

Summary
Spirochetes that cause Lyme borreliosis (also called Lyme disease) possess the vls locus, encoding an elaborate antigenic variation system. This locus contains the expression site vlsE as well as a contiguous array of vls silent cassettes, which contain variations of the central cassette region of vlsE. The locus is present on one of the many linear plasmids in the organism, e.g. plasmid lp28-1 in the strain B. burgdorferi B31. Changes in the sequence of vlsE occur continuously during mammalian infection and consist of random, segmental, unidirectional recombination events between the silent cassettes and the cassette region of vlsE. These gene conversion events do not occur during in vitro culture or the tick portion of the infection cycle of Borrelia burgdorferi or the other related Borrelia species that cause Lyme disease. The mechanism of recombination is largely unknown, but requires the RuvAB Holliday junction branch migrase. Other features of the vls locus also appear to be required, including cis locations of vlsE and the silent cassettes and high G+C content and GC skew. The vls system is required for long-term survival of Lyme Borrelia in infected mammals and represents an important mechanism of immune evasion. In addition to sequence variation, immune selection also results in significant heterogeneity in the sequence of the surface lipoprotein VlsE. Despite antigenic variation, VlsE generates a robust antibody response, and both full length VlsE and the C6 peptide (corresponding to invariant region 6) are widely used in immunodiagnostic tests for Lyme disease.
Antigenic variation is defined as a hereditable, reversible variation in an antigenic structure that occurs during the course of infection at a rate higher than would be expected for standard recombination or mutation mechanisms. Many bacterial and protozoal pathogens have developed antigenic variation systems in which surface antigens can be continually altered as a means of evading the constant onslaught of adaptive antibody and T cell responses (1). In 1997, an elaborate antigenic variation system was identified in B. burgdorferi B31 (2). Because of sequence similarity between this system and the previously characterized Variable Major Protein (VMP) system of relapsing fever bacteria, it was termed the VMP-like sequence (vls) locus. Its expression site, called vls Expressed (vlsE), undergoes remarkable sequence variation involving segmental gene conversion events from vls silent cassettes. This chapter will describe what is currently known about the structure, properties, role in host-pathogen interactions, recombination process, and evolution of the vls system.
Lyme borreliosis
Lyme borreliosis (LB; also called Lyme disease) is a multistage, tick-transmitted infection caused by spirochetes in the genus Borrelia. Borrelia burgdorferi is the principal human pathogen in North America, whereas B. garinii, B. afzelii, and B. burgdorferi all give rise to Lyme borreliosis in Euroasia (3-5). These organisms are transmitted by hard-bodied ticks of the genus Ixodes; I. scapularis and I. pacificus are the transmitting ticks in North America, whereas I. ricinus and I. persulcatus are most active in Europe and Asia, respectively. B. spielmanii, B. bissettii and B. valaisiana have also been associated with rare cases of human infections (6). There are many additional Lyme Borrelia species that are not known to cause human disease. All of the Lyme Borrelia species are referred to collectively as Borrelia burgdorferi sensu lato (in a broad sense), whereas B. burgdorferi sensu stricto (in a strict sense) refers only to the type species of the group. Relapsing fever Borrelia (including B. hermsii, B. crocidurae, and B. recurrentis), although related to Lyme Borrelia, cause an entirely different disease transmitted by soft-bodied, fast-feeding Ornithodoros ticks.
