

Abstract
Monophosphoryl lipid A (MPLA) is a TLR4 agonist that is used as an immunomodulator in human vaccines; additionally, it has been shown to be protective in models of sepsis. As endothelial cells regulate inflammation, we hypothesized that MPLA would decrease activation of human umbilical vein endothelial cells (HUVECs) to LPS. We studied HUVECs challenged with LPS (100 ng/ml), MPLA (0.001–100 μg/ml) or a combination. Secretion of IL-6, RANTES (CCL5) and IP-10 (CXCL10) were assessed by ELISA. Activation of MAPK phosphorylation and cytokine transcription were assessed by Western blot analysis and PCR, respectively. MPLA alone was a weak stimulator of myeloid differentiation primary response protein 88-dependent IL-6 and did not induce TIR-domain-containing adapter-inducing IFN-β (TRIF)-dependent chemokine responses. MPLA significantly reduced LPS-mediated IL-6 production. This inhibitory effect was also conferred for the TRIF-dependent chemokines RANTES and IP-10. Inhibition of LPS-mediated activation by MPLA was associated with reduced p38 phosphorylation and mRNAs encoding inflammatory cytokines. MPLA inhibition of LPS signaling appeared to be at the level of the TLR4 receptor, acting as a receptor antagonist with weak agonistic properties. This study provides evidence of a novel mechanism for the inhibitory effect of MPLA on LPS-induced endothelial activation.
Keywords: Endotoxin, inflammation, monophosphoryl lipid A, endothelial cells, TLR4
Introduction
Endothelial cells are key regulators of the innate immune response.1 In healthy individuals, they aid in vascular homeostasis. However, in diseased states such as those seen with severe bacterial infections, they promote leukocyte trafficking through expression of cellular adhesion molecules, which enhance a prothrombotic state and produce local inflammatory responses that control vascular tone and permeability.2 These inflammatory activities of endothelial cells are controlled by their expression of pattern recognition receptors, an important one being TLR4. TLR4 is expressed on the surface of cells involved in innate immunity, including endothelial cells,3 and its primary ligand is a component of the Gram-negative bacterial envelope, LPS. LPS activation of endothelial cells requires the presence of two serum co-factors, LPS binding protein (LBP) and soluble CD14.4 Once activated, TLR4 has been proposed to initiate two signaling pathways in a sequential fashion, first through a myeloid differentiation primary response protein 88 (MyD88)-dependent pathway and then through the TIR-domain-containing adapter-inducing IFN-β (TRIF)-dependent pathway.5 The MyD88 pathway is activated at the cell surface and induces expression of proinflammatory mediators, such as IL-6,6 through MAPK pathways,7 while the TRIF pathway requires dynamin-mediated,8 CD14-dependent endocytosis to generate primarily expression of type I IFNs such as IFN-β.9 Activation of TLR4 and its signaling pathways vary depending on the conformation of LPS and its lipid A components.10 LPS derived from Escherichia coli can activate both pathways, but preferentially promotes MyD88-mediated expression of proinflammatory cytokines.11 Repeated exposure of leukocytes to LPS down-regulates MyD88-dependent responses and enhances TRIF-dependent responses inducing a tolerance to further LPS challenges—a phenomenon known as endotoxin tolerance.12 However, the toxicity of LPS prevents it from being used in humans as an immunomodulator.13
Monophosphoryl lipid A (MPLA), a TLR4 agonist derived from the Re mutant of Salmonella Minnesota R595, is a unique TLR4 agonist structurally similar to the native lipid A fraction of LPS, except that the (R)-3-hydroxytetradeconoyl group and 1-phosphate have been removed by successive acid and base hydrolysis.14 In contrast to LPS, MPLA is a less potent activator of the MyD88 pathway in leukocytes,15 and is thought to exert its primary effects through activation of the TRIF-dependent signaling pathway.16 It is through this latter pathway that the immunomodulatory effects of endotoxin tolerance are thought to occur,7 thus providing a potential mechanism for the protective effects of MPLA in animal models of Gram-negative sepsis.17,18 Though it is postulated that MPLA acts primarily through the TRIF pathway, which requires CD14, there is evidence supporting that the effects of MPLA are independent of CD14 expression.19 Thus, the role of CD14 in mediating the effects of MPLA is uncertain, as is the role of LBP.20 Furthermore, the activation of intracellular pathways downstream of TLR4, namely MyD88 and TRIF pathways, in the immunomodulation of endothelial cells remains unknown.
Given the absence of data regarding the effects of MPLA in endothelial inflammation, we sought to answer several fundamental questions relating to an overall hypothesis that MPLA would prevent endothelial activation mediated by LPS. First, we wanted to understand the contributions of CD14 and LBP for MPLA signaling through the TLR4 complex compared to LPS. Second, we sought to determine whether MPLA exposure in the presence of LPS derived from E. coli, a toxin commonly employed in models of sepsis, could inhibit LPS-mediated cellular cytokine activation and whether this affected the MyD88 and TRIF pathways of TLR4 signaling. Lastly, we explored whether the attenuated proinflammatory response to LPS in the presence of MPLA was a result of altered MAPK signaling leading to reduced cytokine transcription. By understanding the protective potential of MPLA to alter the inflammatory response of endothelial cells to LPS, we can explore mechanisms through which MPLA might be used to hinder the detrimental effects of endotoxemia on the endothelium.
Materials and methods
Cells and culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (Basel, Switzerland) and grown in Endothelial Growth Media-2 (Lonza) supplemented with 2% FBS. HUVECs were plated at a density of 30,000 cells/cm2 and grown to confluence. Experiments were conducted between the third and eighth passages. Medium was changed every 3 d.
