

Abstract
Preimplantation mouse embryos of many strains become arrested at the 2-cell stage if the osmolarity of culture medium that normally supports development to blastocysts is raised to approximately that of their normal physiological environment in the oviduct. Arrest can be prevented if molecules that serve as “organic osmolytes” are present in the medium, because organic osmolytes, principally glycine, are accumulated by embryos to provide intracellular osmotic support and regulate cell volume. Medium with an osmolarity of 310 mOsM induced arrest of approximately 80% of CF1 mouse embryos at the 2-cell stage, in contrast to the approximately 100% that progressed beyond the 2-cell stage at 250 or 301 mOsM with glycine. The nature of this arrest induced by physiological levels of osmolarity is unknown. Arrest was reversible by transfer to lower-osmolarity medium at any point during the 2-cell stage, but not after embryos would normally have progressed to the 4-cell stage. Cessation of development likely was not due to apoptosis, as shown by lack of external annexin V binding, detectable cytochrome c release from mitochondria, or nuclear DNA fragmentation. Two-cell embryos cultured at 310 mOsM progressed through the S phase, and zygotic genome activation markers were expressed. However, most embryos failed to initiate the M phase, as evidenced by intact nuclei with decondensed chromosomes, low M-phase promoting factor activity, and an inactive form of CDK1, although a few blastomeres were arrested in metaphase. Thus, embryos become arrested late in the G2 stage of the second embryonic cell cycle when stressed by physiological osmolarity in the absence of organic osmolytes.
Keywords: cell cycle; culture; developmental arrest; embryo culture; osmolarity; preimplantation embryo; rodents (rats, mice, guinea pigs, voles); stress
Mouse embryos cultured at similar osmolarity to that found in the oviduct arrest at the 2-cell stage in the absence of organic osmolytes that control cell volume; arrest occurs after DNA synthesis and zygotic genome activation, but before M-phase, and without apoptosis.
INTRODUCTION
Preimplantation (PI) embryos are extremely sensitive to increased osmolarity near the physiological range. One of the major changes made in PI embryo culture media to eliminate the developmental blocks that had previously prevented complete PI development in vitro was that the initial successful media for mouse embryo culture had substantially lower osmolarities than previous traditional culture media [1–5]. The osmolarities of these media (potassium simplex optimized medium [KSOM] and CZB) were in the range of 250–275 mOsM, which is well below that of the physiological environment of the PI embryo in the oviduct, the fluid of which is in the range of 300–310 mOsM [6, 7]. Indeed, simply raising the osmolarity of KSOM medium to approximately 310 mOsM (with glutamine also omitted) resulted in an almost complete block of development of embryos from sensitive mouse strains at the 2-cell stage, whereas somewhat higher osmolarities similarly blocked development even of mouse embryos from strains that had been considered to be resistant to in vitro developmental blocks [8]. Thus, most fertilized mouse eggs became blocked at the 2-cell stage when the osmolarity of the medium was near or just above physiological levels.
Study of this phenomenon has led to a greater understanding of PI embryo physiology. A key observation was that PI embryos would develop in vitro at higher osmolarities if any of several organic compounds, including glycine, glutamine, or N,N,N-trimethylglycine (betaine), was present in the culture media [5, 9, 10]. Lawitts and Biggers [5] as well as Van Winkle et al. [5, 9] proposed that this was because such compounds functioned in PI embryos as organic osmolytes—small, uncharged organic molecules that are accumulated by cells to provide intracellular osmotic support to replace ions that can disrupt cellular physiology at higher concentrations [11, 12].
Although mammalian somatic cells possess several transporters that mediate the uptake of organic osmolytes when cell volume is decreased, none of these was found to function in the uptake of effective organic osmolytes in early PI embryos [13]. Instead, the earliest stages of mouse PI embryos possess novel cell volume-regulatory mechanisms that utilize organic osmolytes. The major one of these mediates the osmoregulated accumulation of glycine, via the glycine transporter GLYT1 (SLC6A9 protein) that transports glycine with high affinity and can also accept glutamine [14, 15]. Volume regulation using glycine and GLYT1 is activated in oocytes upon ovulation and persists through the 2-cell stage [16]. A secondary volume-regulatory mechanism functions by the accumulation of betaine and is mediated by the SIT1 transporter (SLC6A20 protein), which becomes activated at fertilization and remains active through the 2-cell stage [17, 18]. In vivo-derived mouse eggs and embryos up to the 2-cell stage contain very high levels of intracellular glycine [16] and substantial amounts of betaine [18], implying that organic osmolytes are required for their normal development in the oviduct.
Thus, early PI embryos clearly possess at least two systems, apparently unique to embryos, for accumulating the organic osmolytes that play key roles in regulating embryo cell volume [13]. In the absence of their substrates, or if their transport is blocked, mouse embryos cultured at near-physiological osmolarities do not develop and, instead, become arrested at the 2-cell stage [8]. Whereas the arrest at the 2-cell stage that is induced by increased osmolarity and decreased cell volume resembles the “2-cell block” suffered by most strains of mouse embryos in traditional culture media, the etiology of the 2-cell block also remained elusive [19], although it has been reported that such embryos are arrested at the late 2-cell stage [20, 21]. To begin to elucidate the mechanism of developmental arrest induced by physiological osmolarity in the absence of organic osmolytes employed by the early embryo, we tested two separate hypotheses regarding the arrest of embryo development by physiological osmolarity in the absence of organic osmolytes: first, that the cessation of development at the 2-cell stage involves mechanisms common to apoptotic cell death and, second, that embryos arrest at a specific point in the second mitotic cell cycle.
MATERIALS AND METHODS
Chemicals and Media
All chemicals and components of culture media were obtained from Sigma unless otherwise noted, and those used in culture media were embryo tested or cell-culture grade. The embryo culture media were based on KSOM medium [1] with glutamine omitted and bovine serum albumin replaced with polyvinyl alcohol (1 mg/ml) [10]. This modified KSOM (mKSOM) was equilibrated with 5% CO2 in air at 37°C. Glycine was added directly to the medium when specified. Similarly modified Hepes-KSOM (mHepes-KSOM; 21 mM Hepes replacing equimolar NaHCO3, pH 7.4) was used for oocyte and embryo collection [10]. The osmolarity of mKSOM was 250 mOsM (precision, ±5 mOsM). Osmolarity of the medium was increased to 310 or 340 mOsM by adding appropriate amounts of d-(+)-raffinose, and osmolarity was confirmed using a vapor pressure osmometer (model 5520; Wescor).
Embryo Collection and Culture
Embryos were obtained from female CF1 mice (age, 4–6 wk; Charles River) that had been superovulated by intraperitoneal injection of 5 IU of equine chorionic gonadotropin followed 47.5 h later by 5 IU of human chorionic gonadotropin (hCG). Immediately after hCG, the females were caged overnight with B6D2F1 males (Charles River) for mating. One-cell embryos were removed from excised oviducts approximately 19–22 h post-hCG, and 2-cell embryos at approximately 43–44 h post-hCG, by flushing with mHepes-KSOM. One-cell embryos were freed from residual cumulus matrix by brief exposure to 300 μg/ml of hyaluronidase and fertilization confirmed by the presence of two pronuclei.
One-cell embryos were cultured in groups of 10–15 according to standard techniques in microdrop cultures under mineral oil at 37°C in 5% CO2 in air. One-cell embryos were placed into culture (designated as Day 1) and the proportions reaching the 2-cell stage on Day 2, 4-cell-or-greater stage on Day 3, morula on Day 4, and blastocyst on Days 5 and 6 were recorded. Because development may be slower at higher osmolarities, culture was extended one additional day (to Day 6) beyond what is normally sufficient to allow maximal blastocyst formation. Culture procedures specific to each experiment are detailed in Results. Generally, control cultures were carried out in mKSOM (250 mOsM), and embryos were induced to arrest at the 2-cell stage in mKSOM for which the osmolarity was increased with raffinose to 310 or 340 mOsM. Development at 310 or 340 mOsM was rescued by the presence of 1 mM glycine in the medium when specified.
All procedures were approved by the Animal Care Committee of the Ottawa Hospital Research Institute and conducted in accordance with the guidelines of the Canadian Council on Animal Care.