B. burgdorferi and other Lyme Borrelia survive by contiguous transmission between Ixodes ticks and susceptible mammalian hosts. Infection of humans occurs through the bite of an infected tick (usually at the nymphal stage), causing a localized infection and a resulting expanding red rash called erythema migrans (Table 1). The spirochetes multiply locally, but even at these early stages of infection are able to penetrate blood vessels and lymphatics and thereby disseminate to other tissues. The erythema migrans lesion will eventually clear. However, most patients will go on to develop disseminated symptoms, including a variety of musculoskeletal, neurologic, and cardiovascular manifestations. Months to years later, persistent infection causes Lyme arthritis, which is the most prominent late symptom in North American patients infected with B. burgdorferi. B. garinii infection tends to cause neurologic signs, whereas most cases of the skin lesion acrodermatitis chronica atrophicans (ACA) are caused by B. afzelii. The manifestations shown in Table 1 really form a continuum, and the disease properties differ greatly from patient to patient. Lyme Borrelia are present at high concentrations only in erythema migrans skin lesions, and otherwise are typically present in small numbers and can be distributed to almost any tissue. The organisms produce no known toxins; rather, pathogenesis appears to be primarily due to the induction of inflammatory reactions in the infected mammalian host (7). During the transitions between the tick and mammalian hosts, Lyme Borrelia undergo massive changes in gene expression (8), resulting in concomitant shifts in the proteins required for survival and growth in the arthropod or warm-blooded animal environments.
Table 1.
Stages of Lyme borreliosis
| Localized (days to weeks post infection) |
| Erythema migrans skin lesion |
| Headache, malaise, fatigue, muscle and joint pain |
| Disseminated (weeks to months post infection) |
| Secondary annular skin lesions |
| Neuroborreliosis – meningitis, facial palsy, radiculoneuritis |
| Migratory musculoskeletal pain |
| Atrioventricular nodal heart block |
| Lymphocytomaa |
| Eye manifestations |
| Persistent (months to years post infection) |
| Migratory arthritis of large joints |
| Neuroborreliosis – meningitis, encephalitis, facial palsy, radiculoneuropathy, polyneuritis |
| Atrioventricular nodal heart block |
| Acrodermatitis chronica atrophicans skin lesionsa |
Information from Steere et al. (3) and Müllegger et al. (5)
Found in Eurasia
While it is not known how long humans can be infected with Lyme Borrelia, viable spirochetes can be cultured from almost any tissue of experimentally infected mice throughout their lifetimes; thus it is likely that, without treatment, humans can carry viable organisms for years. Lyme Borrelia thus fall in a category of persistent, nontoxigenic pathogens that also includes the syphilis spirochete Treponema pallidum subsp. pallidum (9).
Persistence requires mechanisms for evading host immune responses, particularly the adaptive immune response. Immune evasion mechanisms that have been described in Lyme Borrelia include Complement Regulator-Acquiring Surface Proteins (CRASPs), which bind Factor H and Factor H-like protein 1 (FHL-1) and thus inhibit the activation of the complement cascade (10,11). Another mechanism involves the down-regulation of the antigenic tick phase-associated outer surface lipoproteins OspA and OspB, as well as OspC, which is required for survival of B. burgdorferi during the early phase of mammalian infection (12,13).
vls system of B. burgdorferi B31
The B31 strain of B. burgdorferi was isolated from I. scapularis (then called I. dammini) ticks collected on Shelter Island, New York, and was the first strain of Lyme Borrelia described(14). In early studies of this organism, it was determined that its genome consisted of a linear chromosome and multiple linear and circular plasmids ranging in size from 56 kbp to 5 kbp. Many of these plasmids were easily lost during in vitro culture, and absence of some of these plasmids correlated with loss of infectivity in animal models (15-23). The availability of the genome sequence of B. burgdorferi B31 showed that the organism contained 12 linear and 9 circular plasmids (with a total of over 600 kbp) as well as a 972 kbp linear chromosome (24,25). The linear replicons of Borrelia were found to have covalently closed, hairpin telomeres in which a 5’-3’ bond is present between the positive and negative strands (25-27). Replication of these linear molecules occurs through the formation of a circular intermediate that undergoes asymmetric single-stranded cleavage by the plasmid-encoded telomere resolvase ResT at specific sites near the telomere, followed by separation of the two plasmid copies, snap-back hybridization, and ligation (28).