Treatment conditions
To assess the effect of MPLA on HUVEC proinflammatory responses, HUVECs grown to >90% confluence were exposed to 10μg/ml of MPLA (S. enterica serotype Minnesota Re 595; Sigma, St. Louis, MO, USA) or vehicle control (0.2% triethylamine; Sigma) for 16 h in media containing 0% or 2% FBS, with or without soluble cofactors: 1μg/ml CD14 (ProSpec-Tany Techno Gene Ltd., Rehovot, Israel), 1μg/ml LBP (Sigma). Doses of soluble co-factors were based upon previous literature examining the proinflammatory effects of LPS in HUVECs,4 as well as internal dose titration effects (data not shown).To examine the role of endocytosis in the MPLA-mediated cytokine activation, cells were pre-treated with Dynasore (15 μMl Enzo Life Sciences, Farmingdale, NY, USA) or vehicle control 30 min prior to agonist exposure in 0% FBS conditions. This dose of Dynasore was based on previously published literature showing the effect of inhibited endocytosis on endothelial inflammation.21
To determine the effect of MPLA exposure on LPS-mediated cytokine activation, HUVECs were exposed to 10μg/ml of MPLA and co-incubated in fresh media containing 0% or 2% FBS with Ultra-Pure LPS from E. coli 0111:B4 (100 ng/ml; List Biological Laboratories, Inc. Campbell, CA, USA) or vehicle control and CD14 (1 μg/ml) for 16 h. Poly (I:C) (10 μg/ml; Sigma) served as a positive control for TRIF-dependent activation. For these experiments the dose of MPLA was based on cytokine activation extinguishing studies (see ‘Results’) as well as doses previously reported in the literature to induce maximal activation in human peripheral mononuclear cells.22 The dose of LPS was based on previous literature using dose titration of LPS with regard to proinflammatory responses in HUVECs.4
Cytokine production
Culture supernatants were collected at the end of experimental conditions, 16 h post-agonist exposure, and stored at −80°C until used in the assays. The concentrations of supernatant IL-6 (eBioScience, San Diego, CA, USA), RANTES and IP-10 (R&D Systems, Minneapolis, MN, USA) were assessed using a commercially available ELISA kit and performed according to the manufacturer’s specifications.
Western blot analysis
HUVECs were exposed to agonist for 1 or 16 h. Afterwards, protein extracts (40–60 μg) were separated by electrophoresis on a polyacrylamide gel (10%) and transferred to nitrocellulose membranes. Non-specific binding was inhibited by washing the membranes with 5% skimmed milk in Tris-buffered saline solution with Tween 20 (0.1%) for 1 h at room temperature. Membranes were then incubated with primary Abs overnight at 4°C on a rocker. Abs were as follows: phosphorylated c-Jun N-terminal kinase (c-Jun), p-p38, p38, phosphorylated extracellular signal-regulated kinase (ERK), ERK and p-IRF3 (all Cell Signaling Technology, Danvers, MA, USA); IRF3 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA); and Tubulin (Vanderbilt Ab Core, Nashville, TN, USA). Afterwards, membranes were incubated with fluorescent secondary Abs and developed using the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE, USA) and quantified by densitometry. Data were normalized as a ratio of expressed phosphorylated protein to its respective protein control.
RNA isolation and real-time PCR
RNA was isolated from cultured cells via a Gen Elute Mammalian Total RNA Miniprep Kit (Sigma) following the manufacturer’s instructions. For TLR4-specific experiments, RNA was isolated at 1, 6 and 16 h after exposure. Afterwards, 1.5 μg of total RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Life Technologies, Grand Island, NY, USA) according to the manufacturer’s protocol. Efficiency of the PCR reactions was tested by amplification of the target from serially diluted cDNA generated from reverse transcription to achieve an efficiency of 95% ± 5%. Real-time PCR was performed using the TaqMan Fast Advanced Master Mix on a Step One Plus Real Time PCR System (Applied Biosystems, Life Technologies). PrimeTime® qPCR 5′ Nuclease Assays (Integrated DNA Technologies, Inc., Coralville, IA, USA) were used to amplify the target mRNAs containing the following sequences: 5′-GAAACTGAAGATCTCCTAGCCT and 5′-GCCATCAGTCACTTAAACAGC for IFN-p; 5′-GCAGATGAGTACAAAAGTCCTGA and 5′-TTCTGTGCCTGCAGCTTC for IL-6; 5′-GAGTATACATTGCTGTTTCCTGTTG and 5′-ACCCCATTAATTCCAGACACA for TLR-4; 5′-ACATCGCTCAGACACCATG and 5′-TGTAGT TGAGGTCAATGAAGGG for GAPDH. Data were normalized as a ratio of threshold cycle of target mRNA to GAPDH and corrected for efficiency using the StepOne software.
Statistical analysis
Data are expressed as means ± SE of a single, replicated experiment. Comparisons of treatment groups and conditions were done via one-way ANOVA, with Bonferroni correction for multiple-group comparisons. For dose–response curves, inhibition and stimulation were expressed as a percentage of the cytokine levels relative to baseline samples without MPLA. Concentration–response curves were fitted using a nonlinear interactive fitting program and analyzed using two pharmacological parameters: the slope of the regression curve and the log IC50 (concentration of agonist producing 50% of the inhibitory response). All analyses were done using GraphPad Prism 5.03 statistical software (GraphPad Software Inc., La Jolla, CA, USA). A P-value of < 0.05 was considered statistically significant.
Results
MPLA utilizes both CD14 and LBP and creates a serum-dependent cytokine response
MPLA is known to have a mild inflammatory effect. The more potent agonist of TLR4, LPS, is known to require both CD14 and LBP for receptor activation in endothelial cells.4 Furthermore, CD14 is known to be instrumental in TLR4 endocytosis to further enhance inflammatory signals.9 Whether these soluble co-factors affect MPLA-mediated cytokine activation in endothelial cells is unknown. To test this, we examined the response of HUVECs to MPLA in 0% serum media conditions with various combinations of CD14 and LBP. Culture supernatant levels of IL-6 were measured as an indication of HUVEC activation. The addition of MPLA (10 μg/ml) to HUVECs in isolation did not induce a cytokine response (data not shown). However, as shown in Figure 1a, the addition of either CD14 (1 μg/ml) or LBP (1 μg/ml) increased IL-6 in the supernatant compared with MPLA alone, albeit weakly. Only in combination did CD14 and LBP produce any significant potentiation of IL-6 secretion by HUVECs exposed to MPLA (P < 0.05). Inhibition of endocytosis with Dynasore (15 μM) did not significantly alter the amount of IL-6 produced compared with vehicle controls (n = 4–6 per group). The cytokine response to MPLA in 2% serum was significantly increased compared with MPLA in 0% serum alone, but not compared with MPLA in 0% serum with the addition of CD14 and LBP with or without the presence of Dynasore.
Figure 1.
