

Abstract
Poly(ADP-ribose) polymerases (PARPs) modify target proteins post-translationally with poly(ADP-ribose) (PAR) or mono(ADP-ribose) (MAR) using NAD+ as substrate. The best-studied PARPs generate PAR modifications and include PARP1 and the tankyrase PARP5a, both of which are targets for cancer therapy with inhibitors in either clinical trials or preclinical development. There are 15 additional PARPs, the majority of which modify proteins with MAR, and their biology is less well understood. Recent data identify potentially cancer relevant functions for these PARPs, indicating that we need to understand more about these PARPs in order to target them effectively.
Introduction
The 17 member poly (ADP-ribose) polymerase (PARP) family of proteins, also known as ADP-ribosyltransferase diphtheria-toxin-like proteins (ARTD1-17; referred to here as PARPs according to standard nomenclature for the cancer field) has garnered much attention over the past decade as a target for cancer therapy due to the success of PARP inhibitors in preclinical trials1. The primary function of PARPs is to post-translationally modify target proteins with ADP-ribose using NAD+ as substrate2. The four best-studied family members, PARP1 and PARP5a along with their close functional homologs PARP2 and PARP5b respectively, all generate poly (ADP-ribose) (PAR). However, most PARPs do not generate PAR, and instead attach ADP-ribose as a monomer (MAR) onto target proteins3. Recent data has shown that many of these MAR-generating PARPs might have cancer relevant functions (Table 1). Therefore, understanding the distinction between PAR and MAR synthesis is important as they function by different mechanisms that will likely impact the efficacy of current and novel PARP inhibitors.
Table 1.
Enzymatic activity and cancer relevant functions of PARPs.
| PARP | Other Names | Demonstrated Activity* | Predicted Activity | ADPr Binding Domains | Cancer Related Functions | Cancers to target |
|---|---|---|---|---|---|---|
| 1 | “PARP” ARTD1 | PAR10 | DNA Repair14, ERK/ NF-kB signaling118, Heat shock response119 | HR deficient Elevated ERK/NF-kB signaling | ||
| 2 | ARTD2 | PAR120 | DNA Repair120 | HR deficient | ||
| 3 | ARTD3 | MAR121 | DNA Repair122 | |||
| 4 | vPARP ARTD4 | MAR61 | ||||
| 5a | TNKS1 ARTD5 | PAR123 | Telomere Maintenance124, Wnt Signaling125, Proteasome Regulation126, Stress Granule Assembly17, Cell Division127 | Elevated Wnt Signaling Telomerase Dependent Stress Granule Positive Solid Tumors | ||
| 5b | TNKS2 ARTD6 | PAR128 | Telomere Maintenance128, Wnt Signaling125 | Elevated Wnt Signaling, Telomerase Dependent | ||
| 6 | ARTD17 | MAR3 | Negative Regulator of Proliferation62 | Potential Tumor Suppressive Functions | ||
| 7 | tiPARP ARTD14 | MAR64 | WWE | |||
| 8 | ARTD16 | MAR3 | ||||
| 9 | BAL1 ARTD9 | No automodification activity113 | Inactive3 | Macro (2) | Cell Migration60 | Metastatic Cancers |
| 10 | ARTD10 | MAR3 | Inhibits Myc88 and NF-kB57 signaling Pro-apoptotic91 | Potential Tumor Suppressive Functions | ||
| 11 | ARTD11 | MAR3 | WWE | |||
| 12 | ARTD12 | MAR17 | WWE | Stress Granule Assembly17 | Stress Granule Positive Solid Tumors | |
| 13 | ZAP ZC3HAV1 ARTD13 | No automodification activity3 | Inactive3 | WWE | Stress Granule Assembly17, miRNA-RISC regulation17, 56 | Stress Granule Positive Solid Tumors |
| 14 | BAL2 ARTD8 | MAR3 | Macro (3) WWE | B cell survival102, Cell Migration52, Stress Granule Assembly17 | Hematopoeitic malignancies, Metastatic Cancers | |
| 15 | BAL3 ARTD7 | MAR17 | Macro (1) | Stress Granule Assembly17 | Stress Granule Positive Solid Tumors | |
| 16 | ARTD15 | MAR51, 70 | ER Unfolded Protein Response51 | UPR dependent |
Catalytic activity is based on ability of PARPs to automodify when incubated with NAD+. PAR: poly(ADP)-ribose; MAR: mono(ADP-ribose); HR: Homologous Recombination; ER: endoplasmic reticulum
In this Perspective Opinion article, we describe the key functional differences between MAR and PAR and discuss recently discovered PARP functions that may be cancer-related. The majority of these functions involve MAR-generating PARPs, identifying this class of PARP proteins as potentially important targets for cancer therapy.
MAR versus PAR
Multiple characteristics of the PARP catalytic domain are important in determining whether a PARP generates PAR or MAR modifications. These include the specific amino acid residues that bind to NAD+ and catalyze the transfer reaction as well as structural elements that define the substrate and acceptor binding pockets (Figure 1 and Table 1). PAR-generating PARPs contain an H-Y-E motif in which the histidine and tyrosine are involved in NAD+ binding and coordination whereas the glutamate is required for PAR transfer and elongation activity4. The majority of PARP family members lack this glutamate and instead contain leucine, isoleucine or valine and are predicted or experimentally demonstrated to generate MAR using automodification reactions containing purified PARP and labeled NAD+ (Table 1). In addition, PARPs 9 and 13, which lack the histidine, are predicted to be enzymatically inactive and do not exhibit automodification activity (Table 1)3, 5. Structural characteristics of the substrate and acceptor binding pockets that impact enzymatic activity include the Donor loop (D-loop) which makes contacts with the substrate NAD+ and is thought to act as a “lid” to hold NAD+ within the catalytic pocket6 (Figure 1). Additionally, the acceptor pocket is partly lined by the loop between β sheets 4 and 5, referred to as the acceptor loop (Figure 1). This loop is implicated in the binding of protein substrate for MAR- and PAR-generating PARPs, or an incoming ADP-ribose unit for PAR-generating PARPs7, 8.
Figure 1. Sequence and Structural Elements of the PARP catalytic domain.

The Donor (yellow) and acceptor (orange) loops of PARP1 (3L3M117), which shape the substrate and acceptor binding pockets respectively, are indicated. H-Y-E motif is shown in magenta. A co-crystallized NAD+ analog inhibitor (A927929) is shown in cyan.