Annexin V Staining
Zonae pellucidae were removed with acid Tyrode solution and embryos incubated at 37°C for 1 h in the dark with 1 μg/ml of annexin V-fluorescein isothiocyanate (annexin V-FITC; hereafter referred to as annexin V) and 2.5 μg/ml of propidium iodide in mKSOM according to the manufacturer's directions (ApoAlert Annexin V Apoptosis Kit; Clontech). Embryos were washed with mHepes-KSOM and immediately observed using a conventional fluorescence microscope. Embryos were considered to be positive if they were clearly labeled with annexin V on their plasma membranes (excluding punctuate fluorescence from external debris adhering to the membrane) but with nuclei not stained by propidium iodine (lack of cell-impermeant propidium iodine staining confirmed that the plasma membrane was intact). Two-cell embryos cultured in mKSOM with 1 mM H2O2 at 37°C for 1.5 h before annexin V staining served as a positive control, as previously described [22]. Only embryos that remained impermeable (i.e., negative for propidium iodine) after H2O2 treatment were used as examples of annexin V-positive embryos for comparisons.
Immunocytochemistry for Localization of Cytochrome c
Cytochrome c was detected using immunocytochemistry as described previously [22] with minor modifications. All procedures were performed at 37°C unless otherwise noted. Briefly, embryos were incubated in 400 nM MitoTracker (MitoTracker Red CMXRos; Invitrogen/Molecular Probes) for 1 h, washed 3 times in Dulbecco PBS containing 1 mg/ml of polyvinylpyrrolidone (D-PBS/PVP), fixed in 2% formaldehyde in D-PBS/PVP for 30 min, and permeabilized in 0.5% Triton X-100 in D-PBS/PVP for 1 h. After washing, embryos were incubated in blocking solution (D-PBS/PVP containing 5% goat serum) for 2 h and then with a mouse monoclonal anti-cytochrome c antibody (0.5 mg/ml; BD Pharmingen) overnight at 4°C, followed by goat anti-mouse immunoglobulin (Ig) G-FITC secondary antibody (200 μg/ml; Santa Cruz Biotechnology) for 1 h in the dark. Embryos were washed four times in D-PBS/PVP and mounted in SlowFade mounting medium (Invitrogen/Molecular Probes). Images were obtained using laser-scanning confocal microscopy with a 522- to 532-nm band-pass filter and a 600-nm long-pass filter for cytochrome c and MitoTracker, respectively.
Detection of DNA Fragmentation by TUNEL
The TUNEL was performed using the In Situ Cell Death Detection Kit, Fluorescein (Roche Applied Science). All procedures were performed at 37°C unless otherwise noted. Embryos were fixed and permeabilized in 2% formaldehyde containing 0.02% Triton X-100 in D-PBS/PVP for 30 min, washed 3 times for 10 min each in blocking solution (100 mM glycine, 2% fetal bovine serum, 2% bovine serum albumin, and 0.01% Triton X-100 in Tris-buffered saline), and then incubated in blocking solution overnight at 4°C before being washed 3 times with Tris-buffered saline containing 0.01% Triton (TBS-T) and incubated in the TUNEL reaction mixture for 1 h in the dark, according to the manufacturer's instructions. After TUNEL labeling, embryos were treated with 100 μg/ml of RNase A for 1 h in the dark. Nuclei were counterstained with 500 μg/ml of propidium iodine for 1 h in the dark. Embryos were washed four times in TBS-T and mounted in SlowFade mounting medium. Positive-control embryos were treated with 1 U/ml of DNase I for 10 min before TUNEL labeling. Images were obtained using laser-scanning confocal microscopy with 488-nm excitation/522- to 532-nm band-pass emission for detection of FITC and 568-nm excitation/585-nm long-pass emission for propidium iodine.
5-Bromo-2′-Deoxyuridine Incorporation
The DNA replication was assessed by 5-bromo-2′-deoxyuridine (BrdU) incorporated into newly synthesized DNA strands. All procedures were performed at 37°C unless otherwise noted. Embryos were incubated with 1 mM BrdU for the periods specified in Results. Negative-control embryos were incubated with the DNA polymerase α-inhibitor aphidicolin (20 μg/ml) during exposure to BrdU. After incubation, embryos were immediately fixed in 4% paraformaldehyde in D-PBS/PVP for 30 min and permeabilized in 0.5% Triton X-100 in D-PBS/PVP for 1 h. After washing in D-PBS/PVP, DNA was denatured using 2 N HCl for 1 h. Embryos were washed three times in D-PBS/PVP and blocked in D-PBS with 5% goat serum for 2 h. Embryos were then incubated with monoclonal anti-BrdU antibody (3 μg/ml; Sigma B8434) overnight at 4°C, followed by goat anti-mouse IgG-FITC secondary antibody (200 μg/ml; Santa Cruz Biotechnology) for 1 h in the dark. Embryos were washed four times in D-PBS/PVP and mounted in SlowFade mounting medium. Images were obtained by laser-scanning confocal microscopy using a 522- to 532-nm band-pass filter.
Assessment of Zygotic Genome Activation by Quantitative RT-PCR
A total of 30 embryos were pooled for each sample of in vitro- or in vivo-developed embryos and stored at −80°C until RNA extraction. Total RNA was extracted from each set of 30 embryos using the RNeasy Micro Kit (Qiagen) and reverse transcribed into cDNA using RETROscript Kit (Ambion) as described previously [18]. Quantitative RT-PCR (Q-RT-PCR) was performed with a LightCycler thermocycler (Roche Applied Science).
We assessed mRNA expression of two genes for which transcription increases greatly during zygotic genome activation (ZGA) and remains elevated—namely, Eif1a (eukaryotic translation initiation factor 1a) and H47 (histocompatibility 47) [23]—and one gene that is expressed transiently during ZGA—namely, Zscan4d (zinc finger and SCAN domain containing 4D) [24]. Hprt (hypoxanthine guanine phosphoribosyl transferase) served as a reference gene, as previously validated for PI mouse embryos [25].
The PCR primer pairs (Table 1) were designed based on mouse mRNA reference sequences spanning an exon-exon border using OligoPerfect software (Invitrogen). Zscan4d is one of six paralogous genes expressed in the mouse (Zscan4a through Zscan4f) [24], all of which could be amplified by the primer pair used here. However, Zscan4d accounts for almost all expression (>90%) in embryos [24] and, thus, was detected here. To confirm the PCR products, conventional RT-PCR was performed, and the purified amplicons were sequenced by the Ontario Genomics Innovation Centre (Ottawa, Canada).
TABLE 1.
Primer sequences for quantitative PCR.

Each reaction mixture consisted of 0.35 embryo equivalent of cDNA template, 500 nM of each primer, and 1× LightCycler 480 SYBR Green I Master Mix (Roche Applied Science) in a 20-μl reaction volume. A negative control (H2O replacing cDNA template) and a positive control (liver cDNA) were always included in each run. An initial 5 min at 95°C was followed by 50 cycles of 95°C for 10 sec, 60°C for 20 sec, and 72°C for 20 sec. At the end of the run, melting-curve analysis was performed to confirm the PCR product. Two replicates of each sample were simultaneously amplified and the results averaged to constitute one independent replicate.
A threshold cycle (CT) standard curve was constructed using serial 10-fold dilutions of a known concentration of template (prepared by PCR from liver or embryo cDNA). The number of mRNA copies per embryo was determined using the absolute quantification method according to the manufacturer's instructions.
DNA and Microtubule Imaging
To image DNA in arrested 2-cell embryos, the embryos were first fixed in 2% formaldehyde and 0.02% Triton X-100 in D-PBS at 30°C for 30 min. DNA was then labeled with Sytox Green (2 μM; Invitrogen/Molecular Probes) and F-actin labeled with Alexa 594-phalloidin (5 U/ml; Invitrogen/Molecular Probes) by incubation at 4°C overnight, as previously described [26]. F-actin was labeled to allow visualization of the cells.
To image DNA together with tubulin, the embryos were first fixed in 2% formaldehyde, 1 μM paclitaxel, 0.5% Triton X-100, and 10 μg/ml of aprotinin in TBS. Tubulin was labeled with rat anti-α-tubulin monoclonal antibody YL1/2 (20 μg/ml; Millipore) at 4°C overnight, washed, and incubated with Alexa 594 goat anti-rat secondary antibody (10 μg/ml; Invitrogen/Molecular Probes) at 37°C for 2–3 h. DNA was then labeled with Sytox Green as described above. This labeling protocol is essentially as previously described [26].
Embryos were mounted in SlowFade mounting medium. Confocal imaging was performed using 488-nm excitation with 522- to 532-nm band-pass for Sytox Green and 568-nm excitation with 600-nm long-pass for Alexa 594, obtaining a z-series of 0.5-μm optical sections through the portion of the embryo encompassing the DNA.
