity Res. 2000;8:249–254. doi: 10.1038/oby.2000.29. [DOI] [PubMed] [Google Scholar]
- 58.Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH, Mao FC. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J Pharmacol Sci. 2009;109:496–503. doi: 10.1254/jphs.08215fp. [DOI] [PubMed] [Google Scholar]
- 59.Chang GR, Wu YY, Chiu YS, Chen WY, Liao JW, Hsu HM, Chao TH, Hung SW, Mao FC. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Bas Clin Pharmacol Toxicol. 2009;105:188–198. doi: 10.1111/j.1742-7843.2009.00427.x. [DOI] [PubMed] [Google Scholar]
- 60.Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, Leibowitz G. mTOR inhibition by rapamycin prevents β-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 62.Fang Y, Westbrook R, Hill C, Boparai RK, Arum O, Spong A, Wang F, Javors MA, Chen J, Sun LY, Bartke A. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. 2013;17:456–462. doi: 10.1016/j.cmet.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caro-Maldonado A, Gerriets VA, Rathmell JC. Matched and mismatched metabolic fuels in lymphocyte function. Semin Immunol. 2012;24:405–413. doi: 10.1016/j.smim.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernandez DR, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discov Med. 2010;9:173–178. [PMC free article] [PubMed] [Google Scholar]
- 67.Moulton VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arth Res Ther. 2010;13 doi: 10.1186/ar3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tomasoni R, Basso V, Pilipow K, Sitia G, Saccani S, Agresti A, Mietton F, Natoli G, Colombetti S, Mondino A. Rapamycin-sensitive signals control TCR/CD28-driven Ifng, Il4 and Foxp3 transcription and promoter region methylation. Eur J Immunol. 2011;41:2086–2096. doi: 10.1002/eji.201041130. [DOI] [PubMed] [Google Scholar]
- 69.Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16:178–187. doi: 10.1038/ni.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ray JP, Craft J. PTENtiating autoimmunity through Treg cell deregulation. Nat Immunol. 2015;16:139–140. doi: 10.1038/ni.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sieling PA, Porcelli SA, Duong BT, Spada F, Bloom BR, Diamond B, Hahn BH. Human Double-Negative T Cells in Systemic Lupus Erythematosus Provide Help for IgG and Are Restricted by CD1c. J Immunol. 2000;165:5338–5344. doi: 10.4049/jimmunol.165.9.5338. [DOI] [PubMed] [Google Scholar]
- 72.Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double-negative (CD4−/CD8−) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol. 1989;143:103–112. [PubMed] [Google Scholar]
- 73.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arth Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 74.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arth Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 75.Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, Bruce IN, Isenberg D, Wallace DJ, Nived O, Sturfelt G, Ramsey-Goldman R, Bae SC, Hanly JG, Sánchez-Guerrero J, Clarke A, Aranow C, Manzi S, Urowitz M, Gladman D, Kalunian K, Costner M, Werth VP, Zoma A, Bernatsky S, Ruiz-Irastorza G, Khamashta MA, Jacobsen S, Buyon JP, Maddison P, Dooley MA, van Vollenhoven RF, Ginzler E, Stoll T, Peschken C, Jorizzo JL, Callen JP, Lim SS, Fessler BJ, Inanc M, Kamen DL, Rahman A, Steinsson K, Franks AG, Sigler L, Hameed S, Fang H, Pham N, Brey R, Weisman MH, McGwin G, Magder LS. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arth Rheum. 2012;64:2677–2686. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, Groot PGDE, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).[see comment]. [136 refs] J Thromb Haemost. 2006;4:295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 77.Canaud G, Bienaimé F, Tabarin F, Bataillon G, Seilhean D, Noël L-H, Dragon-Durey MA, Snanoudj R, Friedlander G, Halbwachs-Mecarelli L, Legendre C, Terzi F. Inhibition of the mTORC Pathway in the Antiphospholipid Syndrome. N Engl J Med. 2014;371:303–312. doi: 10.1056/NEJMoa1312890. [DOI] [PubMed] [Google Scholar]
- 78.Giannakopoulos B, Krilis SA. The Pathogenesis of the Antiphospholipid Syndrome. N Engl J Med. 2013;368:1033–1044. doi: 10.1056/NEJMra1112830. [DOI] [PubMed] [Google Scholar]