Both PAR and MAR act as traditional post-translational modifications that can alter the function of target proteins. However, PAR has a unique chemistry and structure compared to MAR that further influences its biological function (Figure 2). PAR is composed of ADP-ribose residues connected via glycosidic bonds, imparting characteristics of both nucleic acids and polysaccharides. PAR can contain both linear and branched linkages although branched linkages are less frequent (~one per 20–50 linear linkages)9 (Figure 2). When generated in vitro, PAR can be sizeable, containing up to 200 ADP-ribose residues10. In cells, polymers of such size are likely not found constitutively since PAR purified from tissue contains a maximum of 30 residues11. However, they can exist transiently as treatment with DNA damaging agents results in lengths comparable to those produced in vitro12. Thus the chemistry, high negative charge density, and size of PAR results in a molecule that can function as a high density protein binding scaffold13 (Figure 2). Several examples of this function exist. DNA repair proteins are recruited to sites of DNA damage by binding directly to PAR attached to PARP1 and histones14. Similarly, PAR scaffolds have been implicated in regulation of NF-κB signaling15, cajal body function16 and cytoplasmic stress granule assembly17, and they are also present at the mitotic spindle pole18.
Figure 2. Two forms of ADPr modifications.

PARPs synthesize either mono(ADP-ribose) (MAR) or poly(ADP-ribose) (PAR) modifications. Both can alter target protein function through covalent modification. PAR can also function as a scaffold to recruit proteins containing macro, WWE, PBZ and PBM domains. In contrast, MAR is only recognized by macro domains and does not act as a scaffold since it only contains a single binding site for proteins. Therefore, the structural distinction between MAR and PAR has important consequences of the mechanism of function of the modification.
Although both PAR and MAR can bind proteins, the complexity of the recruitment signal, the number of binding proteins, the number of identified binding domains, and the amount of protein that can bind to PAR greatly exceeds that of MAR. There are four known high affinity PAR binding domains – tryptophan-tryptophan-glutamate (WWE)19, poly(ADP-ribose) binding zinc finger (PBZ)20, and ‘macro’21 domains as well as a loosely defined PAR binding motif (PBM)22 (Figure 2; see REFs 13 and 23 for reviews); collectively, these domains are found in over 800 proteins some of which include the PARPs themselves (Table 1)20, 22, 24, 25. In contrast, the only MAR binding domain identified thus far is the macro domain, shown to bind both free21 and protein-attached26, 27 MAR and PAR, although binding affinities for the two types of ADP-ribose modifications varies among different macro domains28. However, although many functions for MAR are now identified29, the biological relevance of protein binding to MAR has not been extensively studied and is therefore unclear.
As a regulatory molecule, MAR is thought to be evolutionarily ancient because viruses such as T4 bacteriophage and certain pathogenic bacteria encode mono ADP-ribosyltransferases (mARTs) that modify host proteins with MAR to mediate pathogenicity30–32. In addition to PARPs, eukaryotes contain multiple MAR-generating enzymes, all of which require NAD+ as substrate. These include two members of the sirtuin family, SIRT433 and SIRT634, and the eukaryotic ADP-ribosyltransferase (ART) family35. In mammals, 3 of the 5 ARTs are active as arginine-ADP-ribosyltransferases35. Although sirtuins can generate MAR intracellularly, the ARTs found in humans likely do not since they are either glycosylphosphatidylinositol-anchored or secreted ecto-enzymes35.
Intracellular MAR modifications in humans have long been identified, although in many cases the specific enzymes responsible for their synthesis are unknown. Known targets of MAR modification include the cytoskeletal proteins actin36, 37 and desmin38, the protein folding chaperone GRP78 (also known as BiP)39–42 and heterotrimeric G-proteins43–47. Each of these proteins is MARylated on arginines instead of the canonical lysine, glutamate or aspartate residues known to be PARylated by PARPs48–50. However, as the amino acid targets of most MARylating PARPs have not been identified, it is possible that among these PARPs are the proteins responsible for arginine modification of the above targets. In fact, several new functions ascribed to MAR-generating PARPs, including actin cytoskeletal regulation, membrane regulation and regulation of the unfolded protein response (UPR) could involve modification of the above proteins51, 52 (Table 1).
New PARP functions relevant to cancer
Recently several new functions have been identified for PARPs that demonstrate their importance in diverse physiological and stress-dependent pathways. (summarized in Figure 3 and reviewed in REFs13, 29, 53–55). The majority of these functions involve MAR-generating PARPs (Table 1), making the development of MAR-generating PARP inhibitors an important priority as current PARP inhibitors primarily target PAR-generating PARPs6. Here we focus on new functions that are cancer relevant, including the regulation of the endoplasmic reticulum (ER) unfolded protein response (UPR)51, the cytoplasmic stress response17, miRNA-mediated post-transcriptional gene regulation17, 56, cancer-related signal transduction pathways57–59 and cell migration52, 60 (Figure 3). Additional functions identified for PARPs 461, 662, 763, 64 and 852 will not be discussed since less is known about their direct relevance to cancer.
Figure 3. Cellular Functions of the PARP Family.
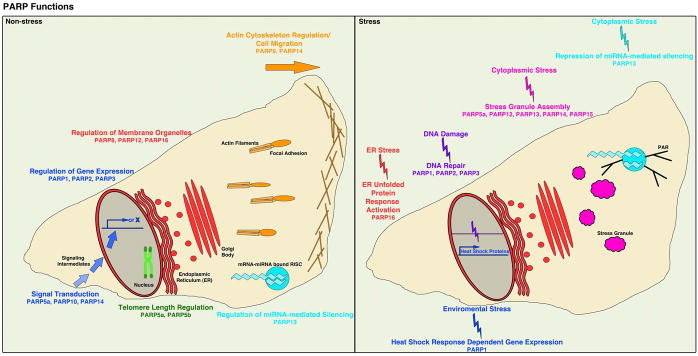
PARPs have multiple diverse in functions in physiological pathways including cell migration, transcriptional regulation, signal transduction, miRNA-mediated gene silencing, regulation of membrane organelles and telomere length regulation. Additionally, PARPs function in stress-responsive cellular pathways upon DNA damage, cytoplasmic stress, environmental stress and ER stress, activating DNA damage repair, stress granule assembly, the heat shock response and the ER unfolded protein response pathways in response. Many of these physiological and stress response pathways are misregulated in cancer, raising the possibility that inhibition of these PARP functions could have therapeutic benefits.