M-Phase Promoting Factor Assay
Activity of M-phase promoting factor (MPF) was measured using the method developed by Moos et al. [27] for oocytes, modified as previously described [28], except that myelin basic protein (used for simultaneous detection of mitogen-activated protein kinase activity) was omitted here. MPF activity was detected as phosphorylation of histone H1. Briefly, 10 embryos in 1–1.5 μl of medium were added to 3.5 μl of lysis buffer [28] and the kinase reaction initiated by addition of 5 μl of kinase buffer [28] containing [γ32P]ATP (500 μCi/m; PerkinElmer) and histone H1 (2 mg/ml; Sigma). The reaction was stopped after 30 min by addition of 10 μl of Laemmli sample buffer and boiling for 5 min. The extent of histone H1 phosphorylation was determined by SDS-PAGE quantified by PhosphorImager (Typhoon 8600 with ImageQuant software; Molecular Dynamics, Inc.). MPF activity was measured in embryos that were arrested at the 2-cell stage by culture with medium of 310 mOsM at 67 h post-hCG. Negative controls consisted of 2-cell embryos cultured from the 1-cell stage at 250 mOsM and processed for MPF measurements at 43 h post-hCG, when they are in interphase (early G2). For positive controls, culture was continued and the embryos treated with nocodazole (0.2 μg/ml, as described above) from 51 to 67 h post-hCG to arrest them in metaphase, after which they were processed for MPF activity measurements. Because absolute band densities vary between experiments, intensities were normalized to the positive control included in each experiment, which was set arbitrarily to a value of 1.0 [28].
CDK1 Detection
CDK1 was detected by Western blot analysis using a protocol similar to that described by Ohashi et al. [21]. Briefly, proteins in lysates of 25 pooled embryos were separated on a 12% polyacrylamide gel by SDS-PAGE, transferred to a nitrocellulose membrane, blocked for 1 h at room temperature with 5% nonfat dry milk in TBS with 0.1% Tween-20 (TBS-T), and then incubated overnight at 4°C with 1:200 anti-CDK1 antiserum (sc-54; Santa Cruz Biotechnology). After washing 3 times for 10 min each with TBS-T, the membrane was incubated for 1 h with 1:1000 horseradish peroxidase (HRP) goat anti-mouse antiserum (170-6516; Bio-Rad), washed 3 times as above, and bands detected with ECL Plus (GE Healthcare) for 30 sec, according to the manufacturer's instructions. The blots were then stripped and reblocked, reprobed using 1:200 anti-glyceraldehyde phosphate dehydrogenase (GAPDH) antiserum (sc-25778; Santa Cruz Biotechnology) and 1:5000 HRP goat anti-rabbit antiserum (170-6515; Bio-Rad), and the bands detected using ECL Plus for 10 sec. For quantitation, CDK1 bands were normalized to GAPDH in the same lane.
Data Analysis
Data are expressed as the mean ± SEM. Graphs were created using Prism 5.0 (GraphPad Software), which was also used for statistical analysis. Comparisons between means were made by ANOVA followed by the Tukey-Kramer multiple comparisons test (the latter only performed if the ANOVA indicated overall significant difference) for three or more groups, or Student t-test (normal distribution) or Mann-Whitney test (nonnormal distribution) for two groups. Comparisons between proportions were made using Fisher exact test. Significance was considered to be at P < 0.05. In cases when all values in a row of the contingency table were zero, no analysis was performed, because the tests were invalid. Embryo development data are presented as the mean percentage of embryos reaching the stated developmental stage and were arcsine transformed before calculating means for statistical comparisons. Fluorescence confocal images were analyzed using Confocal Assistant software (Version 4.02; freeware by Todd Clarke Brelje) and Image J (Version 1.4d; National Institutes of Health). Within a given experiment, all images used for figures were subjected to identical brightness and contrast adjustments to optimize visualization, and in no case did this change the visibility of cellular structures.
RESULTS
Effect of Increased Osmolarity on Development of Embryos from the 1-Cell Stage in the Presence or Absence of Glycine
It was previously shown that arrest at the 2-cell stage can be induced in CF1 mouse embryos cultured in mKSOM by increasing osmolarity to the normal physiological level of oviductal fluid and that development is rescued by adding glycine [8, 10]. To confirm this for the present experiments, 1-cell embryos were cultured for 6 days at 250 mOsM or at 310 or 340 mOsM in the absence or presence of 1 mM glycine, a concentration previously shown to confer maximal osmoprotection on embryos [10].
Essentially all embryos reached the 2-cell stage in each group (Fig. 1). Nearly all then progressed to the blastocyst stage at 250 mOsM but became arrested at the 2-cell stage at 310 or 340 mOsM, with fewer than 15% and 5%, respectively, reaching the 4-cell stage and correspondingly low development to the morula or blastocyst stages. As expected, the presence of glycine at 310 or 340 mOsM rescued development past the 2-cell stage. For subsequent experiments, we compared only 250 and 310 mOsM, because the latter approximates in vivo osmolarity in the oviduct [6, 7] and is sufficient to arrest most embryos.
FIG. 1.

Effect of osmolarity and the presence of glycine on in vitro development of mouse embryos from the 1-cell stage. One-cell embryos were cultured in media of 250, 310, or 340 mOsM either without glycine (closed symbols) or in the presence of 1 mM glycine (+gly; open symbols) at 310 or 340 mOsM. The mean percentages developing to the 2-cell stage on Day 2 post-hCG (2c), 4-cell-or-greater stage on Day 3 (4c), morula stage on Day 4 (M), and blastocyst stage on Day 5 (B5) and Day 6 (B6) are shown. On Day 3, approximately 26%–28% of embryos had three cells in the 310 and 340 mOsM groups and were not counted in the 4-cell-or-greater total. Statistical comparisons were made between treatments within each stage (i.e., vertically, not between stages) after an arcsine transform of the percentage data. The significance of difference was then assessed for the transformed data by ANOVA followed by Tukey-Kramer multiple comparisons test; significant differences are indicated (points not sharing letters are significantly different; P < 0.01, except for comparisons between 340 mOsM+gly and other groups at the 4-cell, morula, and blastocyst [Day 5] stages, which are P < 0.05; where symbols are not clearly separated, significance is indicated in descending order from the top: 310+gly, 250; 310, and 340). Each point represents the mean percentage development of three complete sets of replicates with 15–17 embryos per treatment in each replicate (45–47 embryos total per group). Error bars indicate the SEM of percentages from the three independent replicates, which is shown as a measure of variability but was not used for statistical comparisons.
Rescue of Development by Transfer to 250 mOsM after Culture at 310 mOsM
Embryos cultured at 310 mOsM from the 1-cell stage apparently develop normally to the 2-cell stage but then arrest as 2-cell embryos and do not progress to the 4-cell stage. We examined how late this arrest could be rescued by transfer to 250 mOsM.
One-cell embryos were cultured from 19 h post-hCG at 310 mOsM and then transferred to 250 mOsM at 43, 48, 53, or 66 h post-hCG. On average, CF1-derived embryos cultured from fertilized eggs in mKSOM medium (250 mOsM) cleave to the 2-cell stage at approximately 35 h post-hCG (range, 34–40 h) and to the 4-cell stage at approximately 60 h post-hCG (range, 58–64 h) (Phillips and Baltz, unpublished results). Thus, the first three time points (43, 48, and 53 h) spanned much of the period when all embryos are at the 2-cell stage, whereas at the last time point (66 h), all normal embryos should have completed the transition to the 4-cell stage. Groups of embryos were also continuously cultured at either 250 or 310 mOsM in parallel with the groups transferred from 310 to 250 mOsM, as controls.
Essentially all embryos developed to the 2-cell stage regardless of treatment (not shown). As in previous experiments, approximately 90% of embryos cultured at 250 mOsM developed to the 4-cell stage and approximately 70%–80% to the blastocyst stage, whereas at 310 mOsM, fewer than 20% developed to the 4-cell stage and very few to the blastocyst stage (Fig. 2). Transferring embryos from 310 to 250 mOsM, however, restored normal proportions of development to the 4-cell stage and beyond when the transfer was done at 43, 48, or 53 h post-hCG (Fig. 2, A–C). In contrast, transfer at 66 h, when the embryos should have reached the 4-cell stage, was completely ineffective (Fig. 2D). Thus, transfer of 2-cell embryos from 310 to 250 mOsM fully rescued development if the transfer was done during the period when embryos are normally at the 2-cell stage, but embryos that had been maintained at 310 mOsM until they should have cleaved to the 4-cell stage could not be rescued.
FIG. 2.