- 79.Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 80.Doherty E, Oaks Z, Perl A. Increased Mitochondrial Electron Transport Chain Activity at Complex I is Regulated by N-acetylcysteine in Lymphocytes of Patients with Systemic Lupus Erythematosus. Antiox Redox Signal. 2014;21:56–65. doi: 10.1089/ars.2013.5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7:274ra18. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat Rev Rheumatol. 2013;9:674–686. doi: 10.1038/nrrheum.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev. 2013;252:52–77. doi: 10.1111/imr.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Inducible Factor-Dependent Extension of the Replicative Life Span during Hypoxia. Mol Cell Biol. 2007;27:5737–5745. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential Dependence of Hypoxia-inducible Factors 1-α and 2-α on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495–34499. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perl A, Hanczko R, Lai Z-W, Oaks Z, Kelly R, Borsuk R, Asara JM, Phillips PE. Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: implications for activation of the mechanistic target of rapamycin. Metabolomics. 2015 doi: 10.1007/s11306-015-0772-0. in press: http://link.springer.com/article/10.1007/s11306-015-0772-0. [DOI] [PMC free article] [PubMed]
- 89.Breton J, Avanzi N, Magagnin S, Covini N, Magistrelli G, Cozzi L, Isacchi A. Functional characterization and mechanism of action of recombinant human kynurenine 3-hydroxylase. Eur J Biochem. 2000;267:1092–1099. doi: 10.1046/j.1432-1327.2000.01104.x. [DOI] [PubMed] [Google Scholar]
- 90.Hanczko R, Fernandez D, Doherty E, Qian Y, Vas Gy, Niland B, Telarico T, Garba A, Banerjee S, Middleton FA, Barrett D, Barcza M, Banki K, Landas SK, Perl A. Prevention of hepatocarcinogenesis and acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. 2009;119:1546–1557. doi: 10.1172/JCI35722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perl A, Hanczko R, Telarico T, Oaks Z, Landas S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol Med. 2011;7:395–403. doi: 10.1016/j.molmed.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Singh N, Birkenbach M, Caza T, Perl A, Cohen PL. Tuberous sclerosis and fulminant lupus in a young woman. J Clin Rheumatol. 2013;19:134–137. doi: 10.1097/RHU.0b013e318289c033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30:908–921. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schenone S, Brullo C, Musumeci F, Radi M, Botta M. ATP-competitive inhibitors of mTOR: An update. Curr Med Chem. 2011;18:2995–3014. doi: 10.2174/092986711796391651. [DOI] [PubMed] [Google Scholar]
- 95.Kwiatkowski DJ. Tuberous sclerosis: From tubers to mTOR. Ann Hum Genet. 2003;67:87–96. doi: 10.1046/j.1469-1809.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- 96.Li C, Lee PS, Sun Y, Gu X, Zhang E, Guo Y, Wu CL, Auricchio N, Priolo C, Li J, Csibi A, Parkhitko A, Morrison T, Planaguma A, Kazani S, Israel E, Xu KF, Henske EP, Blenis J, Levy BD, Kwiatkowski D, Yu JJ. Estradiol and mTORC2 cooperate to enhance prostaglandin biosynthesis and tumorigenesis in TSC2-deficient LAM cells. J Exp Med. 2014;211:15–28. doi: 10.1084/jem.20131080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 99.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The Regulation of AMPKβ1, TSC2, and PTEN Expression by p53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-mTOR Pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 100.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR, Lu Y, You H, Kwiatkowski D, Zhang H. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Warburg O. On the Origin of Cancer Cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 102.Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. The mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Qi WX, Huang YJ, Yao Y, Shen Z, Min DL. Incidence and Risk of Treatment-Related Mortality with mTOR Inhibitors Everolimus and Temsirolimus in Cancer Patients: A Meta-Analysis. PLoS ONE. 2013;8:e65166. doi: 10.1371/journal.pone.0065166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharm Sci. 2015 doi: 10.1016/j.tips.2014.11.004. In press. [DOI] [PubMed] [Google Scholar]