Unfolded Protein Response
The UPR is a cellular adaptation to ER stress triggered by an increase in misfolded proteins within the ER lumen, or by extracellular stressors such as nutrient or oxygen deprivation65. Because of the cytoprotective function of the UPR, its up-regulation is a hallmark of many cancers, due in part to the highly oxidative tumor microenvironment and the high rate of protein synthesis found in cancer cells66. During severe stress or prolonged UPR activation, the UPR activates a distinct transcriptional program to induce UPR-dependent apoptosis65. Activation of this apoptotic program makes the UPR a particularly attractive target for cancer therapies54.
In humans two highly homologous transmembrane kinases – protein kinase RNA-like endoplasmic reticulum kinase (PERK) and inositol-requiring enzyme 1α (IRE1α) - act as ER stress sensors that regulate separate but interconnected signaling networks in the UPR65. A third ER stress sensor, ATF6 also exists but is not regulated by PARP activity. UPR activation by these kinases results in a general decrease in translation and while transcription and translation of stress-specific proteins is increased to restore ER homeostasis65. Both PERK and IRE1α are targets for cancer therapies67–69.
PARP16 is an ER transmembrane protein with a cytoplasmic catalytic domain that exhibits MAR activity51, 70. During UPR activation, PARP16 enzymatic activity is highly upregulated resulting in modification of IRE1α, PERK and PARP16 itself with MAR51. Karyopherin-β1, part of the nuclear trafficking machinery, has also been identified as a target of PARP-1670 suggesting that PARP16 has additional non UPR dependent functions. MARylation of IRE1α and PERK is sufficient to activate these enzymes in vitro and knockdown of PARP16 in HeLa cells results in defective UPR activation with PERK and IRE1α signaling dramatically reduced51. Together these results suggest that PARP16 is critical for activating these enzymes in vertebrates and/or maintaining their “on” state51. In addition, PARP16 is phosphorylated by PERK in vitro51 suggesting that PARP16 activation could be regulated by PERK phosphorylation, similar to PARP1 activation by ERK phosphorylation71, 72 and PARP5a activation by GSK3 phosphorylation73. Thus PERK and PARP16 appear to regulate each other via positive feedback.
Since PARP16 regulates UPR activation and PERK and IRE1α signaling, it is an attractive candidate for the therapeutic inhibition of UPR signaling in cancers54 with or without known UPR activating agents such as HSP9074 and 26S proteasome inhibitors75. Data suggests that this approach is feasible as siRNA-mediated knockdown of PARP16 renders HeLa cells highly sensitive to UPR activation resulting in increased cell death of UPR activated cells relative to non-treated PARP16 knockdowns51. Therefore, the requirement of PARP16 function for UPR activation makes it an attractive candidate to target cancers that upregulate UPR (Table 1).
Cytoplasmic stress response
Cytoplasmic stressors including viral infection, oxidative stress, heat shock, hypoxia, and ER stress result in eIF2α phosphorylation, inhibition of cap dependent mRNA translation, and the assembly of stress granules (SG) — large ribonucleoprotein complexes that contain mRNA, RNA binding proteins and 40S ribosomal subunits76. PAR, 5 different PARPs and poly(ADP-ribose) glycohydrolase (PARG), an enzyme specific for hydrolysis of PAR, are enriched in SGs on stress induction and PAR synthesis and turnover dynamics regulate the kinetics of SG formation and disassembly17. Of the SG-PARPs, only PARP5a has PAR synthesis activity whereas PARPs 12, 14 and 15 generate MAR and PARP13 is inactive, but present3, 17.
Under hypoxic or oxidative stress, SGs inhibit the induction of apoptosis through the JUN N-terminal kinase (JNK)-MAPK signaling pathway via the sequestration of RACK1, a mediator of the MAPK pathway77. Similarly, sequestration of the mTORC1 component Raptor in SGs in the presence of oxidative stress prevents apoptosis induced by mTORC1-hyperactivation78. Astrin, a protein that is upregulated in cancer cells, mediates the localization of Raptor to SGs and oxidative stress induced-apoptosis is increased in cancer cells in which SG assembly is inhibited or expression of Astrin is knocked down using siRNAs78.
Cancer cells in solid tumors are subject to multiple SG-inducing stresses, such as hypoxia and oxidative stress, both of which are associated with chemoresistance79. For example, solid tumors are largely resistant to apoptosis induction mediated by bortezomib, a 26S proteasome inhibitor which is effective in treatment of multiple myelomas and hematological tumors80. Interestingly, bortezomib treatment was shown to induce SG assembly in multiple cancer cells lines and inhibition of SG formation promoted bortezomib-mediated apoptosis, further supporting the protective effects of SGs in cancer cell survival81. Therefore, targeting the PARPs that function in SG assembly could be a strategy to sensitize solid tumors to chemotherapy (Table 1).
microRNA-Ago2 silencing pathway
MicroRNAs are small, non-coding RNAs that regulate gene expression via post-transcriptional mechanisms. MicroRNA function is mediated by the RNA induced silencing complex (RISC), core components of which are Argonaute proteins that bind to the microRNA–mRNA duplex and mediate post-transcriptional silencing82. Argonaute proteins are modified by PAR during normal conditions, but the level of PAR modification increases during stress conditions including viral infection and oxidative stress, resulting in decreased microRNA-dependent silencing activity17, 56. PARP13 seems to be important for regulation of Ago2 function, as PARP13 knockdown upregulates Ago2 silencing activity under stress and non-stress conditions17, 56. Although PARP13 is enzymatically inactive, it contains 4 RNA-binding CCCH Zinc finger domains and binds to Ago2 in an RNA dependent manner suggesting that it either binds to RNA attached to Ago2, or that RNA binding to Ago2 results in a conformational change that mediates PARP13 binding. PARP13 is a target for PAR modification and therefore could also target Ago2 for modification by recruiting other PARPs17.
The role of Ago in cancer progression is complex. Profiling of Ago family members in human colon cancer tissue identified overexpression of Ago2 in cancerous tissue compared with adjacent non-cancer tissue83 and Ago2 was similarly found to be upregulated in human hepatocellular carcinoma, promoting tumor growth and metastasis84. In contrast, other reports show that Ago2 overexpression inhibits tumorigenesis by silencing genes required for proliferation85, 86 and that Ago2 expression is downregulated in melanoma87 and lung adenocarcinoma86. Thus, the role of Ago2 and other argonaute proteins in tumorigenesis may depend on tumor specificity, the tumor microenvironment and the genetic alterations of the cancers in question. Because of the complex role of Ago2 in cancer, further investigation will be required to determine if inhibition of Ago2 ADP-ribosylation will have therapeutic benefits. Perhaps one of the most straight-forward benefits of modulating miRNA activity via PARP inhibition might be to increase the effectiveness of siRNA or miRNA therapies, since PAR modification of Ago2 inhibits its miRNA-mediated silencing activity.