Rescue of 1-cell mouse embryo development past the 2-cell stage by transfer from 310 to 250 mOsM. One-cell mouse embryos were cultured at 250 mOsM (250; black bars), at 310 mOsM (310; open bars), or at 310 mOsM and then transferred to 250 mOsM (310→250; gray bars) at 43 h (A), 48 h (B), 53 h (C), or 66 h (D) post-hCG. For each time of transfer, the top two panels (250 and 310 mOsM without transfer) represent control cultures done as part of the same replicates. The fraction developing to the 4-cell-or-greater stage was assessed on Day 3 post-hCG (4c; indicated at bottom of graphs), morula stage on Day 4 (M), and blastocyst stage on Day 5 (B5) and Day 6 (B6). Statistical comparisons were made between treatments within a given stage (i.e., vertically) after an arcsine transformation of the percentage data. The significance of differences was assessed by ANOVA followed by Tukey-Kramer multiple comparisons test (vertically aligned bars not sharing letters are significantly different, P < 0.05). Each bar represents the mean of five to six replicates consisting of 7–15 embryos in a single culture drop per treatment group, for a total of 81–90 embryos per replicate. Error bars indicate the SEM of percentage data from the replicates, which is shown as a measure of variability but was not used for statistical comparisons.
Assessment of Apoptosis in Embryos Arrested at the 2-Cell Stage in 310 mOsM
Failure to develop beyond the 2-cell stage could be due to the induction of programmed cell death as a consequence of the stress induced by increased osmolarity. To determine if increased osmolarity induced apoptosis, we cultured 1-cell embryos at 250 or 310 mOsM and then assessed for several well-established markers of apoptosis at 48 h post-hCG (mid-2-cell stage) and 72 h post-hCG (normally mid-4-cell stage, and after embryos could no longer be rescued by transfer to 250 mOsM). We used annexin V binding to live embryos to determine whether phosphatidylserine had translocated to the external cell surface as an indicator of early apoptosis, cytochrome c immunolocalization to detect whether cytochrome c was released from mitochondria as a marker of mitochondrial-mediated apoptotic events, and TUNEL labeling to detect DNA fragmentation, a late event of apoptosis [29, 30].
Annexin V binding was assessed by fluorescence microscopy in live embryos cultured from the 1-cell stage to 48 h post-hCG at 250 or 310 mOsM or to 72 h post-hCG at 310 mOsM. Three independent replicates were performed. At 48 h post-hCG, no 2-cell embryos at either 250 or 310 mOsM were positive for annexin V labeling (Table 2 and Fig. 3, A and B). Similarly, no embryos at 310 mOsM that were still arrested at the 2-cell stage at 72 h post-hCG were positive for annexin V (Table 2 and Fig. 3C). Costaining with propidium iodide confirmed plasma membrane integrity in all three culture groups (data not shown). In contrast, extensive annexin V labeling was observed in many in vivo-derived, H2O2-treated (1 mM, 1.5 h, 37°C) embryos that were confirmed to be impermeable by lack of propidium iodide staining (Fig. 3D). These provided examples of embryos with external annexin V binding for comparison.
TABLE 2.
Incidence of markers of apoptosis in 2-cell embryos cultured at 250 or 310 mOsM.
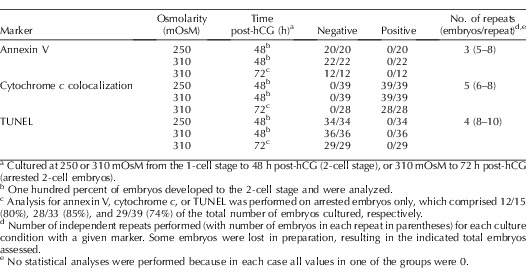
FIG. 3.

Annexin V staining after culture at 250 and 310 mOsM. Representative differential interference contrast images of living embryos are shown (left), as are the fluorescence images from the same embryo to reveal any annexin V binding to phosphatidylserine in the external plasma membrane (right). The images shown here are representative images from three independent replicates of embryos cultured at 250 mOsM assessed at 48 h post-hCG (A), 310 mOsM at 48 h post-hCG (B), or 310 mOsM at 72 h post-hCG (C) (data in Table 2). To obtain positive controls for comparison, embryos were treated with 1 mM H2O2 as indicated in the text. Of those that remained negative for propidium iodide staining and, therefore, had intact, impermeable plasma membranes, 59% (10/17 embryos, from five replicates) were annexin V positive, an example of which is presented (D). Bar = 20 μm.
In contrast to the lack of annexin V binding in embryos, clear annexin V binding was observed in polar bodies of 21% and 27% of embryos 48 h after culture at 250 or 310 mOsM, respectively, and in 50% of polar bodies in embryos arrested at the 2-cell stage after culture for 72 h at 310 mOsM (data not shown).
Cytochrome c localization by immunofluorescence and mitochondrial localization by MitoTracker were assessed with confocal imaging. Five independent replicates were performed with embryos cultured from the 1-cell stage to 48 h post-hCG at 250 or 310 mOsM or to 72 h post-hCG at 310 mOsM. In all 2-cell embryos from each of the three groups, MitoTracker labeling displayed a punctuate pattern, and cytochrome c was colocalized with MitoTracker (Table 2 and Fig. 4, A–C), including 2-cell embryos arrested at the 2-cell stage after culture at 310 mOsM to 72 h post-hCG (Table 2 and Fig. 4C). Thus, loss of mitochondrial localization of cytochrome c in arrested embryos was not observed. In positive-control embryos treated with 1 mM H2O2, which has been shown previously to induce cytochrome c release from mitochondria [22], punctuate cytochrome c staining was not observed (0/16 embryos in four replicates) (Fig. 4D).
FIG. 4.

Distributions of cytochrome c and mitochondria in 2-cell embryos after culture at 250 or 310 mOsM. Embryos with mitochondria labeled with MitoTracker (red) are shown (left), as is cytochrome c immunolocalization (green; middle) and merged images (right), as indicated (top). Treatments in rows A, B, C, and D are indicated (right; as in Fig. 3). Shown are representative examples (confocal slices) of 2-cell embryos assessed at 48 h at each of 250 mOsM (A) and 310 mOsM (B) and arrested 2-cell embryos at 310 mOsM at 72 h (C), as described in the text (data in Table 2). An example of a control embryo treated with H2O2 (D) shows an absence of punctuate cytochrome c staining (faint, nonspecific staining of the zona pellucida is visible). MitoTracker staining is not shown for H2O2-treated embryos, because this marker accumulates in active mitochondria and did not stain mitochondria in H2O2-treated cells. Bar = 20 μm.
Finally, nuclear DNA fragmentation was assessed by TUNEL. Four independent replicates were performed with embryos cultured from the 1-cell stage to 48 h post-hCG at 250 or 310 mOsM or to 72 h post-hCG at 310 mOsM. TUNEL-positive nuclei were not detected in any embryos at either 250 or 310 mOsM at 48 h post-hCG (Table 2 and Fig. 5, A and B), nor in the embryos at 310 mOsM at 72 h post-hCG that remained arrested at the 2-cell stage (Table 2 and Fig. 5C). Two-cell embryos preincubated with DNase I as a positive control displayed the TUNEL reaction in all nuclei (14/14 embryos in three replicates of four to five embryos each) (Fig. 5D). After more prolonged culture, some TUNEL-positive nuclei were first detected in embryos cultured at 250 mOsM at the blastocyst stage, primarily in the inner cell mass (not shown), as previously reported [31], further validating the assay. In contrast to the absence of TUNEL-positive embryonic nuclei, intense TUNEL-positive staining was detected in polar bodies in approximately 30% of 2-cell embryos at 250 and 310 mOsM at 48 h post-hCG and approximately 85% of 2-cell embryos at 310 mOsM at 72 h post-hCG (Fig. 5, A–C).
FIG. 5.

DNA fragmentation assessed by TUNEL labeling in 2-cell embryos after culture at 250 or 310 mOsM. Embryos were labeled with propidium iodide (PI) for DNA (left) and TUNEL labeled for fragmented DNA (middle), with merged images also shown (right), as indicated (top). Treatments in rows A, B, and C are indicated (right; as in Fig. 3). Shown are representative examples (confocal slices) for each treatment as described in the text (data in Table 2). Positive-control embryos (D) were treated with DNase I (1 U/ml at 37°C for 10 min). The small, bright-green structures in A–D are TUNEL-positive DNA in polar bodies. Bar = 20 μm.
Effect of Increased Osmolarity in Culture Media on DNA Replication During the 2-Cell Stage
We next sought to determine the point in the cell cycle at which 2-cell stage embryos cultured at 310 mOsM became arrested. The second mitotic cell cycle (2-cell stage) (Fig. 6) consists of a very short (approximately 1 h) G1 phase followed by DNA synthesis in an S phase that lasts about 6 h and then a very long (>15 h) G2 phase [32]. To determine whether the S phase occurred during the 2-cell stage, we assessed DNA synthesis by BrdU incorporation.
FIG. 6.