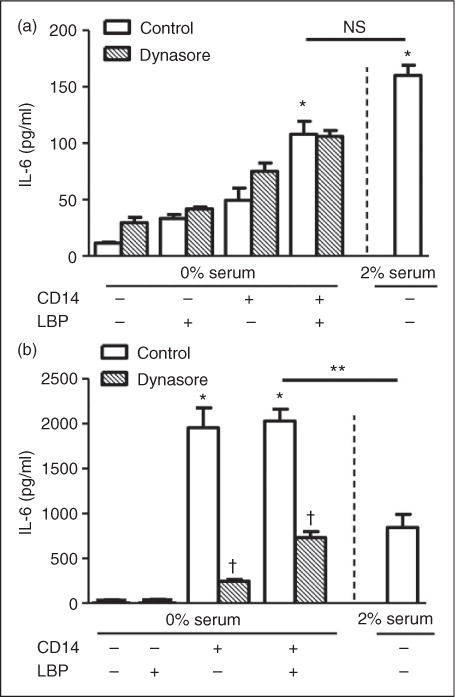
MPLA and LPS enhance IL-6, but with a differential role of endocytosis. (a) Primary HUVECs were incubated under control conditions or in the presence of Dynasore (15 μM) for 30 min in 0% FBS. Then HUVECs were exposed to MPLA (10μg/ml) with or without CD14 (1 μg/ml), LBP (1 μg/ml), or both for 16 h. Additionally, HUVECs were incubated with MPLA in 2% FBS alone for 16 h. (b) Primary HUVECs were incubated with Dynasore for 30 min in 0% FBS. HUVECs were then exposed to LPS (100 ng/ml) with or without CD14, LBP or both for 16 h. Additionally, HUVECs were incubated with LPS in 2% FBS alone. IL-6 was determined from culture supernatant by ELISA. Data are expressed as means ± SE of a single, replicated experiment (n = 4–6). *P < 0.05 between agonist at baseline vs. agonist with supplements. **P <0.05 between agonist with supplements in 0% serum vs. agonist alone in 2% serum. †P < 0.05 between control vs. Dynasore groups. NS, not significant.
These results are contrasted with the effect of LPS on IL-6 production in HUVECs under the same conditions. As shown in Figure 1b, LPS (100 ng/ml) in combination with CD14 greatly enhanced IL-6 production in HUVECs. The addition of LBP only added minimally to the response (n = 4 per group; not significant). Dynasore significantly reduced the amount of IL-6 produced (P<0.001). The cytokine response to LPS in 2% serum conditions was stronger than LPS in 0%, but significantly weaker than LPS in 0% serum with the addition of CD14 or CD14 and LBP. These results show that the weak proinflammatory effects of MPLA are enhanced by CD14 and LBP, as well as serum, and likely occur on the cell surface, as the inhibition of endocytosis did not alter the cytokine response to MPLA.
Expressed endothelial cytokine responses to LPS are reduced by exposure to MPLA
While it is known in other cells of innate immunity, such as macrophages, that MPLA can activate both MyD88- and TRIF-dependent pathways, the majority of the inflammatory effects of MPLA have been postulated to be mediated through TRIF.9,16 To test whether the effects of MPLA on endothelial cells were via similar mechanisms, we exposed HUVECs to LPS in the presence or absence of MPLA. For these studies, we added only CD14 as it appeared sufficient to promote full LPS-induced activation (Figure 1b). We examined cytokine and chemokine production related to MyD88-(IL-6) and TRIF-dependent (IP-10 and RANTES) pathway activation to look at differences in downstream cellular signaling. Similar to our previous findings, under 0% serum conditions, LPS required CD14 to produce the MyD88-dependent cytokine, IL-6 (Figure 2a). However, when MPLA was added to LPS/CD14, the amount of IL-6 produced after 16 h dropped by 75% (P<0.001). Because serum alone enhanced the MPLA-induced inflammatory response, these tests were also carried out in 2% serum conditions. With the addition of serum, LPS induced IL-6 production; however, the addition of CD14 greatly enhanced IL-6 production. Again, under 2% conditions in which MPLA was co-incubated with LPS/CD14, the amount of IL-6 produced by HUVECs compared to LPS/CD14 alone was reduced by 79% (n = 4–8 per group; P < 0.001). Under both 0% and 2% serum conditions, MPLA was a weak activator of IL-6 compared with LPS.
Figure 2.
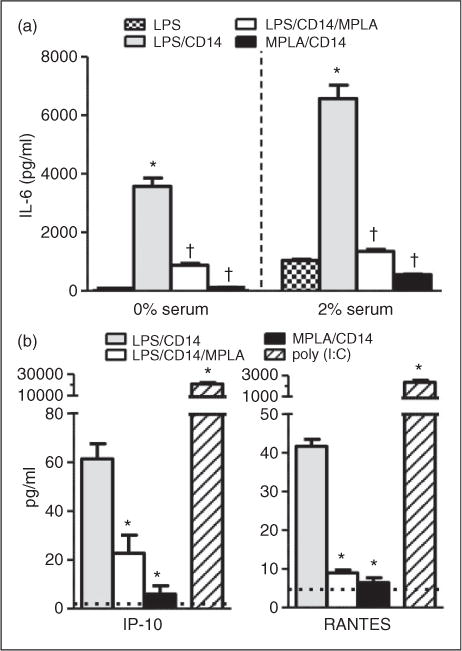
MPLA protects against LPS-mediated MyD88 and TRIF activation. (a) HUVECs were exposed to LPS (100ng/ml) or MPLA (10μg/ml) or both, with or without CD14 (1 μg/ml) for 16 h in 0% and 2% FBS conditions. IL-6 levels were measured in culture supernatant via ELISA. *P<0.05 compared with LPS group alone. †P<0.05 compared with LPS/CD14 group. (b) Primary HUVECs were incubated with LPS or MPLA or both, with CD14 for 16 h in 2% FBS. Poly I:C (10μg/ml) served as a positive control. IP-10 (CXCL10) and RANTES (CCL5) supernatant levels were determined by ELISA. Dotted line (− −) indicates average baseline levels in the absence of agonist. *P < 0.05 compared with LPS/CD14 group. Data are expressed as means ± SE of a single, replicated experiment (n = 4–8).
Next, we sought to determine whether this inhibitory effect of MPLA applied to the other proinflammatory pathway of TLR4 activation, the TRIF pathway. For this purpose, we examined HUVEC culture supernatant for IP-10 (CXCL10) and RANTES (CCL5) after exposure to LPS or MPLA, or a combination of both (Figure 2b). Cells were cultured in 2% serum. Compared with the TLR3 agonist poly (I:C), LPS was a weak activator of the TRIF-dependent chemokines RANTES and IP-10, even in the presence of CD14 and 2% serum. Both responses were endocytosis-dependent as the addition of Dynasore inhibited their production by LPS (data not shown). Despite evidence that MPLA preferentially activates TRIF-dependent pathways in leukocytes, the addition of MPLA to LPS reduced RANTES and IP-10 in HUVECs by 79% and 63%, respectively (n = 4 per group; P < 0.01). In fact, in the presence of 2% serum and CD14, MPLA was unable to induce any significant amount of RANTES or IP-10 from HUVECs. Poly (I:C), which is known to activate the TRIF pathway via TLR3, served as a positive control and potently induced production of IP-10 and RANTES, showing that TRIF-dependent cytokines are capable of being produced by HUVECs. The addition of MPLA to poly (I:C) did not inhibit poly (I:C) induced RANTES or IP-10 production (data not shown). These results demonstrate that MPLA can reduce the proinflammatory response in HUVECs exposed to LPS for both MyD88 and TRIF-dependent pathways.