Signal Transduction
PARP10, a MAR-generating PARP3 with RNA and ubiquitin binding domains, has functions in multiple signaling pathways. It was initially identified as a MYC interacting protein that inhibits transformation of rat embryo fibroblasts when coexpressed with MYC and HRAS, but its effect was independent of its ADP-ribosylase activity88. This was the fist indication of a potential tumor suppressive role for PARP10 because MYC, a transcription factor that regulates many cellular processes including cell proliferation, is frequently deregulated in cancer cells and is associated with tumor progression89.
Recently, PARP10 was also shown to regulate NF-κB signaling in a manner dependent both on PARP10 interacting with poly-ubiquitin chains on proteins that regulate NF-κB activity and its ADP-ribosylase activity. Exogenous PARP10 expression in both HeLa and U20S cells resulted in the inhibition of downstream NF-κB target gene expression in response to interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα) by altering the poly-ubiquitylation state of several NF-κB signaling intermediates and preventing the translocation of the NF-κB transcription factor p65-RelA to the nucleus57. Additionally, PARP10 overexpression in HeLa cells inhibited cell proliferation through the induction of apoptosis90, 91, although the contribution of altered NF-κB and/or MYC signaling to apoptosis induction was not investigated. PARP10 function in NF-κB signaling regulation is physiological relevant as shRNA and siRNA mediated knock down of endogenous PARP10 increased expression of NF-κB targets in HeLa and U2OS cell lines57.
PARP10 is enriched in cytoplasmic poly-ubiquitin containing foci that can interact with autophagosomes marked by p62, a ubiquitin-binding autophagy adaptor protein92. Although the role of PARP10 in autophagy has not been investigated, this association is particularly interesting because autophagy is activated in cancer cells and might represent another mechanism to cope with cellular stress93–95. Inhibition of autophagy can sensitize cancer cells to chemotherapy96 or inhibit tumour growth97. The autophagy pathway exerts cytoprotective effects on cancer cells by inhibiting apoptosis and necrosis in response to metabolic stress98, 99. Since NF-κB signaling can regulate autophagy, PARP10 may represent a important link between these two pathways100. Further study of the PARP10 function in NF-κB signaling will be important to understand its role in normal physiology and disease and determine if it does indeed have a function in autophagy regulation.
PARP14 (also known as BAL2 on the basis of its homology to PARP9 (BAL1)), is a MAR-generating PARP that contains three macro domains (Table 1). PARP14 regulates interleukin-4 (IL-4) signaling by acting as a transcriptional co-activator of the transcription factor signal transducer and activator of transcription 6 (STAT6) in a mechanism that is dependent on its catalytic activity59, 101. B cell proliferation and survival is compromised in splenocytes from Parp14−/− mice due to an impaired response to IL-4102. Furthermore, whereas IL-4 exerts an anti-apoptotic effect on ex vivo cultured B cells from wild type mice, the response is attenuated in B cells from Parp14 −/− mice102. Moreover, Parp14−/− mice have delayed MYC-induced B cell lymphomagenesis, highlighting the role of PARP14 in B cell lymphoma development102, 103. Recently, PARP14 has also been implicated in mediating JNK pro-survival signaling in multiple myeloma cells and is highly expressed in these cells compared with normal plasma cells58. Therefore, PARP14 is a new candidate for therapeutic intervention in hematological malignancies due to its functions in B cell development (Table 1).
Cell Migration
PARP9 and PARP14 are members of the macro PARP subfamily, containing 2 and 3 macro domains respectively. The macro domains of PARP9 can bind to both free MAR and PAR whereas PARP14 macro domains specifically bind to MAR-modified proteins21, 27. Both are involved in the regulation of cell migration52, 60, a highly complex process requiring the coordinated activity of multiple proteins that is necessary for the development of metastases104, 105. The discovery of regulatory functions for PARPs 9 and 14 in cell migration suggests that they could be important targets for cancer therapy.
PARP9 was originally identified as BAL1 (B-aggressive lymphoma 1) because it is expressed at higher levels in high-risk diffuse large B-cell lymphomas when compared to low risk tumors60. PARP9 overexpression promotes the migration of B-cell lymphoma cells, suggesting a function in regulation of cell motility60. Consistent with this observation, PARP9 knockdown results in defects in the actin cytoskeleton52. Although PARP9 is constitutively expressed in cells, its expression can be induced by interferon-γ (IFN-γ)106, an immunostimulatory cytokine which has been previously implicated in activating B cell motility107, 108. IFN-γ has tumor suppressive effects by increasing tumor immunogenicity and is used clinically as a cancer treatment, however many studies also report pro-tumorigenic effects of IFN-γ treatment109. This contradiction seems to be a consequence of dosage. Treatment of a low grade bladder cancer cell line with high concentrations of IFN-γ had anti-proliferative effects, whereas low doses of IFN-γ resulted in resistance to TNFα-mediated cytotoxicity and was associated with an increase in cell migration110. Additionally, mammary adenocarcinoma cells expressing low levels of IFN-γ were more metastatic than those expressing high levels when evaluated in BALB/c mice after tail vein injections of cells111. Finally, low surface expression of IFN-γR2, a component of the IFN-γ receptor, on T cells results in a proliferative effect on treatment with IFN-γ, which switches to an apoptotic effect if the levels of IFN-γ R2 are increased through exogenous expression112. One possible model that integrates these disparate findings is that low amounts of IFN-γ induce PARP9 expression, resulting in upregulation of cell motility and metastasis, whereas higher levels overcome the effects of PARP9 expression and result in anti-proliferative and pro-apoptotic effects. Therefore, the expression levels of PARP9 and other IFN-γ dependent genes following low or high doses of IFN-γ treatment should be evaluated.
PARP9 is catalytically inactive based on automodification activity3, 113 and how PARP9 functions in cell migration is unknown. It will be important to determine whether binding to MAR or PAR regulates PARP9 function. If PARP9 is a contributing factor to the pro-metastatic effects of IFN-γ treatment, inhibition of PARP9 function in conjunction with IFN-γ could potentially overcome the tumorigenic effects of low levels of IFN-γ.