BrdU incorporation during the S phase in embryos cultured at 250 or 310 mOsM. BrdU was added to embryos cultured from the 1-cell stage for the intervals indicated in the schematic diagram of embryonic cell-cycle timing (top) as described in the text. Times (h post-hCG) indicated for key events are derived from published data [32], detailed observations of timing in culture under our conditions (Phillips and Baltz, unpublished results), and experiments to determine the timing of the end of the S phase at the 1-cell stage (see text). BrdU was present from 33 to 42 h post-hCG to be incorporated during the second cell cycle, as indicated schematically by the upper box labeled BrdU, with corresponding examples shown (A–D). Examples of the patterns of BrdU immunofluorescence at 42 h post-hCG are shown for 2-cell embryos cultured at 250 mOsM (A), 310 mOsM (B), 310 mOsM in the presence of 1 mM glycine (C), and at 250 mOsM with the DNA synthesis inhibitor aphidicolin (20 μg/ml) present as a negative control (D). BrdU was present from 51 to 67 h post-hCG to be incorporated during the third cell cycle, as indicated schematically by the lower box labeled BrdU, with corresponding examples shown (E–G). Examples of the patterns of BrdU immunofluorescence at 67 h post-hCG are shown for 4-cell embryos cultured at 250 mOsM (E), embryos arrested at the 2-cell stage by culture at 310 mOsM (F), and 4-cell embryos cultured at 310 mOsM in the presence of 1 mM glycine (G). Replicates and number of embryos are as described in the text. Bar = 20 μm.
We introduced BrdU at the end of the 1-cell stage to ensure that DNA synthesis occurring during the 2-cell stage would be detected, given that the S phase begins almost immediately after cleavage to two cells. To determine the appropriate timing of BrdU introduction at the end of the 1-cell stage, we first determined when the S phase during the 1-cell stage ended, to ensure that the BrdU was added after this point and, therefore, would only be incorporated into DNA synthesized during the 2-cell stage. One-cell embryos cultured either in 250 or 310 mOsM were collected after BrdU had been present for 1 h starting at 24, 26, 29, or 32 h post-hCG, and then BrdU incorporation was assessed by immunofluorescence with anti-BrdU antibody. This showed that almost all embryos were in the S phase of the first cell cycle during the first two intervals, approximately 50% during the period from 29 to 30 h, but few (<17%) during the period from 32 to 33 h post-hCG, for 1-cell embryos cultured at either osmolarity (three replicates each; data not shown). The latest time point we could test was 32–33 h, because cleavage occurs on average at 34–35 h post-hCG. Thus, to specifically detect DNA synthesized during the 2-cell stage, we added BrdU to 1-cell embryos in culture at 33 h post-hCG (Fig. 6).
To determine whether DNA replication occurred during the 2-cell stage, embryos were cultured in the continuous presence of BrdU from 33 to 42 h post-hCG, and then BrdU incorporation in the resulting 2-cell embryos was assessed. Four independent replicates were performed, with 9–10 embryos per replicate imaged at each osmolarity. BrdU incorporation was observed at 42 h post-hCG in 100% of 2-cell embryos cultured at either 250 mOsM (40/40 embryos) (Fig. 6A) or 310 mOsM (40/40 embryos) (Fig. 6B). The same pattern was observed in embryos cultured at 310 mOsM in the presence of glycine (39/39 embryos) (Fig. 6C). As a negative control, we showed that embryos cultured at 250 mOsM in the presence of the DNA synthesis inhibitor aphidicolin never exhibited any BrdU incorporation (0/26 embryos in three replicates) (Fig. 6D). Thus, all embryos cultured at either 250 or 310 mOsM had progressed into the S phase by 42 h post-hCG during the 2-cell stage.
To determine if another round of DNA replication occurred in embryos arrested at the 2-cell stage by culture in 310 mOsM at the time when the S phase in the 4-cell stage (third mitotic cell cycle) would normally occur, embryos that had been cultured at either 250 or 310 mOsM from the 1-cell stage were exposed to BrdU beginning at 51 h post-hCG (toward the end of the G2 phase of the 2-cell stage [32]) (Fig. 6). BrdU incorporation was then assessed at 67 h post-hCG (corresponding to the mid-4-cell stage). Three independent replicates were performed at 250 and 310 mOsM, with 7–10 embryos per replicate. At 250 mOsM, 93% (28/30 embryos) developed to the 4-cell stage, and 100% (28/28 embryos) of these had incorporated BrdU at 67 h post-hCG (Fig. 6E) and, thus, had undergone the third round of DNA replication. At 310 mOsM, 96% (25/26 embryos) were arrested at the 2-cell stage, and no BrdU incorporation was found in any of these arrested embryos (0/25) (Fig. 6F). Addition of glycine (1 mM) to 310 mOsM, which rescued development to the 4-cell stage (23/27 embryos), restored the ability of all 4-cell embryos to incorporate BrdU by 67 h post-hCG at 310 mOsM (23/23 embryos) (Fig. 6G). Taken together, these data suggest that increasing osmolarity to 310 mOsM did not inhibit DNA replication in the second cell cycle. In contrast, it did prevent a third round of DNA replication in mouse embryos cultured in 310 mOsM that were arrested at the 2-cell stage.
ZGA in Embryos at 310 mOsM in the Presence or Absence of Glycine
It has been well documented that the major ZGA occurs after the S phase during the 2-cell stage in mouse embryos [33, 34]. We therefore determined whether increased osmolarity prevents ZGA that normally occurs at the 2-cell stage. Eif1a, H47, and Zscan4d genes were chosen as markers of ZGA as described in Materials and Methods, with the first two increasing greatly during ZGA and the last being transiently expressed during ZGA [23, 24].
We first confirmed the expected expression patterns of Eif1a, H47, and Zscan4d genes for in vivo-developed embryos using Q-RT-PCR, with 1-cell embryos (27 h post-hCG) and early (33 h post-hCG), middle (40 h post-hCG), and later (48 h post-hCG) 2-cell embryos. Consistent with previous reports [23, 24], the expression of Eif1a and H47 transcripts was low at the 1-cell stage and increased substantially (∼9-fold and 230-fold, respectively) by the later 2-cell stage (Fig. 7, A and B, in vivo), whereas Zscan4d transcripts were also low in 1-cell embryos but became abundant at the mid-2-cell stage (a 47-fold increase over the 1-cell stage), then decreased again by the late 2-cell stage (Fig. 7C). Expression of the reference gene Hprt did not vary significantly (Fig. 7D).
FIG. 7.

Expression profiles of ZGA markers. Q-RT-PCR was used to measure the mRNA expression of three ZGA markers (Eif1a [A], H47 [B], and Zscan4d [C]) and one reference gene (Hprt [D]) during embryo development in vivo and in vitro from the 1- to 2-cell stage. Gene expression assessed in embryos that developed in vivo (left) and were removed from oviducts at the 1-cell stage at 27 h post-hCG (1c 27 h), the early 2-cell stage at 33 h post-hCG (E2c 33 h), the mid-2-cell stage at 40 h post-hCG (M2c 40 h), and the late 2-cell stage at 48 h post-hCG (L2c 48 h) are shown. Negative controls were run with water only, and no transcripts were detected (no detection for <40 cycles; not shown). Gene expression assessed in embryos cultured in vitro (right) from the 1-cell stage to the later 1-cell stage at 29 h post-hCG (1c 29 h), the mid-2-cell stage at 43 h post-hCG (M2c 43 h), or the late 2-cell stage at 48 h post-hCG (L2c 48 h) are also shown. Embryos were cultured in media of 250, 310, or 310 mOsM with 1 mM glycine added as indicated. Data are presented as the mean ± SEM for number of copies of mRNA expressed per embryo (n = 3 independent replicates). Each replicate consisted of a pool of 30 embryos reverse transcribed together. Two quantitative PCR (Q-PCR) replicates were averaged for each cDNA pool for each replicate. Each Q-PCR reaction contained 0.35 embryo equivalent. Statistical comparisons were made within each of the 16 panels shown, comparing expression levels for each gene between time points within the in vivo group or within each of the three in vitro conditions separately. Bars that share the same letter are not significantly different (ANOVA followed by Tukey-Kramer multiple comparisons test, P < 0.05). Means in panels with no letters were not significantly different; for these, the overall P value is shown for the ANOVA. For Zscan4d, the in vitro data at 250 and 310 mOsM approached significance, with P values as indicated.
