

Abstract
Lectins from different sources have been shown to interfere with HIV infection by binding to the sugars of viral envelope glycoproteins. Three-dimensional atomic structures of a number of HIV-inactivating lectins have been determined, both as free proteins and in glycan-bound forms. However, details on the mechanism of recognition and binding to sugars are elusive. Here we focus on the anti-HIV lectin from Oscillatoria agardhii (OAA): we show that in the absence of sugar in solution, both sugar-free and sugar-bound protein conformations that were observed in the X-ray structures exist as conformational sub-states. Our results suggest that glycan recognition occurs via conformational selection within the ground state, differing from the popular “excited” state model. Our findings provide further insight into molecular recognition of the major receptor on the HIV virus by OAA, details which can potentially be used for optimization and/or development of preventive anti-HIV therapeutics.
Keywords: Anti-HIV lectins, conformational selection, ground state, NMR, protein dynamics
Oscillatoria agardhii agglutinin (OAA) possesses potent and broad-spectrum anti-HIV activity, mediated by binding the high-mannose glycans on the viral protein gp120 and thereby interfering with viral entry.[1–5] In particular, the OAA carbohydrate recognition epitope on a high-mannose glycan was identified as 3α,6α–mannopentaose (the branched core unit of Man-9), which is a unique recognition element to OAA family lectins compared to all other anti-HIV lectins.[2,6–7] The X-ray crystal structures of both free and glycan-bound OAA revealed a β-barrel-like structure with two symmetrically positioned glycan binding sites at opposite ends of the barrel.[3] In the presence of sugar, the protein structure in binding site 2 shows a conformational change, namely the orientation of the peptide bond between W77 and G78 is flipped by ~180°. The equivalent peptide bond in binding site 1 (W10-G11), in contrast, is essentially identical in the absence and in the presence of sugar, i.e. adopts the bound conformation even without any glycan present, possibly due to protein-protein contacts within the crystal.[3] In order to elucidate the general mechanism underlying glycan recognition by OAA, we investigated the conformational properties sampled by the protein in solution using Nuclear Magnetic Resonance spectroscopy (NMR).
Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments[8–9] have become popular tools for studying conformational exchange on the microsecond-millisecond timescale, providing thermodynamic (relative populations), kinetic (rates of exchange) and structural (chemical shift differences) information on sparsely populated and transient conformers[10], termed excited states, that are structurally different from the most populated state, termed ground state. In order to investigate whether the conformers associated with sugar binding are sampled in solution already in the absence of sugar we measured 15N CPMG relaxation dispersion on sugar-free OAA. At 298 K, we found that 22 residues undergo conformational exchange. However, at this temperature, the conformational exchange is fast on the chemical shift scale and therefore chemical shift differences between the ground and the excited states could not be extracted. By lowering the temperature to 277 K, amide resonances of 60 amino acids undergo conformational exchange (Figure 1A, B), with the affected amino acids spread throughout the entire protein and not restricted to the binding sites (as highlighted by the blue spheres in Figure 1A). In contrast, residues associated with significant amide 15N chemical shift changes upon sugar interaction are confined to the binding sites (Figure 1C). More importantly, for 22 amides, exchange is slow on the chemical shift time scale at this temperature (see Methods), and structural information about the exchanging or “excited” state can be extracted in the form of chemical shift differences (Δω) obtained from the analysis of the dispersion profiles (Table S1). These can directly be compared to the chemical shift differences extracted from 1H-15N HSQC spectra of sugar-free and sugar-saturated OAA samples. If they were identical, the excited state sampled by the sugar-free protein would correspond to the sugar-bound state. This, however, is not the case for OAA: as illustrated in Figure 1D and Figure S1, the chemical shift differences between the ground and excited states, derived from CPMG relaxation dispersion, differ from those measured in the sugar-free and -bound 1H-15N HSQC spectra. Both the diverse spatial distribution of residues associated with CPMG relaxation dispersion and of resonances undergoing chemical shift changes upon sugar addition, as well as their non-correlation, suggest that the excited state that is sampled by the sugar-free protein in solution does not correspond to the sugar-bound conformation, and may indicate a transient state not related to binding. Of note, the excited state also does not correspond to the unfolded state (Figure S2).
Figure 1. Sugar-free OAA samples a high-energy state that does not resemble the sugar-bound state.