PARP14 associates with focal adhesions, identified by both biochemical purification of focal adhesion complexes from human foreskin fibroblast cells114 and by co-immunostaining with focal adhesion proteins in HeLa cells52. PARP14 knockdown results in defects in cell migration in adherent cells in which cells are unable to effectively retract protrusions and have increased adhesiveness to a fibronectin substrate52. These findings indicate that PARP14 regulates focal adhesion turnover52. Although metastasis requires the loss of cell-cell contacts, metastatic cells are able to bind to extracellular matrix components that are not bound by cells in primary tumors115. Fibronectin and integrin interactions have long been implicated in the promotion of tumor cell invasion and metastasis116. Further mechanistic investigation of the function of PARP14 in focal adhesion regulation will be important to determine whether PARP14 is a useful target for the inhibition of metastasis.
Concluding Remarks
Recent analysis examining the binding of 185 known PARP inhibitors to bacterially expressed catalytic domains of 14 of the 17 human PARPs showed that almost none of the inhibitors bind to MAR-generating PARPs and the handful that bind do so with low affinity6. These results suggest that selective inhibition of MAR generating PARPs is possible, and provide an explanation as to why MAR dependent phenotypes do not occur on treatment with current PARP1 and 2, and PARP5a and 5b inhibitors.
Within the past several years, data identifying important cellular functions for MAR generating PARPs have emerged. Many of these functions are disease relevant and could be attractive targets for the therapeutic inhibition of cancer. Much remains unanswered regarding the mechanism of MAR function, the potential functional interactions between MAR and PAR and the potential regulatory interactions between PARPs. Identifying PARP-specific activating signals and targets, and determining the manner in which protein function is altered upon modification will be critical steps for our understanding of MAR. This information will also allow us to better evaluate the therapeutic relevance of MAR inhibition for the treatment of cancer.
Acknowledgments
We thank Frank Solomon, Matthew Vander Heiden, John Pascal and Florian Bock for their helpful comments and discussion about the manuscript. This work is supported by a grant to P.C. from the National Institute of Health (ROI GM087465) and was partially supported by Cancer Center Support (core; grant P30-CA14051).
References
- 1.Rouleau M, Patel A, Hendzel M, Kaufmann S, Poirier G. PARP inhibition: PARP1 and beyond. Nature reviews. Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bürkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+ The FEBS journal. 2005;272:4576–4589. doi: 10.1111/j.1742-4658.2005.04864.x. [DOI] [PubMed] [Google Scholar]
- 3.Kleine H, et al. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Molecular cell. 2008;32:57–69. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Marsischky GT, Wilson BA, Collier RJ. Role of glutamic acid 988 of human poly-ADP-ribose polymerase in polymer formation. Evidence for active site similarities to the ADP-ribosylating toxins. The Journal of biological chemistry. 1995;270:3247–54. doi: 10.1074/jbc.270.7.3247. [DOI] [PubMed] [Google Scholar]
- 5.Otto H, et al. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs) BMC genomics. 2005;6:139. doi: 10.1186/1471-2164-6-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wahlberg E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nature biotechnology. 2012;30:283–288. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 7.Ruf A, Rolli V, de Murcia G, Schulz G. The mechanism of the elongation and branching reaction of poly(ADP-ribose) polymerase as derived from crystal structures and mutagenesis. Journal of molecular biology. 1998;278:57–65. doi: 10.1006/jmbi.1998.1673. [DOI] [PubMed] [Google Scholar]
- 8.Han S, Tainer JA. The ARTT motif and a unified structural understanding of substrate recognition in ADP-ribosylating bacterial toxins and eukaryotic ADP-ribosyltransferases. International journal of medical microbiology : IJMM. 2002;291:523–9. doi: 10.1078/1438-4221-00162. [DOI] [PubMed] [Google Scholar]
- 9.D’Amours D, Desnoyers S, D’Silva I, Poirier G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. The Biochemical journal. 1999;342 ( Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez-Gonzalez R, Jacobson M. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry. 1987;26:3218–3224. doi: 10.1021/bi00385a042. [DOI] [PubMed] [Google Scholar]
- 11.Sakura H, et al. Natural occurence of a biopolymer, poly (adenosine diphosphate ribose) Nucleic acids research. 1977;4:2903–2915. doi: 10.1093/nar/4.8.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Affar E, et al. Immunological determination and size characterization of poly(ADP-ribose) synthesized in vitro and in vivo. Biochimica et biophysica acta. 1999;1428:137–146. doi: 10.1016/s0304-4165(99)00054-9. [DOI] [PubMed] [Google Scholar]
- 13.Gibson B, Kraus W. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature reviews. Molecular cell biology. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 14.Malanga M, Althaus F. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2005;83:354–364. doi: 10.1139/o05-038. [DOI] [PubMed] [Google Scholar]
- 15.Stilmann M, et al. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Molecular cell. 2009;36:365–378. doi: 10.1016/j.molcel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 16.Kotova E, Jarnik M, Tulin A. Poly (ADP-ribose) polymerase 1 is required for protein localization to Cajal body. PLoS genetics. 2009;5 doi: 10.1371/journal.pgen.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung A, et al. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular cell. 2011;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang P, Coughlin M, Mitchison TJ. Interaction between Poly(ADP-ribose) and NuMA contributes to mitotic spindle pole assembly. Molecular biology of the cell. 2009;20:4575–85. doi: 10.1091/mbc.E09-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, et al. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes & development. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahel I, et al. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008;451:81–85. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 21.Karras G, et al. The macro domain is an ADP-ribose binding module. The EMBO journal. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pleschke J, Kleczkowska H, Strohm M, Althaus F. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. The Journal of biological chemistry. 2000;275:40974–40980. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 23.Barkauskaite E, Jankevicius G, Ladurner A, Ahel I, Timinszky G. The recognition and removal of cellular poly(ADP-ribose) signals. The FEBS journal. 2013;280:3491–3507. doi: 10.1111/febs.12358. [DOI] [PubMed] [Google Scholar]