Given that MPLA inhibited both the MyD88 and TRIF-dependent pathways induced by LPS, we hypothesized that the effect of MPLA was exerted at the level of the TLR4 receptor, proximal to the separation of these distinct signaling pathways. To test this hypothesis, we examined the effect of escalating doses of MPLA on LPS-induced IL-6 and RANTES production in the presence of CD14. As shown in Figure 3a, with increasing concentrations of MPLA from 0.001 μg/ml to 100 μg/ml, the amount of IL-6 and RANTES produced with exposure to LPS was decreased in a dose-dependent manner. By comparing the amount of IL-6 and RANTES produced by LPS and CD14 alone against production in the presence of escalating doses of MPLA, we were able to generate dose-response relationships for MPLA in extinguishing the inflammatory response of LPS (Figure 3b). Though the dose-response curves generated for IL-6 and RANTES secretion had similar slopes and overall appearances, they did have small, albeit significant, differences in the log IC50 values (log IC50 −0.79 for IL-6 vs. −1.22 for RANTES, n = 4 per group; P< 0.001) with an average log IC50 of both pathways being 0.1 μg/ml. In concordance, using fixed concentrations of MPLA (10 μg/ml) and escalating doses of LPS, LPS was able to produce elevated amounts of IL-6 at a dose of 100ng/ml and higher compared with MPLA alone, again suggesting a competitive inhibitory effect of MPLA in LPS-induced cytokine production that is dose dependent (see Supplementary Figure 1).
Figure 3.
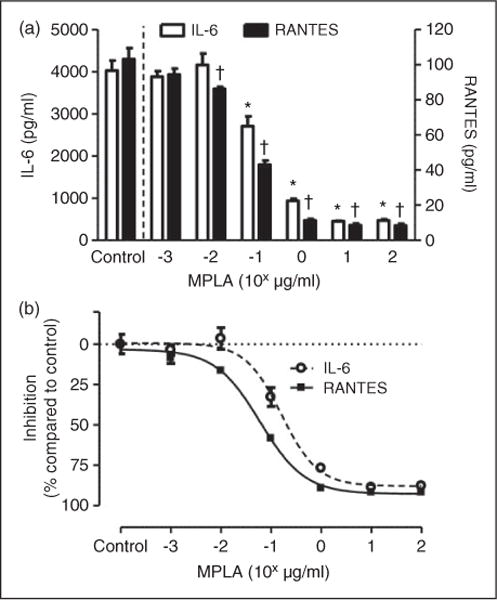
MPLA serves as a competitive antagonist to LPS. (a) Primary HUVECs were exposed to a fixed dose of LPS (100ng/ml) and CD14 (1 μg/ml) with escalating doses of MPLA (range 0.001–100 μg/ml) for 16 h. Control indicates LPS and CD14 in the absence of MPLA. Exposure of LPS, MPLA and CD14 occurred concurrently for escalating MPLA concentrations. Culture supernatant were collected afterwards and measured to levels of IL-6 and RANTES by ELISA. *P < 0.05 compared with control with LPS and CD14 in the absence of MPLA with regard to IL-6. †P < 0.05 compared with controls with regard to RANTES. (b) Estimated dose–inhibition curves for IL-6 and RANTES. Cytokine levels in the absence of MPLA were considered to be baseline at 0% inhibition. Inhibition was expressed as percentage of cytokine levels compared with baseline level among escalating doses of MPLA. Data are expressed as means ± SE of a single, replicated experiment (n = 4 per group).
As MPLA appeared to require a slightly higher concentration to inhibit IL-6 production by LPS compared with RANTES, we wanted to examine if this difference existed between the pathways with MPLA stimulation. Compared with control levels in the absence of MPLA, increasing log doses of MPLA significantly enhanced the amount of IL-6 production at a dose of 0.1 μg/ml up to the top of the range tested (Figure 4a). In contrast, the addition of MPLA had no discernible effect on RANTES production at all doses tested compared to baseline levels. As shown in Figure 4b, when plotted as a dose-response of pathway activation compared with baseline levels, MPLA produced a dose-dependent increase in IL-6 production within the range tested, while the amount of RANTES production compared with baseline remained nearly unchanged (n = 3 per group; P< 0.001). As we were unable to achieve a peak IL-6 with regard to MPLA in the doses tested, a log EC50 was unable to be calculated. These results show that while MPLA can act as an antagonist to LPS-mediated TRIF and MyD88 activation, it can only significantly activate the MyD88-pathway and does not activate the TRIF-pathway by itself.
Figure 4.
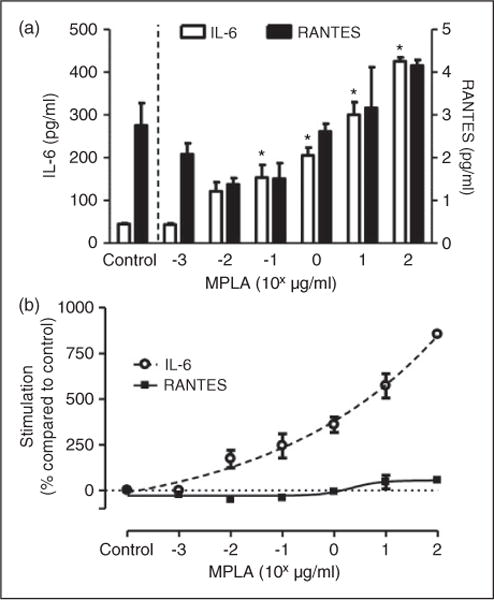
MPLA preferentially activates MyD88. (a) Primary HUVECs were exposed to escalating doses of MPLA (range 0.001–100 μg/ml) for 16 h. Culture supernatant were collected afterwards and measured to levels of IL-6 and RANTES by ELISA. *P < 0.05 compared with controls in the absence of MPLA with regard to IL-6. (b) Estimated dose-stimulation curves for IL-6 and RANTES. Cytokine levels in the absence of MPLA were considered to be baseline at 0% stimulation. Stimulation was expressed as percentage of cytokine levels compared with baseline level among escalating doses of MPLA. Data are expressed as means ± SE of a single, replicated experiment (n = 3 per group).