A, Residues that undergo conformational exchange on the μs - ms time scale (in green) are located within and outside (blue) of the sugar binding sites. B, 15N relaxation dispersion curves of M51 (triangles), W90 (circles) and T117 (squares) amide resonances. Solid lines are the best fits to a two-state exchange model on a per residue basis. All relaxation dispersion data was recorded at two fields (800 MHz, empty symbols; 600 MHz, full symbols). Experimental errors were estimated based on duplicate measurements and are within the size of each symbol. C, Residues whose amide resonances are significantly perturbed upon addition of sugar (yellow) are in general confined to the binding sites. D, The 15N chemical shift differences (|Δδ|) between the ground and the excited state sampled by sugar-free OAA as extracted from the CPMG experiment (green circles) are different from those measured directly from spectra of the sugar-free and sugar-bound OAA protein (yellow circles). The white circles represent all residues that do not exhibit any significant chemical shift perturbation upon addition of sugar. Areas associated with sugar binding are shaded yellow.
Conformational fluctuations in solution, potentially relevant for molecular recognition, can occur not only between a ground and an excited state but also within the ground state itself[11] (i.e., on a timescale faster than the one probed by CPMG relaxation dispersion). In order to explore this possibility we first determined the solution structure of free OAA. The NMR ensemble of free OAA was determined based on complete assignment[12] and a large number of experimental constraints summarized in Table S2, amounting an average of ~19 constraints per residue. The structure is well defined as judged by atomic r.m.s.d. values relative to the mean coordinates of 0.7 ± 0.1 Å and 0.33 ± 0.05 Å for all backbone atoms and those in secondary structure elements, respectively. No violations of experimental constraints are present in the final ensemble. The solution structure of OAA exhibits a β-barrel fold made up from 10 anti-parallel β-strands comprising residues 3-9, 18-24, 33-40, 46-53, 58-65, 70-76, 84-91, 100-106, 113-120 and 126-132 (Figure 2A), closely resembling the sugar-free and the sugar-bound X-ray structures, as evidenced by backbone r.m.s.d. values of 0.7 and 0.67 Å, respectively. The two previously identified carbohydrate binding sites[2–3], encompassing the loops between β1–β2, β7–β8 and β9–β10 (site 1) and β2–β3, β4–β5 and β6–β7 (site 2) are highlighted in Figure 2B, C, respectively.
Figure 2. Solution structure of the anti-HIV protein OAA samples the bound conformation.
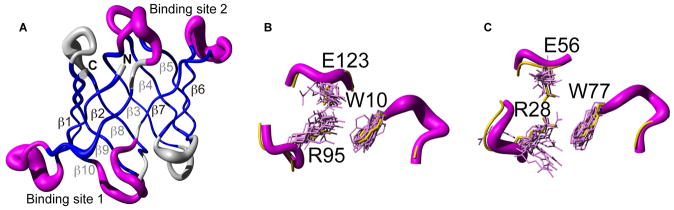
A, The overall fold of the sugar-free solution structure of OAA comprises 10 anti-parallel β-strands (blue) that form a β-barrel, very similar to the sugar-free and sugar-bound X-ray structures (PDB code 3S5V and 3S5X, [3] respectively). The two sugar binding sites (magenta; details of binding site 1 and 2 conformations are shown in B and C, respectively), resemble the sugar-bound conformation of the X-ray structure (yellow). Side chains, directly involved in the carbohydrate binding (W10, R95 and E123 in site 1 and R28, E56 and W77 in site 2) are shown in stick representation. The mean position of the backbone Cα atoms is shown in tube representation, with the radius of the tube corresponding to the average deviation of all conformers with respect to the mean.