- 24.Schultz J, Milpetz F, Bork P, Ponting C. SMART, a simple modular architecture research tool: identification of signaling domains. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic acids research. 2012;40:5. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dani N, et al. Combining affinity purification by ADP-ribose-binding macro domains with mass spectrometry to define the mammalian ADP-ribosyl proteome. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4243–4248. doi: 10.1073/pnas.0900066106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forst A, et al. Recognition of mono-ADP-ribosylated ARTD10 substrates by ARTD8 macrodomains. Structure (London, England : 1993) 2013;21:462–475. doi: 10.1016/j.str.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Neuvonen M, Ahola T. Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. Journal of molecular biology. 2009;385:212–225. doi: 10.1016/j.jmb.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feijs K, Verheugd P, Lüscher B. Expanding functions of intracellular resident mono-ADP-ribosylation in cell physiology. The FEBS journal. 2013;280:3519–3529. doi: 10.1111/febs.12315. [DOI] [PubMed] [Google Scholar]
- 30.Deng Q, Barbieri J. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annual review of microbiology. 2008;62:271–288. doi: 10.1146/annurev.micro.62.081307.162848. [DOI] [PubMed] [Google Scholar]
- 31.Tiemann B, et al. ModA and ModB, two ADP-ribosyltransferases encoded by bacteriophage T4: catalytic properties and mutation analysis. Journal of bacteriology. 2004;186:7262–7272. doi: 10.1128/JB.186.21.7262-7272.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Depping R, Lohaus C, Meyer H, Rüger W. The mono-ADP-ribosyltransferases Alt and ModB of bacteriophage T4: target proteins identified. Biochemical and biophysical research communications. 2005;335:1217–1223. doi: 10.1016/j.bbrc.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 33.Haigis M, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 34.Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. The Journal of biological chemistry. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 35.Glowacki G, et al. The family of toxin-related ecto-ADP-ribosyltransferases in humans and the mouse. Protein science : a publication of the Protein Society. 2002;11:1657–1670. doi: 10.1110/ps.0200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terashima M, Yamamori C, Shimoyama M. ADP-ribosylation of Arg28 and Arg206 on the actin molecule by chicken arginine-specific ADP-ribosyltransferase. European journal of biochemistry / FEBS. 1995;231:242–249. [PubMed] [Google Scholar]
- 37.Terashima M, et al. ADP-ribosylation of actins by arginine-specific ADP-ribosyltransferase purified from chicken heterophils. European journal of biochemistry / FEBS. 1992;204:305–311. doi: 10.1111/j.1432-1033.1992.tb16638.x. [DOI] [PubMed] [Google Scholar]
- 38.Huang H, Graves D, Robson R, Huiatt T. ADP-ribosylation of the intermediate filament protein desmin and inhibition of desmin assembly in vitro by muscle ADP-ribosyltransferase. Biochemical and biophysical research communications. 1993;197:570–577. doi: 10.1006/bbrc.1993.2517. [DOI] [PubMed] [Google Scholar]
- 39.Gregory HL, Barry EL. ADP-ribosylation of the 78-kDa glucose-regulated protein during nutritional stress. European Journal of Biochemistry. 1989;186 doi: 10.1111/j.1432-1033.1989.tb15196.x. [DOI] [PubMed] [Google Scholar]
- 40.Laitusis A, Brostrom M, Brostrom C. The dynamic role of GRP78/BiP in the coordination of mRNA translation with protein processing. The Journal of biological chemistry. 1999;274:486–493. doi: 10.1074/jbc.274.1.486. [DOI] [PubMed] [Google Scholar]
- 41.Ledford B, Leno G. ADP-ribosylation of the molecular chaperone GRP78/BiP. Molecular and cellular biochemistry. 1994;138:141–148. doi: 10.1007/BF00928456. [DOI] [PubMed] [Google Scholar]
- 42.Chambers J, Petrova K, Tomba G, Vendruscolo M, Ron D. ADP ribosylation adapts an ER chaperone response to short-term fluctuations in unfolded protein load. The Journal of cell biology. 2012;198:371–385. doi: 10.1083/jcb.201202005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lupi R, Corda D, Di Girolamo M. Endogenous ADP-ribosylation of the G protein beta subunit prevents the inhibition of type 1 adenylyl cyclase. The Journal of biological chemistry. 2000;275:9418–9424. doi: 10.1074/jbc.275.13.9418. [DOI] [PubMed] [Google Scholar]
- 44.Lupi R, et al. Endogenous mono-ADP-ribosylation of the free Gbetagamma prevents stimulation of phosphoinositide 3-kinase-gamma and phospholipase C-beta2 and is activated by G-protein-coupled receptors. The Biochemical journal. 2002;367:825–832. doi: 10.1042/BJ20020660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bondarenko VA. Residues within the Polycationic Region of cGMP Phosphodiesterase gamma Subunit Crucial for the Interaction with Transducin alpha Subunit. IDENTIFICATION BY ENDOGENOUS ADP-RIBOSYLATION AND SITE-DIRECTED MUTAGENESIS. Journal of Biological Chemistry. 1997;272 doi: 10.1074/jbc.272.25.15856. [DOI] [PubMed] [Google Scholar]
- 46.Bondarenko V, Yamazaki M, Hayashi F, Yamazaki A. Suppression of GTP/T alpha-dependent activation of cGMP phosphodiesterase by ADP-ribosylation by its gamma subunit in amphibian rod photoreceptor membranes. Biochemistry. 1999;38:7755–7763. doi: 10.1021/bi990106a. [DOI] [PubMed] [Google Scholar]
- 47.Dani N, et al. Mono-ADP-ribosylation of the G protein betagamma dimer is modulated by hormones and inhibited by Arf6. The Journal of biological chemistry. 2011;286:5995–6005. doi: 10.1074/jbc.M110.112466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altmeyer M, Messner S, Hassa P, Fey M, Hottiger M. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic acids research. 2009;37:3723–3738. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Messner S, et al. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic acids research. 2010;38:6350–6362. doi: 10.1093/nar/gkq463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tao Z, Gao P, Liu H-w. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. Journal of the American Chemical Society. 2009;131:14258–14260. doi: 10.1021/ja906135d. [DOI] [PubMed] [Google Scholar]
- 51.Jwa M, Chang P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nature cell biology. 2012;14:1223–30. doi: 10.1038/ncb2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vyas S, Chesarone-Cataldo M, Todorova T, Huang YH, Chang P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nature communications. 2013;4:2240. doi: 10.1038/ncomms3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo X, Kraus W. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes & development. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scarpa E, Fabrizio G, Di Girolamo M. A role of intracellular mono-ADP-ribosylation in cancer biology. The FEBS journal. 2013;280:3551–3562. doi: 10.1111/febs.12290. [DOI] [PubMed] [Google Scholar]