MPLA alters LPS-induced intracellular signaling and transcription
In endothelial cells, activation of TLR4 by LPS is known to stimulate MAPK signaling pathways, including the ERK, c-Jun N-terminal kinase (c-Jun) and p38 pathways.23 The balance of these MAPK signaling pathways can alter the phenotype of endothelial cells, inducing cytokine activation or promotion of cell death (p38, c-Jun) or proliferation and survival (ERK). To examine whether MAPK pathways were altered by the presence of MPLA during LPS stimulus, we examined phosphorylation of p38, c-Jun and ERK by LPS, MPLA or the combination of the two at 1 h after exposure (Figure 5a). LPS-enhanced phosphorylation of c-Jun by 1.8 fold (P < 0.05). MPLA also increased c-Jun phosphorylation compared with controls, and MPLA with LPS was reduced compared with LPS alone, though neither of these changes were statistically significant. No significant changes in ERK phosphorylation were observed in any of the treatment groups. In contrast to these findings, levels of phosphorylated p38 were significantly enhanced by exposure to LPS compared with baseline conditions by >5 fold (n = 4 per group; P < 0.05). MPLA did not enhance phosphorylation of p38 by itself compared with control, but the addition of MPLA to LPS inhibited LPS-induced phosphorylation of p38 by roughly 50% (n = 4 per group; P < 0.05), suggesting an antagonist effect of MPLA on LPS-induced p38 pathway activation. This reduction in LPS-mediated phosphorylation of p38 by MPLA persisted up to 16 h of agonist exposure (data not shown). To examine if LPS or MPLA exposure modulated phosphorylation of IRF3, a protein activated by TRIF signaling and a promoter of IFN-β transcription,24 we attempted to detect phosphorylated IRF in HUVECs after 1 h of exposure to agonists. As shown in Figure 5b, while there was an abundant presence of IRF3 in HUVECs, exposure to LPS, MPLA or a combination induced no significant phosphorylation of IRF3, suggesting that IFN-β is not produced in response to LPS or MPLA.
Figure 5.
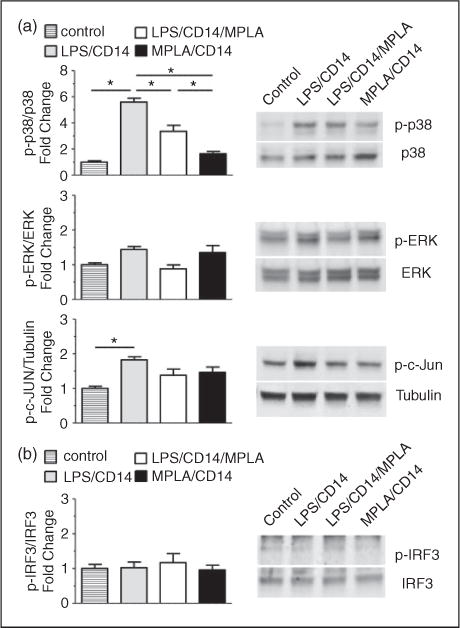
MPLA inhibits p38 phosphorylation induced by LPS. HUVECs were exposed to LPS (100ng/ml) or MPLA (10 μg/ml), both or vehicle control, with CD14 (1 μg/ml) for 1 h. Cell lysates were harvested and analyzed by Western blots. (a) Blots were probed for total and phosphorylated MAPK proteins. (Left) Bar graphs showing the relative abundance of phosphorylated proteins after normalization to total protein expression. (Right) Representative images showing phosphorylated (p-p38, p-ERK, p-c-Jun) and total proteins (p38, ERK, Tubulin). (b) Blots were probed for total and phosphorylated IRF3 protein. (Left) Bar graphs showing the relative abundance of phosphorylated proteins after normalization to total protein expression. (Right) Representative images showing phosphorylated and total IRF3. Data are expressed as means ± SE of a single, replicated experiment (n = 4) after normalization to control quantities.*P < 0.05 between groups as shown with lines between compared groups.
As the p38 pathway is linked to transcription of proinflammatory factors,25 we next wanted to test whether the inhibitory effect of MPLA was exerted via down-regulation of LPS-induced intracellular transcription of proinflammatory cytokines. As shown in Figure 6a, we confirmed that LPS was a poor stimulator of TRIF-dependent pathways in HUVECs as production of IFN-β mRNA was not induced, and this was also true for MPLA stimulation. However, contrary to TRIF cytokine transcription, LPS induced high levels of IL-6 mRNA at 16 h of exposure compared with baseline. When MPLA was used in combination with LPS, IL-6 transcription was significantly reduced by approximately 80% (n = 3–4 per group; P<0.001). MPLA alone did not induce significant transcription of IL-6 compared with controls at 16 h.
Figure 6.
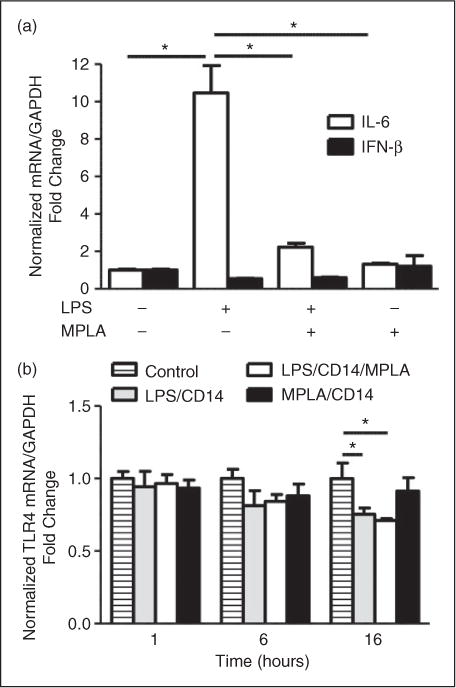
LPS and MPLA have differential effects on nuclear transcription. (a) HUVECs were exposed to LPS (100ng/ml) or MPLA (10 μg/ml), both or vehicle control, with CD14 (1 μg/ml) for 16 h. Cellular RNA was transcribed into cDNA copies and amplified using real-time PCR with primers for IL-6 or IFN-β. Data are expressed as means ± SE of a single, replicated experiment after normalization to GAPDH expression (n = 3–4). *P < 0.05 compared with normalized control. (b) HUVECs were exposed to with LPS (100 ng/ml) or MPLA (10 μg/ml) or both or vehicle control, with CD14 (1 μg/ml) for 1, 6 and 16 h. Cellular RNA was transcribed into cDNA copies and amplified using real-time PCR with primer pairs for TLR4. Bar graphs showing the relative abundance of TLR4 expression at the indicated time points (1, 6 and 16 h). Data are expressed as means ± SE of a single, replicated experiment after normalization to GAPDH expression at each time point (n = 3–4 per group). *P < 0.05 compared with normalized control.