Next, we determined the effect of increased osmolarity on Eif1a, H47, and Zscan4d expression in embryos cultured in vitro at 250 or 310 mOsM from the 1-cell stage (starting ∼22 h post-hCG). Somewhat different time points were used for in vitro assessment, because development is slowed in vitro relative to in vivo, with 1-cell embryos collected at 29 h and 2-cell embryos at 43 and 48 h post-hCG. In embryos cultured at 250 mOsM, both Eif1a and H47 expression increased markedly from the 1-cell to the 2-cell stage, reaching approximately the same level of transcripts as in vivo embryos (Fig. 7, A and B, in vitro). Zscan4d also was expressed at levels similar to those found in vivo, but variability appeared to be higher (Fig. 7C). Again, Hprt expression did not vary significantly with stage (Fig. 7D).
When cultured at 310 mOsM, the level at which embryos would become arrested, similar patterns were evident. Expression of both Eif1a and H47 increased significantly during the 2-cell stage, with patterns resembling those at 250 mOsM or in vivo (Fig. 7, A and B), although the levels of transcripts appeared to be somewhat decreased. Zscan4d was also clearly expressed during the 2-cell stage, showing an expression pattern and transcript levels similar to, but apparently more variable than, those seen in vivo (Fig. 7C). When glycine was present at 310 mOsM, expression of all four genes resembled those in vivo (Fig. 7, A–D). Thus, it appears that markers of ZGA increased at the 2-cell stage in cultured embryos not only at 250 mOsM but also at 310 mOsM, indicating that ZGA had occurred.
Nuclear Status of Arrested Embryos
Because embryos arrested at the 2-cell stage in 310 mOsM have passed through the S phase and express genes associated with ZGA, which takes place in the G2 phase, we assessed the nuclear status of arrested embryos to determine whether they became arrested in interphase (G2) or had progressed into the M phase before arresting. Five independent sets of 15 one-cell embryos (75 embryos total) were cultured at 310 mOsM, of which 88% (66/75 embryos) were arrested at the 2-cell stage on Day 3. Confocal imaging of the arrested embryos for which DNA was labeled with Sytox Green (63 embryos total; the remaining three were lost in processing) indicated that 93% of blastomeres (117/126 blastomeres) in the 63 embryos that were arrested at the 2-cell stage on Day 3 had decondensed chromosomes consistent with interphase nuclei, whereas 7% (9/126 embryos) had condensed chromosomes consistent with their having progressed into metaphase (Fig. 8). Thus, the large majority of blastomeres appeared to be in interphase, whereas a few had progressed into metaphase before arresting.
FIG. 8.

Nuclear status of arrested 2-cell embryos. Embryos were cultured at 310 mOsM from the 1-cell stage until Day 3, when they normally would have progressed to the 4-cell stage, and were then stained for DNA with Sytox Green (green) and for F-actin with phalloidin (red). They were then assessed for whether, in each arrested 2-cell embryo, the two blastomeres had condensed chromosomes and no interphase nuclei (“0 nuclei”; A), one blastomere with condensed chromosomes and one with a nucleus (“1 nucleus”; B), or both blastomeres with nuclei (“2 nuclei”; C), as indicated below the graph (D). In total, 63 arrested embryos with 126 blastomeres were assessed. The graph (D) shows the percentage of arrested embryos in each class (numbers in parentheses over the bars indicate the number of embryos in that class out of the total of 63). Error bars indicate the SEM of the percentage data, which is shown as a measure of variability between the five independent replicates (11–15 embryos per replicate). Quantitative analysis by blastomere is given in the text. Bar = 50 μm.
It is possible that embryos arrested at the 2-cell stage on Day 3 that are apparently in interphase had actually progressed into the M phase and their chromosomes had condensed but subsequently decondensed and had nuclei reformed, particularly given that a few arrested blastomeres were found to be in metaphase, as shown above. Thus, we used a protocol to trap cells in the M phase once they had entered. On Day 2 (∼50 h post-hCG, when 2-cell embryos are in the mid-G2 phase), nocodazole (0.2 μg/ml) was added to embryos in culture to disrupt microtubules and stop cell-cycle progression at prometaphase [35]. On Day 3 (∼65 h post-hCG, when embryos would have progressed to the mid-4-cell stage), they were washed free of nocodazole and transferred to identical medium containing the proteosome inhibitor MG132 (2 μM) and incubated for 20 min. For cells trapped in prometaphase, the removal from nocodazole allows the chromosomes to condense, but MG132 prevents cyclin B degradation and traps any cells that had entered the M phase in metaphase. This protocol also allows the quantitative comparison of the fractions of embryos entering metaphase between those cultured at 250 versus 310 mOsM.
The DNA and microtubules were visualized by confocal imaging after labeling for DNA and microtubules. Three independent replicates were performed, each starting with 15 one-cell embryos (45 embryos total at each of 250 and 310 mOsM). At both 250 and 310 mOsM, 100% of embryos (45/45 embryos in each group) were at the 2-cell stage on Day 3 (as expected, because they were prevented from leaving metaphase by the nocodazole/MG132 treatment), with 37 and 38 embryos (11–14 embryos per replicate) imaged, respectively (others were lost during staining).
When embryos had been cultured at 250 mOsM, 92% of blastomeres exhibited condensed chromosomes (Fig. 9, A and B). When cultured at 310 mOsM, however, 77% of blastomeres had decondensed DNA and no evident spindle microtubules, whereas 23% had condensed chromosomes (Fig. 9C). Thus, most embryos arrested by culture at 310 mOsM had not entered metaphase.
FIG. 9.

Arrested 2-cell embryos had not entered the M phase. Any embryos entering the M phase were trapped in prometaphase using nocodazole, and then chromosome condensation was induced by removal of nocodazole in the presence of MG132 to prevent exit from metaphase (see text). A) Labeling the embryos for DNA with Sytox Green (green) and tubulin (red) revealed those that had entered the M phase and exhibited clearly condensed chromosomes and often metaphase spindles (left) contrasted with those that remained in interphase with a clear nucleus (right). B) Nearly all embryos cultured at 250 mOsM and assessed on Day 3 had apparently entered metaphase, whereas most of those cultured in 310 mOsM had not. Numbers in parentheses over the bars indicate the number of embryos in that class out of the total assessed. Error bars indicate the SEM of the percentage data, which is shown as a measure of variability between the three independent replicates. The labels 0, 1, and 2 (bottom) represent the number of blastomeres with condensed chromosomes within the same 2-cell embryo. The statistical analysis of the incidence of interphase nuclei versus condensed chromosomes/spindles after culture at each osmolarity is shown (C). Because a few embryos at each osmolarity had one blastomere with condensed chromosomes and one with a nucleus (4/37 embryos at 250 mOsM and 5/38 embryos at 310 mOsM; i.e., the bars labeled 1 in B), we performed the statistical analysis on the same data as in B, but we compared the proportions of blastomeres in each class (i.e., exhibiting condensed chromosomes vs. a nucleus) out of the total of 74 blastomeres (37 embryos × 2 blastomeres each) at 250 mOsM and 76 blastomeres (38 embryos × 2 blastomeres each) at 310 mOsM. The difference between the proportions at 250 and 310 mOsM was highly significant (Fisher exact test, P < 0.0001). Bar = 50 μm.
MPF in Arrested 2-Cell Embryos
To determine whether embryos arrested at the 2-cell stage by culture at 310 mOsM had activated the MPF that regulates entry into the M phase, MPF activity in embryo lysates was measured. Little MPF activity was present in control interphase 2-cell embryos collected at 43 h post-hCG around the beginning of the G2 phase, whereas high MPF activity was detected in control 2-cell embryos arrested in metaphase by nocodazole and assessed at 67 h post-hCG, as expected (five replicates each). For the embryos cultured at 310 mOsM, we selected only those with a nucleus visible in each blastomere, to avoid assaying embryos from the minority with blastomeres that had progressed into metaphase before arresting (see Fig. 8). The mean MPF activity in embryos cultured at 310 mOsM and remaining arrested with nuclei at the 2-cell stage at 67 h post-hCG was low and not significantly different from that in control interphase 2-cell embryos (five replicates) (Fig. 10A). However, the variability of measured MPF activity was higher in arrested embryos. In interphase (G2) embryos, normalized MPF activities fell in a narrow range, whereas the replicates of embryos with nuclei arrested after culture at 310 mOsM had a wider range of relative MPF activities (variance significantly different from that of G2 embryos by F test, P = 0.02) (Fig. 10A).
FIG. 10.