- 55.Dani N, Barbosa AJ, Del Rio A, Di Girolamo M. ADP-ribosylated proteins as old and new drug targets for anticancer therapy: the example of ARF6. Curr Pharm Des. 2013;19:624–33. [PubMed] [Google Scholar]
- 56.Seo G, et al. Reciprocal Inhibition between Intracellular Antiviral Signaling and the RNAi Machinery in Mammalian Cells. Cell host & microbe. 2013 doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verheugd P, et al. Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nature communications. 2013;4:1683. doi: 10.1038/ncomms2672. [DOI] [PubMed] [Google Scholar]
- 58.Barbarulo A, et al. Poly(ADP-ribose) polymerase family member 14 (PARP14) is a novel effector of the JNK2-dependent pro-survival signal in multiple myeloma. Oncogene. 2013;32:4231–4242. doi: 10.1038/onc.2012.448. [DOI] [PubMed] [Google Scholar]
- 59.Goenka S, Boothby M. Selective potentiation of Stat-dependent gene expression by collaborator of Stat6 (CoaSt6), a transcriptional cofactor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4210–4215. doi: 10.1073/pnas.0506981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aguiar R, et al. BAL is a novel risk-related gene in diffuse large B-cell lymphomas that enhances cellular migration. Blood. 2000;96:4328–4334. [PubMed] [Google Scholar]
- 61.Kickhoefer VA, et al. The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase. The Journal of cell biology. 1999;146:917–28. doi: 10.1083/jcb.146.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuncel H, et al. PARP6, a mono(ADP-ribosyl) transferase and a negative regulator of cell proliferation, is involved in colorectal cancer development. Int J Oncol. 2012;41:2079–86. doi: 10.3892/ijo.2012.1652. [DOI] [PubMed] [Google Scholar]
- 63.Ma Q, Baldwin K, Renzelli A, McDaniel A, Dong L. TCDD-inducible poly(ADP-ribose) polymerase: a novel response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochemical and biophysical research communications. 2001;289:499–506. doi: 10.1006/bbrc.2001.5987. [DOI] [PubMed] [Google Scholar]
- 64.MacPherson L, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic acids research. 2013;41:1604–1621. doi: 10.1093/nar/gks1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science (New York, NY ) 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 66.Tsai Y, Weissman A. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes & cancer. 2010;1:764–778. doi: 10.1177/1947601910383011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Atkins C, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer research. 2013;73:1993–2002. doi: 10.1158/0008-5472.CAN-12-3109. [DOI] [PubMed] [Google Scholar]
- 68.Papandreou I, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mimura N, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Di Paola S, Micaroni M, Di Tullio G, Buccione R, Di Girolamo M. PARP16/ARTD15 is a novel endoplasmic-reticulum-associated mono-ADP-ribosyltransferase that interacts with, and modifies karyopherin-β1. PloS one. 2012;7 doi: 10.1371/journal.pone.0037352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kauppinen T, et al. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7136–7141. doi: 10.1073/pnas.0508606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen-Armon M, et al. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Molecular cell. 2007;25:297–308. doi: 10.1016/j.molcel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 73.Yeh TY, Sbodio JI, Chi NW. Mitotic phosphorylation of tankyrase, a PARP that promotes spindle assembly, by GSK3. Biochem Biophys Res Commun. 2006;350:574–9. doi: 10.1016/j.bbrc.2006.09.080. [DOI] [PubMed] [Google Scholar]
- 74.Patterson J, Palombella V, Fritz C, Normant E. IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer chemotherapy and pharmacology. 2008;61:923–932. doi: 10.1007/s00280-007-0546-0. [DOI] [PubMed] [Google Scholar]
- 75.Obeng E, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–50. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 77.Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nature cell biology. 2008;10:1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 78.Kathrin T, et al. Inhibition of mTORC1 by Astrin and Stress Granules Prevents Apoptosis in Cancer Cells. Cell. 2013;154 doi: 10.1016/j.cell.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 79.Wilson W, Hay M. Targeting hypoxia in cancer therapy. Nature reviews. Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 80.Caravita T, de Fabritiis P, Palumbo A, Amadori S, Boccadoro M. Bortezomib: efficacy comparisons in solid tumors and hematologic malignancies. Nature clinical practice Oncology. 2006;3:374–387. doi: 10.1038/ncponc0555. [DOI] [PubMed] [Google Scholar]
- 81.Fournier MJ, Gareau C, Mazroui R. The chemotherapeutic agent bortezomib induces the formation of stress granules. Cancer cell international. 2010;10:12. doi: 10.1186/1475-2867-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin H, Gregory JH. Correction: MicroRNAs: small RNAs with a big role in gene regulation. Nature Reviews Genetics. 2004;5 doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 83.Li L, Yu C, Gao H, Li Y. Argonaute proteins: potential biomarkers for human colon cancer. BMC cancer. 2010;10:38. doi: 10.1186/1471-2407-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cheng N, Li Y, Han ZG. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology (Baltimore, Md) 2013;57:1906–1918. doi: 10.1002/hep.26202. [DOI] [PubMed] [Google Scholar]
- 85.Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nature cell biology. 2012;14:266–275. doi: 10.1038/ncb2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang X, Graves P, Zeng Y. Overexpression of human Argonaute2 inhibits cell and tumor growth. Biochimica et biophysica acta. 2013;1830:2553–2561. doi: 10.1016/j.bbagen.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 87.Völler D, Reinders J, Meister G, Bosserhoff AK. Strong reduction of AGO2 expression in melanoma and cellular consequences. British journal of cancer. 2013 doi: 10.1038/bjc.2013.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu M, et al. PARP-10, a novel Myc-interacting protein with poly(ADP-ribose) polymerase activity, inhibits transformation. Oncogene. 2005;24:1982–1993. doi: 10.1038/sj.onc.1208410. [DOI] [PubMed] [Google Scholar]
- 89.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Seminars in cancer biology. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 90.Chou HYE, Chou H, Lee SC. CDK-dependent activation of poly(ADP-ribose) polymerase member 10 (PARP10) The Journal of biological chemistry. 2006;281:15201–15207. doi: 10.1074/jbc.M506745200. [DOI] [PubMed] [Google Scholar]
- 91.Herzog N, et al. Caspase-dependent cleavage of the mono-ADP-ribosyltransferase ARTD10 interferes with its pro-apoptotic function. The FEBS journal. 2013;280:1330–1343. doi: 10.1111/febs.12124. [DOI] [PubMed] [Google Scholar]