The vls system was first discovered prior to the availability of the genomic sequence as the result of a subtractive hybridization study aimed at identifying sequences that were present in low passage, infectious clones of B. burgdorferi B31 but absent in a high passage, noninfectious B31 clone (2). A single recombinant plasmid called pJRZ53 was found to encode an amino acid sequence with a low but significant sequence identity with the B. hermsii antigenic variation protein Vlp17. The pJRZ53 insert hybridized with several restriction fragments of lp28-1, a 28 kbp linear plasmid. Cloning of the 10 kbp region containing the vls sequences into a lambda phage vector followed by sequencing revealed the presence of a single telomeric copy of the expression site vlsE as well as a contiguous array of 15 sequences that shared 90.0% to 96.1% nucleotide sequence identity and 76.9% to 91.4% encoded amino acid sequence identity with the central “cassette” region of the vlsE gene (Fig. 1A). The 15 unexpressed (silent) cassette regions are 474 to 594 bp in length and are demarcated by 17 bp direct repeats found also at either end of the cassette region of vlsE. They are present in a head to tail arrangement, forming a long contiguous open reading frame interrupted only by one stop codon (in cassette vls11) and two frame shifts (in cassettes vls14 and vls16). Alignment of the nucleotide or encoded amino acid sequences of the vls cassettes reveals the presence of 6 variable regions (VRs) interspersed among 7 relatively invariant regions (IRs) (Fig. 1B). In the variable regions, up to 6 different amino acids can be encoded in some positions of the aligned sequence. Nearly all indels are in multiples of three base pairs, indicating the importance of maintaining an intact open reading frame. Some of the silent cassettes are truncated or have long internal deletions; these regions were excluded from the analysis shown in Fig. 1B, because they do not appear to participate in vlsE recombination events.
Fig. 1.
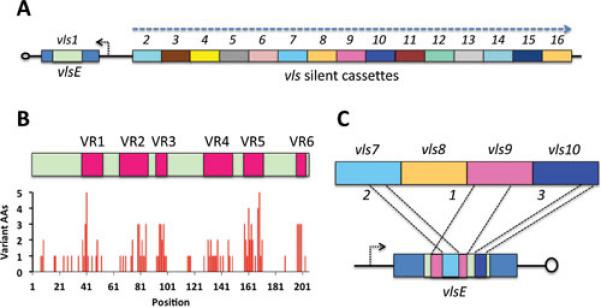
Characteristics of the vls locus of B. burgdorferi B31. A. Arrangement of the vlsE expression site and the 15 silent cassettes near the telomere of the linear plasmid lp28-1. The promoter for vlsE is indicated by the short arrow and the orientation of the silent cassettes is shown by the large arrow. B. The cassette regions contain six variable regions (VR1 through VR6) separated by relatively invariant regions (IRs). The graph indicates the number of different amino acids encoded by the silent cassettes at each codon in the aligned sequences. C. Unidirectional, random, segmental recombination occurs sequentially during mammalian infection, as indicated by this hypothetical example of sequential recombination between vlsE and silent cassettes 9, 7 and 10.
The vlsE gene itself encodes a 36 kDa protein with a lipoprotein leader sequence; further studies verified that the VlsE product is lipidated and localized on the outer surface of the outer membrane (2). The gene has a consensus sigma 70 promoter region, and primer extension analysis showed that transcription is initiated with nearly equivalent efficiency at two adjacent thymidine residues at 13 and 14 bp downstream from the beginning of the −10 sequence(29). Interestingly, inverted repeats that include portions of the vlsE promoter region and the 5’ end of the first silent cassette are predicted to form a 51-bp stem loop structure; this feature is likely involved in vlsE transcription and/or recombination.