Our data suggest that MPLA inhibited LPS-induced proinflammatory signaling in endothelial cells. To determine if this effect of MPLA was secondary to altered LPS-induced TLR4 expression, we examined the effect of MPLA and LPS on TLR4 transcription. Previous data have pointed to LPS-induced changes in TLR4 expression early after exposure.23,26 For this purpose, we tested multiple time points for relative TLR4 mRNA after LPS and MPLA exposure. As shown in Figure 6b, at 1 h after exposure to MPLA, LPS or both, there was no discernible difference in the amount of TLR4 mRNA compared with unexposed cells. At 6 h, there was a trend toward decreased TLR4 expression greatest in the LPS exposed groups, though the difference was not significant. However, at 16 h of exposure, there was a significant decrease in relative TLR4 mRNA in both the LPS alone and LPS with MPLA exposed groups by 25% and 29%, respectively (n = 3–4 per group; P < 0.05). This decrease was not observed in cells treated with MPLA alone at 16 h. These data show that MPLA down-regulates intracellular proinflammatory signaling in HUVECs exposed to LPS by a mechanism that is independent of alterations in TLR4 expression.
Discussion
The central finding of this study was that MPLA was able to mitigate virtually all of the proinflammatory effects of LPS on HUVECs. MPLA itself induced a mild proinflammatory cytokine response. However, unlike both MyD88 and TRIF-dependent responses to LPS, the response to MPLA was not affected by inhibition of endocytosis. Despite evidence in other immune cell types that MPLA preferentially induces the TRIF pathway, MPLA exposure of HUVECs did not induce TRIF-dependent chemokines and, further, was able to inhibit the production of TRIF-dependent chemokines in response to LPS. The concentration-dependence of the effects of MPLA on MyD88 and TRIF-dependent signals suggested enhanced activation of the MyD88 pathway and enhanced inhibition of the TRIF pathway. Intracellular signals activated by LPS were also reduced by co-exposure with MPLA as reflected by reduced levels of phosphorylated p38 and, in turn, decreased expression of proinflammatory cytokine mRNAs. The ability of MPLA to inhibit LPS-induced cytokine activation in HUVECs was not related to changes in TLR4 mRNA, suggesting the mechanism of MPLA was not secondary to alterations in TLR4 expression. These studies indicate that MPLA has a very proximal effect on TLR4 signaling in endothelial cells, acting prior to endocytosis and affecting both of the critical TLR4-dependent signaling pathways, albeit with preferential activation of only the MyD88 pathway. These data provide new insights into the immunomodulatory role of MPLA on LPS-induced endothelial cytokine activation.
The use of MPLA as a modulator of adaptive immunity has been explored previously, with special emphasis on T-cell modulation and Ab production.27,28 Modulation of the immune response by MPLA has been postulated to be an effect of the preferential activation of TRIF-mediated cellular activation over MyD88-mediated mechanisms.16 This preferential activation of adaptive immunity has generated great interest for the role of MPLA as an immunomodulator and has led to its use as a vaccine adjuvant.29 More recently, the immunomodulatory effects of MPLA have been suggested as a protective mechanism for other inflammatory states, such as bacterial infections.17,30 To examine whether this potential protective effect was conferred to other cells of innate immunity, we studied human endothelial cells. When endothelial cells were exposed to MPLA, weak activation of IL-6, a MyD88-mediated cytokine, occurred that was enhanced by LBP and more so CD14, similar to LPS (Figure 1). However, unlike LPS, cytokine activation by MPLA was not dependent upon endocytosis. Additionally, we found that the enhancement of MyD88-mediated IL-6 production by LPS could be abolished when endothelial cells were co-exposed to MPLA (Figure 2a).
Contrary to MyD88-specific pathways, when examining TRIF-specific pathways in endothelial cells, we found that LPS was a poor activator of TRIF-mediated chemokines RANTES and IP-10 compared with the positive control poly (I:C) (Figure 2b). Interestingly, MPLA did not induce TRIF-mediated chemokine production and, furthermore, had an inhibitory effect on the minimal TRIF activation produced by LPS. The ability of MPLA to affect both MyD88 and TRIF-dependent signaling suggests that the mechanism of action of MPLA on both pathways is exerted at the same site, one that is upstream of the association of these proteins with TLR4. These data are most consistent with MPLA acting as a very weak partial agonist and functionally as an antagonist of LPS-induced TLR4 activation (Figures 3 and 4). While these results differ from a previous report suggesting a preferential activation of TRIF by MPLA,16 it is likely that the mechanisms responsible for MPLA-mediated TLR4 activation differ among cell types. Specifically, the TRIF-predominant response of bone marrow-derived macrophages does not appear to be present in endothelial cells. This can also be seen in models of endotoxin tolerance, a TLR4-specific mechanism, where endothelial cells have been suggested to be non-tolerant to repeat LPS exposures as compared with monocytes and macrophages in which tolerance to repeated LPS challenged has been well described.3,31,32 Furthermore, while LPS and MPLA are known to bind to the same receptor, they have been shown to have differential effects on proinflammatory cellular mechanisms.30 Instead, our data suggest that the inhibitory effect of MPLA with regard to endothelial cells is not via preferential activation of the TRIF pathway, but instead via direct interference with LPS activation of TLR4, likely at the cell surface. While we cannot exclude the possibility that the activity of MPLA is exerted via inhibition of receptor endocytosis, the inability of an endocytosis inhibitor to prevent IL-6 activation by MPLA suggests that MPLA acts at TLR4 independent of receptor endocytosis. One postulated mechanism of the attenuated cytokine activation of MPLA is impairment of TLR4/MD2 dimerization,20 a process that is dependent on LBP and independent of CD14.33 While we did not test this hypothesis directly, we did observe that the inhibitory effects of MPLA were independent of LBP, as MPLA in serum-free conditions, with only the addition of CD14, still conferred an effect of reduced cytokine activation to LPS. Determining whether this inhibitory effect altered further intracellular downstream TLR4 signals was our next objective.