- 106.Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, Battistelli M, Falcieri E, Melchionda F, Pession A, Pagliaro P, McCubrey JA, Martelli AM. Activity of the Novel Dual Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin Inhibitor NVP-BEZ235 against T-Cell Acute Lymphoblastic Leukemia. Cancer Res. 2010;70:8097–8107. doi: 10.1158/0008-5472.CAN-10-1814. [DOI] [PubMed] [Google Scholar]
- 107.Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak Ö, Hackl W, Baselga J. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012;23:2399–2408. doi: 10.1093/annonc/mds011. [DOI] [PubMed] [Google Scholar]
- 108.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, Javors MA, Li X, Nadon NL, Nelson JF, Pletcher S, Salmon AB, Sharp ZD, Van Roekel S, Winkleman L, Strong R. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014;13:468–477. doi: 10.1111/acel.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ye L, Widlund AL, Sims CA, Lamming DW, Guan Y, Davis JG, Sabatini DM, Harrison DE, Vang O, Baur JA. Rapamycin doses sufficient to extend lifespan do not compromise muscle mitochondrial content or endurance. Aging. 2013;5:539–550. doi: 10.18632/aging.100576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen C, Liu Y, Liu Y, Zheng P. mTOR Regulation and Therapeutic Rejuvenation of Aging Hematopoietic Stem Cells. Sci Signal. 2009;2:ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Caza TN, Fernandez D, Talaber G, Oaks Z, Haas M, Madaio MP, Lai Z-W, Miklossy G, Singh RR, Chudakov DM, Malorni W, Middleton FA, Banki K, Perl A. HRES-1/RAB4-Mediated Depletion of DRP1 Impairs Mitochondrial Homeostasis and Represents a Target for Treatment in SLE. Ann Rheum Dis. 2014;73:1887–1897. doi: 10.1136/annrheumdis-2013-203794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Syed F, Sanganee HJ, Bahl A, Bayat A. Potent Dual Inhibitors of TORC1 and TORC2 Complexes (KU-0063794 and KU-0068650) Demonstrate In Vitro and Ex Vivo Anti-Keloid Scar Activity. J Invest Dermatol. 2013;133:1340–1350. doi: 10.1038/jid.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stefanini GG, Holmes DR. Drug-Eluting Coronary-Artery Stents. N Engl J Med. 2013;368:254–265. doi: 10.1056/NEJMra1210816. [DOI] [PubMed] [Google Scholar]
- 115.Brattstrom C, Wilczek H, Tyden G, Bottiger Y, Sawe J, Groth CG. Hyperlipidemia in renal transplant recipients treated with sirolimus (rapamycin) Transplantation. 1998;65:1272–1274. doi: 10.1097/00007890-199805150-00023. [DOI] [PubMed] [Google Scholar]
- 116.Trager J, Ward MM. Mortality and causes of death in systemic lupus erythematosus. Curr Opin Rheumatol. 2001;13:345–351. doi: 10.1097/00002281-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 117.Lee R, Margaritis M, Channon KM, Antoniades C. Evaluating oxidative stress in human cardiovascular disease: Methodological aspects and considerations. Curr Med Chem. 2012;19:2504–2520. doi: 10.2174/092986712800493057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tepel M, van der Giet M, Statz M, Jankowski J, Zidek W. The Antioxidant Acetylcysteine Reduces Cardiovascular Events in Patients With End-Stage Renal Failure. Circulation. 2003;107:992–995. doi: 10.1161/01.cir.0000050628.11305.30. [DOI] [PubMed] [Google Scholar]
- 119.Franceschini G, Werba JP, Safa O, Gikalov I, Sirtori CR. Dose-related increase of HDL-cholesterol levels after N-acetylcysteine in man. Pharmacological Research. 1993;28:213–218. doi: 10.1006/phrs.1993.1124. [DOI] [PubMed] [Google Scholar]
- 120.Kinscherf R, Cafaltzis K, Roder F, Hildebrandt W, Edler L, Deigner HP, Breitkreutz R, Feussner G, Kreuzer J, Werle E, Michel G, Metz J, Droge W. Cholesterol levels linked to abnormal plasma thiol concentrations and thiol/disulfide redox status in hyperlipidemic subjects. Free Rad Biol Med. 2003;35:1286–1292. doi: 10.1016/j.freeradbiomed.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 121.Ilowite NT, Samuel P, Ginzler E, Jacobson MS. Dyslipoproteinemia in pediatric systemic lupus erythematosus. Arthritis and Rheumatism. 1988;31:859–863. doi: 10.1002/art.1780310706. [DOI] [PubMed] [Google Scholar]
- 122.Lahita RG, Rivkin E, Cavanagh I, Romano P. Low levels of total cholesterol, high-density lipoprotein, and apolipoprotein A1 in association with anticardiolipin antibodies in patients with systemic lupus erythematosus. Arthritis and Rheumatism. 1993;36:1566–1574. doi: 10.1002/art.1780361111. [DOI] [PubMed] [Google Scholar]