MPF activity and CDK1 in arrested embryos. A) Histone H1 phosphorylation was measured to detect MPF activity in 2-cell embryos cultured from the 1-cell stage, in the early G2 phase (G2; 43 h post-hCG), in M phase (M; arrested in metaphase with nocodazole at 67 h post-hCG), and in embryos arrested at the 2-cell stage by culture at 310 mOsM (310; selected with nuclei visible in both blastomeres). Ten 2-cell embryos were pooled for each measurement. An example of one of the five replicates is shown above the graph. Bars indicate the mean ± SEM, and circles show the individual measurements for each of the five replicates. Measurements were normalized to the values for M embryos run within the same replicate (arbitrarily set to 1.0). ***G2 and 310 groups were significantly different from M, P < 0.001 by ANOVA followed by Tukey-Kramer multiple comparisons test; G2 and 310 groups did not differ from each other (P > 0.05). B) CDK1 was detected by Western blot analysis with lysates of 25 two-cell embryos per lane. One example of the three replicates is shown (top; lanes: 3T3 indicates NIH 3T3 cell lysate; G2, M, and 310 two-cell embryos are as in A). The CDK1 from 3T3 cells, G2 embryos, and embryos arrested by culture at 310 mOsM all showed bands running above the band for CDK1 in metaphase embryos. The lowest band (in M) had an apparent molecular weight of approximately 30 kDa, whereas those in the other lanes were approximately 31–32 kDa. Bands in each lane were normalized to GAPDH (not shown) and relative intensities plotted. No significant difference was found in total CDK1 between groups (NS).
The kinase activity of MPF is due to CDK1, which is active when complexed with cyclin B1 and dephosphorylated [36]. We examined the status of CDK1 by Western blot analysis for the same three conditions described above (three replicates each). This showed that 2-cell embryos arrested in metaphase by nocodazole had CDK1 that migrated as a single band, consistent with the dephosphorylated, active state; in contrast, 2-cell embryos in the G2 phase and embryos arrested by culture at 310 mOsM exhibited CDK1 that migrated higher, consistent with the inactive, phosphorylated forms (Fig. 10B). The amount of total CDK1 did not differ between interphase 2-cell embryos and those arrested by culture at 310 mOsM.
DISCUSSION
Culture of CF1-strain mouse embryos from the 1-cell zygote stage at physiological levels of osmolarity in the absence of organic osmolytes causes their arrest at the 2-cell stage. This is reversible throughout most of the 2-cell stage, because transfer of the 2-cell embryo to lower-osmolarity medium (250 mOsM) permitted essentially normal development to the 4-cell stage and beyond. Once the embryo normally would have progressed to the 4-cell stage, however, the arrest became irreversible, suggesting that the point at which embryos become arrested is near the end of the 2-cell stage. Curiously, developmental arrest only occurs in embryos cultured at higher osmolarity starting from the 1-cell stage, whereas those cultured starting from the early 2-cell stage develop normally even at very high osmolarities [8]. Why developmental arrest that can be induced only by culture from the 1-cell stage remains reversible through the 2-cell stage is unknown, but this may imply changes in 1-cell embryos that render 2-cell embryos more susceptible to stress.
Consistent with the reversibility of developmental arrest, we found that the cessation of development likely was not due to programmed cell death, because no evidence of several established markers of apoptosis was found. We did not detect a loss of the normally asymmetric distribution of plasma membrane phosphatidylserine, which is one of the earliest manifestations of apoptosis, in arrested embryos. Similarly, cytochrome c remained colocalized with mitochondria. Finally, no evidence of increased DNA fragmentation was found as assessed by TUNEL labeling. This is consistent with previous reports that cleavage-stage embryos do not normally undergo apoptosis [31, 37], although they are capable of initiating components of apoptotic machinery when challenged by extreme stressors, such as the severe oxidative stress induced by high levels of peroxide [22].
The second mitotic cell cycle of mouse embryogenesis is marked by a very short G1 phase that is quickly followed by DNA replication in the S phase and then a long G2 phase before the M phase and cleavage to the 4-cell stage [32]. Embryos cultured at 310 mOsM progressed through the S phase, as evidenced by DNA synthesis assessed by BrdU incorporation. The next major developmental landmark is ZGA during the G2 phase [34]. We chose three markers of ZGA to assess, two of which (Eif1a and H47) have been shown by a gene array-based screen of gene expression during mouse ZGA to be among those with the greatest increases in expression during ZGA of the thousands of genes assessed [23] whereas the other (Zscan4d) is an embryo-specific gene that increases transiently during ZGA [24]. ZGA apparently occurred in 2-cell embryos at 310 mOsM, because each of the three genes showed patterns of changes in expression compared to in vivo embryos and embryos cultured at 250 mOsM similar to those shown by the embryos that progressed from the 1-cell to the middle and late-2-cell stages. However, ZGA as assessed by this limited number of genes appeared to be not entirely unaffected by culture at 310 mOsM, because two genes (Eif1a and H47) showed apparently lower expression in embryos at 310 mOsM than in embryos at 250 mOsM. Perturbation of gene expression during ZGA was not universal, because the transiently expressed Zscan4d had apparently normal expression levels (mRNA levels of the reference gene Hprt were also not affected). Thus, it appears that ZGA occurs in embryos cultured at 310 mOsM but that the levels of expression may not be entirely normal. This is similar to embryos that exhibited the “2-cell block” in traditional culture media, in which ZGA reportedly occurred but was delayed [38].
Confocal imaging of embryos arrested after culture at 310 mOsM at a time when embryos that had instead been cultured at 250 mOsM or at 310 mOsM with glycine had reached the 4-cell stage showed that most embryos had evident nuclei in which the DNA was diffuse and resembled that of interphase nuclei at the 2-cell stage. However, a minority became arrested with metaphase chromosomes. This raised the possibility that most arrested embryos might have entered the M phase but then failed to progress and reformed nuclei, rather than having failed to enter metaphase. By using a method to trap any embryos that entered the M phase and then induce them to condense their chromosomes, we found that the large majority of arrested embryos did not exhibit condensed DNA or spindles and, therefore, had not entered the M phase during the 2-cell stage. The fraction of embryos at 310 mOsM that did become trapped in metaphase by this treatment (23%) probably represents embryos that escape the developmental block at 310 mOsM (∼10%–20%) (Figs. 1 and 2) and the smaller cohort that progress into metaphase but arrest with condensed chromosomes without cleaving (∼10%) (Fig. 8). When MPF was assessed in embryos that still had nuclei, activity was low, and CDK1 was evidently present but in the inactive, phosphorylated state. However, even though only embryos with nuclei had been selected in the 310 mOsM group, significantly more variability in MPF activity was found, and the mean MPF activity was slightly higher (but not significantly different) in these arrested embryos. The “leakage” of blastomeres in some embryos arrested by culture at 310 mOsM into metaphase, along with the greater variation in measured MPF activity with a trend toward higher activity, implies that the arrest induced by increased osmolarity likely occurs very near the G2/M border at the end of the 2-cell stage.
We conclude, therefore, that mouse embryos cultured at physiological osmolarity in the absence of organic osmolytes become arrested only after they have progressed through most of the second embryonic cell cycle and through ZGA, with most arresting in the late G2 phase, just before beginning to enter the M phase. This is consistent with what had been previously found for the classic 2-cell block in traditional culture media, in which arrest at “a state equivalent to a late 2-cell embryo” was reported [20]. It remains unclear what is occurring at the molecular and cellular levels in 1-cell embryos cultured in the absence of organic osmolytes that renders them susceptible to a failure to progress into the M phase at the end of the 2-cell stage unless rescued by that point. Although the 2-cell block in mice and similar developmental blocks in other mammals have been overcome by improved culture media, it remains important to elucidate the effects of even reduced amounts of stress, because human embryos cultured for the treatment of infertility by assisted reproductive technologies are routinely subjected to similar stresses, with possible long-term consequences [39].
ACKNOWLEDGMENTS
The authors thank Minju Cao, Meirong Du, Junjiu Huang, Zhong Liu, Johnny Ngsee, Richard Schultz, Violetta Siyanov, Alina Tartia, Benjamin Tsang, Zhengbo Wang, and Chenxi Zhou for technical help and advice; Karen P. Phillips for permission to cite her unpublished data; and Kerri Courville for excellent technical support. F.W. especially thanks Dr. Hui Xiang (Sun Yat-sen University) for support during her Ph.D. studies.
Footnotes
Supported by China-Canada Joint Health Research Initiative grant CCI82422, Canadian Institutes of Health Research (CIHR), and National Natural Science Foundation of China (NSFC). F.W. received a 985 Program graduate studentship from the China Scholarship Council (CSC).