Previously we noted that the sugar-free and the sugar-bound X-ray structures exhibited different peptide bond conformations for W77 and G78 in binding site 2, typified by characteristic distances between the backbone amide protons (HN) of these two residues: the very short distance (2.2 Å) in the absence of sugar is increased to 4.4 Å in the bound conformation (Figure 3A). Given the steep distance (d) dependence (1/d6) of the nuclear Overhauser effect (NOE), such distinct difference in distance is uniquely suited to be analyzed by NOE measurements. Indeed, in the NOESY spectrum of OAA saturated with glycan, a very weak cross peak at the noise level is observed between HN W77 - HN G78 (Figure S3), consistent with an interatomic distance larger than 4 Å. In contrast, in the absence of sugar, a sizable NOE cross peak between HN W77 - HN G78 is present (Figure 3B). However, the intensity of the latter is much smaller than expected from the distance in the X-ray structure, compared to the NOE cross peak between HN G78 - HN G79. The experimental peak intensities can be reconciled with the structural data if both the free and the bound conformation are considered to be present with relative populations of ~10% sugar-free and ~90% sugar-bound X-ray conformation at 298 K (Table S3; details as to the estimation of relative populations based on NOE data are provided in the Methods section). At 277 K a precise quantitative analysis is prohibited due to spectral overlap, however, a qualitative evaluation of the NOESY spectrum (Figure 3B) suggests that the equilibrium is likewise skewed towards the sugar-bound X-ray conformation. Interestingly, individual conformers of the solution structure ensemble exhibit backbone conformations resembling the sugar-free and the sugar-bound X-ray structures in both binding sites (Figure 3C).
Figure 3. Backbone amide distances measured in solution by NMR are sensitive measures of a flipped peptide bond.

A, Stick representations of the backbone conformations in the sugar-free and sugar-bound crystal structure (PDB code 3S5V and 3S5X, [3] respectively). A short distance (2.2 Å) between the backbone amide protons (HN) of W77 and G78 in site 2 (dashed red arrows) in the sugar-free conformation is longer (4.4 Å) in the sugar bound conformation. Binding site 1 displays the sugar-bound conformation in the sugar-free crystal structure due to protein-protein contacts within the crystal. B, NOE cross peaks corresponding to the distances depicted in A for different temperatures (298 K, top; 277 K, bottom) between HN W77 and HN G78 (red) and between HN G11/G78 and HN G12/G79 (green). The intensity ratio between these NOE cross peaks indicates that both the sugar-free and the sugar-bound conformations are present. C, The different backbone conformations sampled by OAA in solution (magenta) match the sugar-free (cyan) and sugar-bound (yellow) conformations seen in the X-ray structures in both binding sites.
In addition to NOEs, which report on distances, three-bond J coupling constants (3J) provide complementary structural data, given their dependence on dihedral angles.[13] Of special interest here are 3J(HN,Hα) couplings, which depend on the ϕ dihedral angle, and therefore are exquisitely sensitive to peptide bond flips. For glycines, which possess two Hα protons, two different 3J(HN,Hα) coupling constants can be measured, if non-degenerate Hα resonances exist. These couplings depend on the angle θ[13]:
with θ = ϕ −60° for Hα3 (pro-S) and θ = ϕ + 60° for Hα2 (pro-R), A = 7.13 ± 0.34 Hz, B = −1.31 ± 0.13 and C = 1.56 ± 0.34 Hz.[14] We stereospecifically assigned the Hα glycine resonances of G11 and G78 by comparing both HN-Hα NOE cross peak intensities with the interatomic distances derived from the X-ray structures (Figure S4). As for the HN-HN distances described above, the NOE intensities from sugar-bound OAA are in good agreement with the sugar-complexed X-ray structure, while the NOE intensities in sugar-free OAA are better explained if a mixture of ~10% free, ~90% bound conformation is assumed (Table S3).
For an accurate analysis of 3J couplings between geminal protons, an estimation of the flip rates for the peptide bond is required.[15] Nonetheless, even without this rate, the HN-Hα couplings associated with each ϕ angle can be evaluated qualitatively (Figure S5 and Table S4). Here again, the 3J(HN,Hα) couplings of sugar-bound OAA are in good agreement with the ones predicted based on the sugar-complexed X-ray structure (i.e., the larger 3J(HN,Hα) coupling is associated with Hα2 and the smaller 3J(HN,Hα) is associated with Hα3). In contrast, the experimental 3J(HN,Hα) couplings of G11 and G78 for sugar-free OAA are of opposite size to the ones predicted from the free X-ray structure, suggesting that the conformational equilibrium is skewed towards the X-ray sugar-bound conformation (Table S4).