To date, no vlsE recombination events have been reported in in vitro cultures of Lyme Borrelia or in infected ticks (2,30-32). In contrast, vlsE sequence changes have been detected as early as 4 days post inoculation of C3H/HeN mice with B. burgdorferi; by 28 days of infection, few (if any) organisms expressing the ‘parental’ vlsE sequence are present (30,33). Similar results have been obtained with infected rabbits, with multiple vlsE sequence changes observed at 2 weeks, the earliest post-inoculation time point in these experiments (34). In contrast to relapsing fever Borrelia, in which the typical pattern is the sequential outgrowth of clones expressing a single VMP type, mammalian infection with Lyme Borrelia results in the outgrowth of a myriad of clones each expressing a different VlsE variant. Indeed, the degree of sequence variation is such that it is difficult to find two clones with the same vlsE sequence at time points beyond 4 weeks post inoculation, even in the same tissue biopsy (2,30,33).
Given the presence of 15 silent cassettes that represent variants of the vlsE cassette region sequence, the initial hypothesis was that each of these silent cassettes could replace the vlsE cassette, resulting in 15 different variants. Instead, the sequencing of vlsE cassette regions from over 1,400 B. burgdorferi clones has revealed that sequence variation occurs through the replacement of segments of DNA in the expression site with segments of the corresponding regions of the silent cassettes (Fig. 1C) (2,30,33). These segments can range in size from a few base pairs to nearly the entire length of the vlsE cassette region (33). The recombinations represent gene conversion events, in that the sequences of the silent cassettes are not altered (35), as will be discussed further in the context of recombination mechanisms. vlsE gene conversion events appear to occur continuously during the course of mammalian infection, resulting in a mosaic of overlapping recombinations that are difficult to decipher. An Excel spreadsheet-based Visual Basic program has been developed to help determine which silent cassettes could have been the source of sequence replacements (33). Because the silent cassettes have many regions of sequence identity, it is often not possible to attribute a given recombination event to a single silent cassette; multiple cassettes could have served as potential ‘donors’. For the same reason, the boundaries of the recombination usually cannot be determined with confidence, so recombination events are defined as minimal recombination regions (including the nucleotides that differ from the parental sequence) and maximal recombination regions (encompassing the minimal region plus the surrounding nucleotides that are identical between the parental sequence and the silent cassette donor) (33). The sequence variation is restricted to the vlsE cassette region, in that 5’ and 3’ ends of the gene do not undergo sequence changes (35).
vls systems of other Lyme Borrelia
vls sequences have been identified in every Lyme Borrelia organism for which a complete or draft genomic sequence is available, indicating that this antigenic variation system is ubiquitous among all Lyme disease Borrelia. After the initial description of the B. burgdorferi B31 vls system, the identification of vls sequences in recombinant DNA libraries derived from B. burgdorferi 297 (36), B. garinii IP90 (37), and B. garinii A87SA (38) were reported. Comprehensive analysis of vls sequences of B. garinii IP90 and B. afzelii ACA-1 revealed the near-complete sequence of the silent cassettes of these two strains (39). The intact vlsE genes of these two strains were not isolated, but partial sequences were obtained by the cloning of cDNA products and by PCR analysis (39).
All of the 26 Lyme Borrelia strains from which extensive plasmid DNA sequences have been obtained (24,40-45) contain vls sequences, greatly increasing the available information regarding the characteristics of vls systems. Shotgun sequencing of genomes has yielded nearly complete sequences of vls silent cassette arrays. However, vlsE sequences are typically missing from genomic sequences, probably because of their location near a telomere or predicted stem-loop structures and resulting poor cloning and sequencing efficiency.