Activation of endothelial cell MAPK and gene transcription by LPS are well described phenomena. MAPK activation mediates the inflammatory phenotype in response to LPS. LPS induces phosphorylation of MAPK pathways in a time- and dose-dependent manner.34 Furthermore, inhibition of p38 and ERK1/2 signaling prevents LPS-induced expression of proinflammatory cytokines, such as IL-6, in endothelial cells.35 We examined whether MAPK activation by LPS would be altered by MPLA exposure. While we saw little-to-no significant changes in c-Jun or ERK phosphorylation following LPS exposure, we found that at both 1 and 16 h of exposure, p38 phosphorylation was enhanced (Figure 5a). Furthermore, MPLA inhibited phosphorylation of this pathway. In contrast, we found no evidence of IRF3 phosphorylation after LPS or MPLA exposure, suggesting that TLR4 does not significantly activate TRIF-dependent mechanisms in endothelial cells. Next, we investigated whether inhibition of the p38 pathway correlated with down-regulation of proinflammatory cytokine transcription. We found that LPS greatly enhanced transcription of IL-6 and that enhanced transcription could be abrogated by MPLA (Figure 5b). Likewise, in accordance with our earlier data, we found no enhancement of transcription of the TRIF-dependent gene IFN β by LPS or MPLA. These experiments provide evidence that proinflammatory changes induced by LPS can be inhibited by MPLA with regard to activation of the p38 pathway and further transcription of proinflammatory cytokines. Given that the inhibitory effect of MPLA appeared to be upstream of gene transcription and phosphorylation of MAPK pathway, we next wanted to investigate whether MPLA had any effects on the TLR4 expression receptor itself.
Endothelial cells have an abundance of TLR4 expression compared with other TLRs.36 Furthermore, it is has been shown that LPS stimulation of endothelial cells induces expression of TLR4 in a time-dependent manner, although the exact timing of peak TLR4 expression is unclear.23,26 Owing to this positive feedback effect of LPS on TLR4 expression, we postulated that the inhibitory effects of MPLA would be related to changes in TLR4 expression. Though at 1 and 6 h, there was no discernable difference among the LPS and MPLA groups in relative TLR4 mRNA, at 16 h, the presence of LPS reduced TLR4 expression, independent of MPLA (Figure 6). Interestingly, MPLA itself did not alter TLR4 expression compared with controls, suggesting that despite competitive inhibition of MPLA with regard to proinflammatory signaling and activation, MPLA was not able to prevent entirely all intracellular changes induced by LPS. The suppression of TLR4 transcription by LPS was an unexpected finding as it differs from previous reports showing that LPS induces TLR4 mRNA production.23,26 One explanation could be related to methodology, as other studies have used different baseline controls, such as 18s RNA or β-actin, to normalize TLR4 expression. Additionally, we compared TLR4 mRNA production to various agonists and normalized the values to control endothelial cells at the same time point. This was done to control for the known time- and concentration-dependent effects serum has on endothelial protein production.37 Our results are more consistent with previous literature that used GAPDH as a control, where no significant increases in LPS-induced TLR4 expression occurred until after multiple doses of LPS were given for greater than 24 h.3 Again, different methodologies prevent direct comparison; however, with regard to our treatment conditions, LPS inhibited TLR4 transcription at 16 h and this was independent of MPLA, as MPLA alone did not alter TLR4 transcription. Thus, inhibition of TLR4 expression did not appear to be the mechanism responsible for the observed effects of MPLA exposure. Future studies are warranted to determine the precise effects of MPLA on TLR4 in endothelial cells.
In conclusion, we demonstrate that MPLA exposure of endothelial cells during LPS challenge confers an inhibitory effect related to reduced proinflammatory responses. MPLA impairs both the MyD88- and TRIF-mediated pathways of TLR4 signaling. Further, the effect of MPLA last up to 16 h after exposure and induces down-regulation of p38-specific MAPKs and cytokine mRNAs. Our data suggest that this potentially protective effect of MPLA occurs very proximally in TLR4 receptor signaling. MPLA appears to be a weak partial agonist of TLR4 that acts primarily as a receptor antagonist. The specific mechanism responsible for the inhibitory properties of MPLA in endothelial cells remains to be elucidated.
Supplementary Material
Acknowledgments
Funding: This work was supported by a grant from the National Institute of General Medical Sciences (Grant R01 GM104306, E. Sherwood).
Footnotes
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
References
- 1.Jaffe EA. Cell biology of endothelial cells. Hum Pathol. 1987;18:234–239. doi: 10.1016/s0046-8177(87)80005-9. [DOI] [PubMed] [Google Scholar]
- 2.Volk T, Kox WJ. Endothelium function in sepsis. Inflamm Res. 2000;49:185–198. doi: 10.1007/s000110050579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang W, Deng M, Liu X, et al. TLR4 activation induces non-tolerant inflammatory response in endothelial cells. Inflammation. 2011;34:509–518. doi: 10.1007/s10753-010-9258-4. [DOI] [PubMed] [Google Scholar]
- 4.Pugin J, Schurer-Maly CC, Leturcq D, et al. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc Natl Acad Sci USA. 1993;90:2744–2748. doi: 10.1073/pnas.90.7.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kagan JC, Su T, Horng T, et al. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schilling D, Thomas K, Nixdorff K, et al. Toll-like receptor 4 and Toll-IL-1 receptor domain-containing adapter protein (TIRAP)/myeloid differentiation protein 88 adapter-like (Mal) contribute to maximal IL-6 expression in macrophages. J Immunol. 2002;169:5874–5880. doi: 10.4049/jimmunol.169.10.5874. [DOI] [PubMed] [Google Scholar]
- 7.Biswas SK, Tergaonkar V. Myeloid differentiation factor 88-independent Toll-like receptor pathway: Sustaining inflammation or promoting tolerance? Int J Biochem Cell Biol. 2007;39:1582–1592. doi: 10.1016/j.biocel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 8.Husebye H, Halaas O, Stenmark H, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zanoni I, Ostuni R, Marek LR, et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caroff M, Karibian D, Cavaillon JM, Haeffner-Cavaillon N. Structural and functional analyses of bacterial lipopolysaccharides. Microbes Infect. 2002;4:915–926. doi: 10.1016/s1286-4579(02)01612-x. [DOI] [PubMed] [Google Scholar]