- 123.Formiga F, Meco JF, Pinto X, Jacob J, Moga I, Pujol R. Lipid and lipoprotein levels in premenopausal systemic lupus erythematosus patients. Lupus. 2001;10:359–363. doi: 10.1191/096120301669070811. [DOI] [PubMed] [Google Scholar]
- 124.Svenungsson E, Jensen-Urstad K, Heimbürger M, Silveira A, Hamsten A, De Faire U, Witztum JL, Frostegård J. Risk factors for cardiovascular disease in systemic lupus erythematosus. Circulation. 2001;104:1887–1893. doi: 10.1161/hc4101.097518. [DOI] [PubMed] [Google Scholar]
- 125.de Carvalho JF, Bonfa E, Borba EF. Systemic lupus erythematosus and “lupus dyslipoproteinemia”. Autoimmunity Reviews. 2008;7:246–250. doi: 10.1016/j.autrev.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 126.Telles RW, Lanna CCD, Ferreira GA, Ribeiro AL. Metabolic syndrome in patients with systemic lupus erythematosus: Association with traditional risk factors for coronary heart disease and lupus characteristics. Lupus. 2010;19:803–809. doi: 10.1177/0961203309359781. [DOI] [PubMed] [Google Scholar]
- 127.Ardoin SP, Schanberg LE, Sandborg C, Yow E, Barnhart HX, Mieszkalski KL, Ilowite NT, Von Scheven E, Eberhard A, Levy DM, Kimura Y, Silverman E, Bowyer SL, Punaro L, Singer NG, Sherry DD, McCurdy D, Klein-Gitelman M, Wallace C, Silver R, Wagner-Weiner L, Higgins GC, Brunner HI, Jung LK, Imundo L, Soep JB, Reed AM. Laboratory markers of cardiovascular risk in pediatri SLE: The APPLE baseline cohort. Lupus. 2010;19:1315–1325. doi: 10.1177/0961203310373937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Srivastava R, Yu S, Parks BW, Black LL, Kabarowski JH. Autoimmune-mediated reduction of high-density lipoprotein-cholesterol and paraoxonase 1 activity in systemic lupus erythematosus-prone gld mice. Arthritis Rheum. 2011;63:201–211. doi: 10.1002/art.27764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parker B, Ahmad Y, Shelmerdine J, Edlin H, Yates AP, Teh LS, Bruce IN. An analysis of the metabolic syndrome phenotype in systemic lupus erythematosus. Lupus. 2011;20:1459–1465. doi: 10.1177/0961203311416695. [DOI] [PubMed] [Google Scholar]
- 130.Volkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S, Joyo A, Ornelas L, Samse K, Thuerauf DJ, Gude N, Glembotski CC, Sussman MA. Mechanistic Target of Rapamycin Complex 2 Protects the Heart From Ischemic Damage. Circulation. 2013;128:2132–2144. doi: 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moschella PC, McKillop J, Pleasant DL, Harston RK, Balasubramanian S, Kuppuswamy D. mTOR complex 2 mediates Akt phosphorylation that requires PKCε in adult cardiac muscle cells. Cell Signal. 2013;25:1904–1912. doi: 10.1016/j.cellsig.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Peng L, Zhou Y, Ye X, Zhao Q. Treatment-related fatigue with everolimus and temsirolimus in patients with cancer meta-analysis of clinical trials. Tumor Biol. 2015;36:643–654. doi: 10.1007/s13277-014-2669-3. [DOI] [PubMed] [Google Scholar]
- 133.Akeno N, Miller AL, Ma X, Wikenheiser-Brokamp KA. p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene. 2015;34:589–599. doi: 10.1038/onc.2013.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Weigelt B, Warne PH, Downward J. PIK3CA mutation, but not PTEN loss of function, determines the sensitivity of breast cancer cells to mTOR inhibitory drugs. Oncogene. 2011;30:3222–3233. doi: 10.1038/onc.2011.42. [DOI] [PubMed] [Google Scholar]
- 135.Sudhagar S, Sathya S, Lakshmi BS. Rapid non-genomic signalling by 17[beta]-oestradiol through c-Src involves mTOR-dependent expression of HIF-1[alpha] in breast cancer cells. Br J Cancer. 2011;105:953–960. doi: 10.1038/bjc.2011.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, Panteleyev AA, Okkenhaug K, Cantrell DA. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc. 2003;35:S7–S14. doi: 10.1016/s0041-1345(03)00211-2. [DOI] [PubMed] [Google Scholar]
- 139.Dancey JE. Therapeutic targets: MTOR and related pathways. Cancer Biol Ther. 2006;5:1065–1073. doi: 10.4161/cbt.5.9.3175. [DOI] [PubMed] [Google Scholar]
- 140.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, Hummersone M, Malagu K, Menear K, Jenkins R, Jacobsen M, Smith GCM, Guichard S, Pass M. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In vitro and In vivo Antitumor Activity. Cancer Res. 2010;70:288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]