Currently, complete or nearly complete vlsE sequences are available in GenBank for 16 Lyme Borrelia strains. In addition, the B. burgdorferi 29805 silent cassette vlsS1 is contiguous with a region homologous to the 5’ constant (non-cassette) region of vlsE, so it is also included in this comparison. The phylogenetic tree of the predicted VlsE amino acid sequences with the B. hermsii Vlp sequences as outliers (Fig. 2) reveals the conservation and diversity among the VlsE sequences. VlsE proteins of the German B. burgdorferi isolates PKa2, PAbe, PBoe, and PBre all exhibit a high sequence identity/similarity with the United States isolate B31 (83-93% and 90-95%, respectively), with nearly all the sequence differences existing in the variable regions. This result indicates that a closely related clade of B. burgdorferi clones exists in the North American and European continents, such that the VlsE sequence differences are essentially the same as are observed among B31 antigenic variants (33). Indeed, the availability of several genomic sequences from this clade indicate a pan-genomic clonality, consistent with the recent geographic spread of this group of Lyme Borrelia (43). In contrast, the VlsE sequences of North American B. burgdorferi isolates B31 and 297 exhibit a high degree of divergence, with only 46% identity and 53% similarity. A similar level of divergence is observed between the Lyme Borrelia species (Fig. 2). Again using B. burgdorferi B31 for comparison, the B. garinii, B. afzelii, and B. spielmanii strains have only 35-49% identity and 41-59% similarity, indicative of a high level of diversification in “framework” regions outside of the variable regions. B. valaisiana VlsE was the most divergent from the B31 sequence (33% identity, 41% similarity). A very similar pattern of diversification between strains (not shown here) is evident in the much larger group of available vls silent cassette sequences, and also when nucleotide sequences are used for comparison.
Fig. 2.
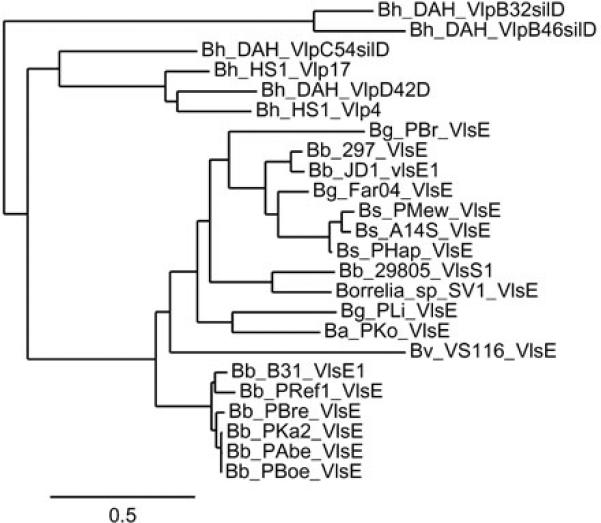
Dendrogram depicting the conservation and diversity of VlsE amino acid sequences and their relatedness to Vlp proteins from relapsing fever organisms. Representative Vlp proteins from B. hermsii (Bh) strains DAH and HS1 are clustered into two groups at the top of the dendrogram. The B31 strain of B. burgdorferi (Bb) and closely related European strains form a distinct grouping at the bottom, whereas the remaining Lyme disease Borrelia VlsE sequences exhibit considerable diversity. Additional species abbreviations: Bg = B. garinii, Bs = B. spielmanii, Ba = B. afzelii, and Bv = B. valaisiana.
The degree of diversity among vlsE sequences is higher than that of any other orthologous gene group in the Lyme Borrelia strains examined. ospC is the next most divergent, with the lowest OspC identity and similarity among available full-length sequences being 68% and 79% respectively (between B. garinii TCLSK and B. valaisiana VS116). The high diversity in vlsE (and ospC) is indicative of a strong selective pressure, most likely driven by mammalian host adaptive immune responses as discussed later in this chapter.