- 11.Zughaier SM, Zimmer SM, Datta A, et al. Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infect Immun. 2005;73:2940–2950. doi: 10.1128/IAI.73.5.2940-2950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biswas SK, Bist P, Dhillon MK, et al. Role for MyD88-independent, TRIF pathway in lipid A/TLR4-induced endotoxin tolerance. J Immunol. 2007;179:4083–4092. doi: 10.4049/jimmunol.179.6.4083. [DOI] [PubMed] [Google Scholar]
- 13.Suffredini AF, Fromm RE, Parker MM, et al. The cardiovascular response of normal humans to the administration of endotoxin. N Engl J Med. 1989;321:280–287. doi: 10.1056/NEJM198908033210503. [DOI] [PubMed] [Google Scholar]
- 14.Ribi E, Cantrell JL, Takayama K, et al. Lipid A and immunotherapy. Rev Infect Dis. 1984;6:567–572. doi: 10.1093/clinids/6.4.567. [DOI] [PubMed] [Google Scholar]
- 15.Cekic C, Casella CR, Sag D, et al. MyD88-dependent SHIP1 regulates proinflammatory signaling pathways in dendritic cells after monophosphoryl lipid A stimulation of TLR4. J Immunol. 2011;186:3858–3865. doi: 10.4049/jimmunol.1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mata-Haro V, Cekic C, Martin M, et al. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 17.Romero CD, Varma TK, Hobbs JB, et al. The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection. Infect Immun. 2011;79:3576–3587. doi: 10.1128/IAI.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bohannon JK, Hernandez A, Enkhbaatar P, et al. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock. 2013;40:451–462. doi: 10.1097/SHK.0000000000000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang Z, Georgel P, Du X, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 20.Tanimura N, Saitoh SI, Ohto U, et al. The attenuated inflammation of MPL is due to the lack of CD14-dependent tight dimerization of the TLR4/MD2 complex at the plasma membrane. Int Immunol. 2014;26:307–314. doi: 10.1093/intimm/dxt071. [DOI] [PubMed] [Google Scholar]
- 21.Choi H, Nguyen HN, Lamb FS. Inhibition of endocytosis exacerbates TNF-alpha-induced endothelial dysfunction via enhanced JNK and p38 activation. Am J Physiol Heart Circ Physiol. 2014;306:H1154–H1163. doi: 10.1152/ajpheart.00885.2013. [DOI] [PubMed] [Google Scholar]
- 22.Puggioni F, Durham SR, Francis JN. Monophosphoryl lipid A (MPL) promotes allergen-induced immune deviation in favour of Th1 responses. Allergy. 2005;60:678–684. doi: 10.1111/j.1398-9995.2005.00762.x. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Zhang MX, Meng X, et al. Atorvastatin suppresses LPS-induced rapid upregulation of Toll-like receptor 4 and its signaling pathway in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;300:H1743–H1752. doi: 10.1152/ajpheart.01335.2008. [DOI] [PubMed] [Google Scholar]
- 24.Han KJ, Su X, Xu LG, et al. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J Biol Chem. 2004;279:15652–15661. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 25.Guan Z, Buckman SY, Pentland AP, et al. Induction of cyclooxygenase-2 by the activated MEKK1 –>SEK1/MKK4 –>p38 mitogen-activated protein kinase pathway. J Biol Chem. 1998;273:12901–12908. doi: 10.1074/jbc.273.21.12901. [DOI] [PubMed] [Google Scholar]
- 26.Sun R, Zhu Z, Su Q, et al. Toll-like receptor 4 is involved in bacterial endotoxin-induced endothelial cell injury and SOC-mediated calcium regulation. Cell Biol Int. 2012;36:475–481. doi: 10.1042/CBI20110535. [DOI] [PubMed] [Google Scholar]
- 27.Johnson AG, Tomai MA. A study of the cellular and molecular mediators of the adjuvant action of a nontoxic monophosphoryl lipid A. Adv Exp Med Biol. 1990;256:567–579. doi: 10.1007/978-1-4757-5140-6_51. [DOI] [PubMed] [Google Scholar]
- 28.Baker PJ, Hiernaux JR, Fauntleroy MB, et al. Ability of monophosphoryl lipid A to augment the Ab response of young mice. Infect Immun. 1988;56:3064–3066. doi: 10.1128/iai.56.12.3064-3066.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson HS, Davies ML, Watts MJ, et al. Enhanced immunogenicity of a recombinant genital warts vaccine adjuvanted with monophosphoryl lipid A. Vaccine. 1998;16:1993–1999. doi: 10.1016/s0264-410x(98)00088-7. [DOI] [PubMed] [Google Scholar]
- 30.Ruchaud-Sparagano MH, Mills R, Scott J, Simpson AJ. MPLA inhibits release of cytotoxic mediators from human neutrophils while preserving efficient bacterial killing. Immunol Cell Biol. 2014;92:799–809. doi: 10.1038/icb.2014.55. [DOI] [PubMed] [Google Scholar]
- 31.del Fresno C, Garcia-Rio F, Gomez-Pina V, et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol. 2009;182:6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- 32.Dobrovolskaia MA, Medvedev AE, Thomas KE, et al. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappa B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 33.Tsukamoto H, Fukudome K, Takao S, et al. Lipopolysaccharide-binding protein-mediated Toll-like receptor 4 dimerization enables rapid signal transduction against lipopolysaccharide stimulation on membrane-associated CD14-expressing cells. Int Immunol. 2010;22:271–280. doi: 10.1093/intimm/dxq005. [DOI] [PubMed] [Google Scholar]
- 34.Schumann RR, Pfeil D, Lamping N, et al. Lipopolysaccharide induces the rapid tyrosine phosphorylation of the mitogen-activated protein kinases erk-1 and p38 in cultured human vascular endothelial cells requiring the presence of soluble CD14. Blood. 1996;87:2805–2814. [PubMed] [Google Scholar]
- 35.Liu HT, He JL, Li WM, et al. Geniposide inhibits interleukin-6 and interleukin-8 production in lipopolysaccharide-induced human umbilical vein endothelial cells by blocking p38 and ERK1/2 signaling pathways. Inflamm Res. 2010;59:451–461. doi: 10.1007/s00011-009-0118-3. [DOI] [PubMed] [Google Scholar]
- 36.Hijiya N, Miyake K, Akashi S, et al. Possible involvement of tolllike receptor 4 in endothelial cell activation of larger vessels in response to lipopolysaccharide. Pathobiology. 2002;70:18–25. doi: 10.1159/000066000. [DOI] [PubMed] [Google Scholar]
- 37.Poimenidi E, Hatziapostolou M, Papadimitriou E. Serum stimulates Pleiotrophin gene expression in an AP-1-dependent manner in human endothelial and glioblastoma cells. Anticancer Res. 2009;29:349–354. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.