Our present results show that not only is the sugar-bound conformation sampled in the absence of sugar but that it is thermodynamically more favorable (larger population) than the sugar-free one. In addition, the X-ray data of sugar-free OAA suggests structural flexibility in binding site 2 (Figure S6), and inspection of additional X-ray structures that are available for several OAA homologues, with high conservation of binding site residues and sugar-protein interactions[6–7], reveals that the sugar-bound conformation is frequently seen in sugar-free structures, reinforcing the notion that the bound conformation is more favorable in solution. This agrees with the observation that the motion between the ground and excited state detected by CPMG relaxation dispersion experiments, which occurs on a slower time scale than what is observed in the NOE data, does not coincide with that between free and bound conformations, extracted from chemical shift differences in the two 1H-15N HSQC spectra. Therefore, these motions are likely associated with structural changes within the ground state.
Altogether, our findings show that in solution the sugar-free and the sugar-bound protein conformations co-exist in equilibrium, and that the recognition of 3α,6α-mannopentaose by OAA, which determines its anti-HIV activity, proceeds via conformational selection from a ground state ensemble. This provides detailed insight into the molecular recognition mechanism associated with OAA’s anti-HIV activity. A thorough understanding of this mechanism may serve to guide further development of this lectin as a potent microbicide: the fact that the sugar-bound conformation is highly populated in the absence of sugar suggests that only minimal changes in conformational entropy upon binding are at play from the side of the protein backbone. However, it should also be noted that backbone conformational fluctuations could affect the conformational entropy of the side-chains through a hierarchy of motions via population shuffling mechanism.[16] Efforts for optimizing OAA’s affinity towards 3α,6α-mannopentaose therefore should primarily be focused on enhancing the enthalpic contribution, without disregarding entropic effects, to the binding energetics.
Supplementary Material
Footnotes
This work was supported by the Humboldt Foundation (postdoctoral fellowship to P.T.M.), by the National Institute of Health grant GM080642 (to A. M. G.), by the Max Planck Society and the EU (ERC grant agreement number 233227 to CG).
The solution NMR structure of free OAA has been deposited in the Protein Data Bank (accession code 2mwh). The NMR data used for structural calculation has been deposited in the Biological Magnetic Resonance Data Bank (accession code 25324).
References
- 1.Sato Y, Okuyama S, Hori K. J Biol Chem. 2007;282:11021–11029. doi: 10.1074/jbc.M701252200. [DOI] [PubMed] [Google Scholar]
- 2.Koharudin LMI, Furrey W, Gronenborn AM. J Biol Chem. 2011;286:1588–1597. doi: 10.1074/jbc.M110.173278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koharudin LMI, Gronenborn AM. Structure. 2011;19:1170–1181. doi: 10.1016/j.str.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huskens D, Schols D. Mar Drugs. 2012;10:1476–1497. doi: 10.3390/md10071476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Férir G, Huskens D, Noppen S, Koharudin LMI, Gronenborn AM, Schols D. J Antimicrob Chemother. 2014;69:2746–2758. doi: 10.1093/jac/dku220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koharudin LMI, Kollipara S, Aiken C, Gronenborn AM. J Biol Chem. 2012;287:33796–33811. doi: 10.1074/jbc.M112.388579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitley MJ, Furey W, Kollipara S, Gronenborn AM. FEBS J. 2013;280:2056–2067. doi: 10.1111/febs.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carr HY, Purcell EM. Phys Rev. 1954;94:630–638. [Google Scholar]
- 9.Meiboom S, Gill D. Rev Sci Instrum. 1958;29:688–691. [Google Scholar]
- 10.Palmer AG, Kroenke CD, Loria JP. Methods Enzymol. 2001;339:204–239. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 11.Lange OF, Lakomek NA, Farès C, Schröder GF, Walter KFA, Becker S, Meiler J, Grubmüller H, Griesinger C, de Groot BL. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 12.Carneiro MG, Koharudin LM, Griesinger C, Gronenborn AM, Lee D. Biomol NMR Assign. 2015 doi: 10.1007/s12104-015-9600-8. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karplus M. J Am Chem Soc. 1963;85:2870–2871. [Google Scholar]
- 14.Habeck M, Rieping W, Nilges M. J Magn Reson. 2005;177:160–165. doi: 10.1016/j.jmr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Vuister GW, Bax A. J Am Chem Soc. 1993;115:7772–7777. [Google Scholar]
- 16.Smith CA, Ban D, Pratihar S, Giller K, Schwiegk C, de Groot BL, Becker S, Griesinger C, Lee D. Angew Chem Int Ed. 2015;54:207–210. doi: 10.1002/anie.201408890. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.