- 92.Kleine H, et al. Dynamic subcellular localization of the mono-ADP-ribosyltransferase ARTD10 and interaction with the ubiquitin receptor p62. Cell communication and signaling : CCS. 2012;10:28. doi: 10.1186/1478-811X-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nature reviews. Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Molecular cancer therapeutics. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sui X, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death and Disease. 2013;4 doi: 10.1038/cddis.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Selvakumaran M, Amaravadi R, Vasilevskaya I, O’Dwyer P. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:2995–3007. doi: 10.1158/1078-0432.CCR-12-1542. [DOI] [PubMed] [Google Scholar]
- 97.Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes & development. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karantza-Wadsworth V, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes & development. 2007;21:1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trocoli A, Djavaheri-Mergny M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. American journal of cancer. 2011 [PMC free article] [PubMed] [Google Scholar]
- 101.Goenka S, Cho S, Boothby M. Collaborator of Stat6 (CoaSt6)-associated poly(ADP-ribose) polymerase activity modulates Stat6-dependent gene transcription. The Journal of biological chemistry. 2007;282:18732–18739. doi: 10.1074/jbc.M611283200. [DOI] [PubMed] [Google Scholar]
- 102.Cho S, et al. PARP-14, a member of the B aggressive lymphoma family, transduces survival signals in primary B cells. Blood. 2009;113:2416–2425. doi: 10.1182/blood-2008-03-144121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cho S, et al. Glycolytic rate and lymphomagenesis depend on PARP14, an ADP ribosyltransferase of the B aggressive lymphoma (BAL) family. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15972–15977. doi: 10.1073/pnas.1017082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nature reviews. Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 105.Roussos E, Condeelis J, Patsialou A. Chemotaxis in cancer. Nature reviews. Cancer. 2011;11:573–587. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Juszczynski P, et al. BAL1 and BBAP are regulated by a gamma interferon-responsive bidirectional promoter and are overexpressed in diffuse large B-cell lymphomas with a prominent inflammatory infiltrate. Molecular and cellular biology. 2006;26:5348–5359. doi: 10.1128/MCB.02351-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilkinson P, Islam L. Recombinant IL-4 and IFN-gamma activate locomotor capacity in human B lymphocytes. Immunology. 1989;67:237–243. [PMC free article] [PubMed] [Google Scholar]
- 108.Clinchy B, Elenstrom C, Moller G. The effect of T cell-derived cytokines on B cell motility in vitro. Cell Immunol. 1993;146:62–70. doi: 10.1006/cimm.1993.1006. [DOI] [PubMed] [Google Scholar]
- 109.Zaidi M, Merlino G. The two faces of interferon-γ in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Champelovier P, et al. Is interferon gamma one key of metastatic potential increase in human bladder carcinoma? Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:4562–4569. [PubMed] [Google Scholar]
- 111.Lollini P, et al. Inhibition of tumor growth and enhancement of metastasis after transfection of the gamma-interferon gene. International journal of cancer. Journal international du cancer. 1993;55:320–329. doi: 10.1002/ijc.2910550224. [DOI] [PubMed] [Google Scholar]
- 112.Bernabei P, et al. Interferon-gamma receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. Journal of leukocyte biology. 2001;70:950–960. [PubMed] [Google Scholar]
- 113.Aguiar R, Takeyama K, He C, Kreinbrink K, Shipp M. B-aggressive lymphoma family proteins have unique domains that modulate transcription and exhibit poly(ADP-ribose) polymerase activity. The Journal of biological chemistry. 2005;280:33756–33765. doi: 10.1074/jbc.M505408200. [DOI] [PubMed] [Google Scholar]
- 114.Kuo JC, Han X, Hsiao CT, Yates J, Waterman C. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nature cell biology. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reticker-Flynn N, et al. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nature communications. 2012;3:1122. doi: 10.1038/ncomms2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Akiyama S, Olden K, Yamada K. Fibronectin and integrins in invasion and metastasis. Cancer metastasis reviews. 1995;14:173–189. doi: 10.1007/BF00690290. [DOI] [PubMed] [Google Scholar]
- 117.Penning T, et al. Optimization of phenyl-substituted benzimidazole carboxamide poly(ADP-ribose) polymerase inhibitors: identification of (S)-2-(2-fluoro-4-(pyrrolidin-2-yl)phenyl)-1H-benzimidazole-4-carboxamide (A-966492), a highly potent and efficacious inhibitor. Journal of medicinal chemistry. 2010;53:3142–3153. doi: 10.1021/jm901775y. [DOI] [PubMed] [Google Scholar]
- 118.Alice NW, Eddy SY. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Frontiers in Oncology. 2013;3 doi: 10.3389/fonc.2013.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Petesch S, Lis J. Activator-induced spread of poly(ADP-ribose) polymerase promotes nucleosome loss at Hsp70. Molecular cell. 2012;45:64–74. doi: 10.1016/j.molcel.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Amé J, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. The Journal of biological chemistry. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 121.Loseva O, et al. PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. The Journal of biological chemistry. 2010;285:8054–8060. doi: 10.1074/jbc.M109.077834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boehler C, et al. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2783–2788. doi: 10.1073/pnas.1016574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Smith S, Giriat I, Schmitt A, de Lange T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science (New York, NY ) 1998;282:1484–1487. doi: 10.1126/science.282.5393.1484. [DOI] [PubMed] [Google Scholar]
- 124.Smith S, de Lange T. Tankyrase promotes telomere elongation in human cells. Current biology : CB. 2000;10:1299–1302. doi: 10.1016/s0960-9822(00)00752-1. [DOI] [PubMed] [Google Scholar]
- 125.Huang SMA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 126.Cho-Park P, Steller H. Proteasome regulation by ADP-ribosylation. Cell. 2013;153:614–627. doi: 10.1016/j.cell.2013.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang P, Coughlin M, Mitchison T. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nature cell biology. 2005;7:1133–1139. doi: 10.1038/ncb1322. [DOI] [PubMed] [Google Scholar]
- 128.Cook B, Dynek J, Chang W, Shostak G, Smith S. Role for the related poly(ADP-Ribose) polymerases tankyrase 1 and 2 at human telomeres. Molecular and cellular biology. 2002;22:332–342. doi: 10.1128/MCB.22.1.332-342.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]