REFERENCES
- Lawitts JA, Biggers JD. Culture of preimplantation embryos. Methods Enzymol 1993; 225: 153 164. [DOI] [PubMed] [Google Scholar]
- Lawitts JA, Biggers JD. Optimization of mouse embryo culture media using simplex methods. J Reprod Fertil 1991; 91: 543 556. [DOI] [PubMed] [Google Scholar]
- Lawitts JA, Biggers JD. Overcoming the 2-cell block by modifying standard components in a mouse embryo culture medium. Biol Reprod 1991; 45: 245 251. [DOI] [PubMed] [Google Scholar]
- Chatot CL, Ziomek CA, Bavister BD, Lewis JL, Torres I. An improved culture medium supports development of random-bred 1-cell mouse embryos in vitro. J Reprod Fertil 1989; 86: 679 688. [DOI] [PubMed] [Google Scholar]
- Lawitts JA, Biggers JD. Joint effects of sodium chloride, glutamine, and glucose in mouse preimplantation embryo culture media. Mol Reprod Dev 1992; 31: 189 194. [DOI] [PubMed] [Google Scholar]
- Collins JL, Baltz JM. Estimates of mouse oviductal fluid tonicity based on osmotic responses of embryos. Biol Reprod 1999; 60: 1188 1193. [DOI] [PubMed] [Google Scholar]
- Fiorenza MT, Bevilacqua A, Canterini S, Torcia S, Pontecorvi M, Mangia F. Early transcriptional activation of the hsp70.1 gene by osmotic stress in one-cell embryos of the mouse. Biol Reprod 2004; 70: 1606 1613. [DOI] [PubMed] [Google Scholar]
- Hadi T, Hammer MA, Algire C, Richards T, Baltz JM. Similar effects of osmolarity, glucose, and phosphate on cleavage past the 2-cell stage in mouse embryos from outbred and F1 hybrid females. Biol Reprod 2005; 72: 179 187. [DOI] [PubMed] [Google Scholar]
- Van Winkle LJ, Haghighat N, Campione AL. Glycine protects preimplantation mouse conceptuses from a detrimental effect on development of the inorganic ions in oviductal fluid. J Exp Zool 1990; 253: 215 219. [DOI] [PubMed] [Google Scholar]
- Dawson KM, Baltz JM. Organic osmolytes and embryos: substrates of the Gly and beta transport systems protect mouse zygotes against the effects of raised osmolarity. Biol Reprod 1997; 56: 1550 1558. [DOI] [PubMed] [Google Scholar]
- Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RK. Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol 2003; 148: 1 80. [DOI] [PubMed] [Google Scholar]
- Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science 1982; 217: 1214 1222. [DOI] [PubMed] [Google Scholar]
- Baltz JM, Tartia AP. Cell volume regulation in oocytes and early embryos: connecting physiology to successful culture media. Hum Reprod Update 2010; 16: 166 176. [DOI] [PubMed] [Google Scholar]
- Dawson KM, Collins JL, Baltz JM. Osmolarity-dependent glycine accumulation indicates a role for glycine as an organic osmolyte in early preimplantation mouse embryos. Biol Reprod 1998; 59: 225 232. [DOI] [PubMed] [Google Scholar]
- Steeves CL, Hammer MA, Walker GB, Rae D, Stewart NA, Baltz JM. The glycine neurotransmitter transporter GLYT1 is an organic osmolyte transporter regulating cell volume in cleavage-stage embryos. Proc Natl Acad Sci U S A 2003; 100: 13982 13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartia AP, Rudraraju N, Richards T, Hammer MA, Talbot P, Baltz JM. Cell volume regulation is initiated in mouse oocytes after ovulation. Development 2009; 136: 2247 2254. [DOI] [PubMed] [Google Scholar]
- Anas MKI, Hammer MA, Lever M, Stanton JA, Baltz JM. The organic osmolytes betaine and proline are transported by a shared system in early preimplantation mouse embryos. J Cell Physiol 2007; 210: 266 277. [DOI] [PubMed] [Google Scholar]
- Anas MK, Lee MB, Zhou C, Hammer MA, Slow S, Karmouch J, Liu XJ, Broer S, Lever M, Baltz JM. SIT1 is a betaine/proline transporter that is activated in mouse eggs after fertilization and functions until the 2-cell stage. Development 2008; 135: 4123 4130. [DOI] [PubMed] [Google Scholar]
- Biggers JD. Reflections on the culture of the preimplantation embryo. Int J Dev Biol 1998; 42: 879 884. [PubMed] [Google Scholar]
- Goddard MJ, Pratt HPM. Control of events during early cleavage of the mouse embryo: an analysis of the ‘2-cell block.' Development 1983; 73: 111 133. [PubMed] [Google Scholar]
- Ohashi A, Minami N, Imai H. Nuclear accumulation of cyclin B1 in mouse two-cell embryos is controlled by the activation of cdc2. Biol Reprod 2001; 65: 1195 1200. [DOI] [PubMed] [Google Scholar]
- Liu L, Trimarchi JR, Keefe DL. Involvement of mitochondria in oxidative stress-induced cell death in mouse zygotes. Biol Reprod 2000; 62: 1745 1753. [DOI] [PubMed] [Google Scholar]
- Zeng F, Schultz RM. RNA transcript profiling during zygotic gene activation in the preimplantation mouse embryo. Dev Biol 2005; 283: 40 57. [DOI] [PubMed] [Google Scholar]
- Falco G, Lee SL, Stanghellini I, Bassey UC, Hamatani T, Ko MS. Zscan4: a novel gene expressed exclusively in late 2-cell embryos and embryonic stem cells. Dev Biol 2007; 307: 539 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamo S, Gal AB, Bodo S, Dinnyes A. Quantitative evaluation and selection of reference genes in mouse oocytes and embryos cultured in vivo and in vitro. BMC Dev Biol 2007; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrunewich MA, Trimarchi JR, Hanlan AK, Hammer MA, Baltz JM. Second meiotic spindle integrity requires MEK/MAP kinase activity in mouse eggs. J Reprod Dev 2009; 55: 30 38. [DOI] [PubMed] [Google Scholar]
- Moos J, Kopf GS, Schultz RM. Cycloheximide-induced activation of mouse eggs: effects on cdc2/cyclin B and MAP kinase activities. J Cell Sci 1996; 109 (pt 4): 739 748. [DOI] [PubMed] [Google Scholar]
- Phillips KP, Petrunewich MAF, Collins JL, Baltz JM. The intracellular pH-regulatory HCO3−/Cl− exchanger in the mouse oocyte is inactivated during first meiotic metaphase and reactivated after egg activation via the MAP kinase pathway. Mol Biol Cell 2002; 13: 3800 3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brison DR. Apoptosis in mammalian preimplantation embryos: regulation by survival factors. Hum Fertil (Camb) 2000; 3: 36 47. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome c-mediated apoptosis. Annu Rev Biochem 2004; 73: 87 106. [DOI] [PubMed] [Google Scholar]
- Kamjoo M, Brison DR, Kimber SJ. Apoptosis in the preimplantation mouse embryo: effect of strain difference and in vitro culture. Mol Reprod Dev 2002; 61: 67 77. [DOI] [PubMed] [Google Scholar]
- Sawicki W, Abramczuk J, Blaton O. DNA synthesis in the second and third cell cycles of mouse preimplantation development: a cytophotometric study. Exp Cell Res 1978; 112: 199 205. [DOI] [PubMed] [Google Scholar]
- Latham KE, Garrels JI, Chang C, Solter D. Quantitative analysis of protein synthesis in mouse embryos. I. Extensive reprogramming at the one- and two-cell stages. Development 1991; 112: 921 932. [DOI] [PubMed] [Google Scholar]
- Schultz RM. Regulation of zygotic gene activation in the mouse. Bioessays 1993; 15: 531 538. [DOI] [PubMed] [Google Scholar]
- Egli D, Sandler VM, Shinohara ML, Cantor H, Eggan K. Reprogramming after chromosome transfer into mouse blastomeres. Curr Biol 2009; 19: 1403 1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G, Taylor W. Control of the G2/M transition. Mol Biotechnol 2006; 32: 227 248. [DOI] [PubMed] [Google Scholar]
- Fabian D, Makarevich AV, Chrenek P, Bukovska A, Koppel J. Chronological appearance of spontaneous and induced apoptosis during preimplantation development of rabbit and mouse embryos. Theriogenology 2007; 68: 1271 1281. [DOI] [PubMed] [Google Scholar]
- Qiu JJ, Zhang WW, Wu ZL, Wang YH, Qian M, Li YP. Delay of ZGA initiation occurred in 2-cell blocked mouse embryos. Cell Res 2003; 13: 179 185. [DOI] [PubMed] [Google Scholar]
- Fleming TP, Kwong WY, Porter R, Ursell E, Fesenko I, Wilkins A, Miller DJ, Watkins AJ, Eckert JJ. The embryo and its future. Biol Reprod 2004; 71: 1046 1054. [DOI] [PubMed] [Google Scholar]