The sequences of only three intact vls systems (including both vlsE and the vls silent cassettes) are currently available (Fig. 3). In each of these, the arrangement is similar to the B31 system, with vlsE and the silent cassettes facing in opposite directions and separated by a short DNA segment. This region includes inverted repeats that could form a stem loop structure; however, these repeats are comprised of different sequences in each case. For example, the possible stem loop structure in strain B31 involves portions of the vlsE promoter on one side and a segment upstream of the first vls silent cassette region on the other. All three strains lack an intact promoter for the silent cassette region. However, both B. burgdorferi JD1 and B. garinii Far04 have sequences identical to the 5’-end of the vlsE open reading frame that are contiguous with the first silent cassette. In JD1, the region complementary with vlsE lacks a start codon but includes a 476 nt region of identity (with 2 nt differences) with the 5’ end vlsE and a portion of the first vls cassette. The Far04 silent cassette region likewise has a 476 nt region of identity to the 5’ end of vlsE and part of the cassette region, but also encompasses the ribosome binding site and −10 sequence. The B31 strain silent cassette region also has a shorter segment of sequence identical to the 5’ end of vlsE. The ‘loop’ region between the complementary ‘stem’ sequences is 300 nt and 299 nt in JD1 and Far04, respectively, but is only 6 nt long in the B31 sequence. The significance and potential functionality of the duplication of the 5’ end of vlsE is not known, but the fact that it is conserved to a varying extent in each organism indicates that its preservation is favored and that ‘purifying’ recombinations may maintain the structure. Of course, there are many additional regions of sequence identity between vlsE and the silent cassettes that likely participate in the vlsE gene conversion events.
Fig. 3.
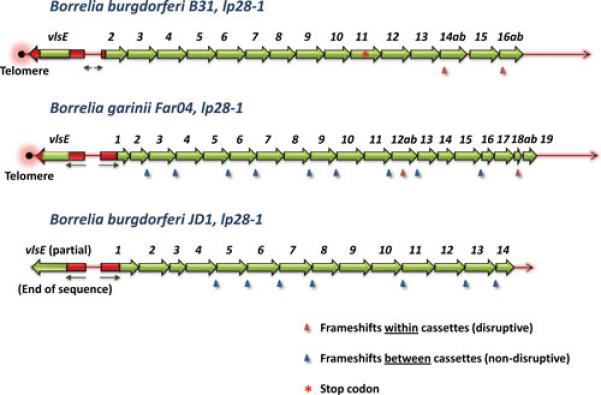
Arrangement of vlsE and the silent cassettes in B. burgdorferi B31, B. garinii Far04, and B. burgdorferi JD1. Cassette region sequences are shown in green, whereas flanking 5’ and 3’ vlsE sequences and homologous 5’ sequences at the beginning of the silent cassette arrays are shown in red. Arrows indicate the locations of inverted repeats. Arrowheads correspond with frameshifts, with the blue arrowheads indicating frameshifts between silent cassettes and red arrowheads showing those located within silent cassettes (as also indicated by the a and b designations of the ORFs before and after the frameshift). The B31 silent cassette 11 contains a stop codon (red asterisk).
Many of the silent cassettes in both B. garinii Far04 and B. burgdorferi JD1 are separated by frame shifts (Fig. 3), and this pattern is found in the vls loci of other Lyme Borrelia organisms as well. Thus the presence of a nearly contiguous open reading frame in the vls cassette region as seen in B. burgdorferi B31 is not required for vlsE recombination and locus functionality. The B. garinii Far04 vls locus is found close to a telomere, as is the B31 locus; the vlsE sequence in JD1 is incomplete, so the nature of the downstream sequence is not known. vls silent cassette regions have been identified in every other complete or near complete genomic sequence from Lyme Borrelia organisms, currently including 2 B. afzelii , 13 B. burgdorferi, 4 B. garinii, 2 B. spielmanii, and 1 B. valaisiana strains (data not shown). In all of these cases, the silent cassette arrangements are similar to those shown in Fig. 3. As mentioned previously, the lack of a contiguous sequence containing vlsE appears to be due to either its telomeric location or the inverted repeats, which interferes with sequencing reactions. Overall, the general properties of vls loci in Lyme Borrelia are well conserved, although substantial sequence diversity is present.
VlsE structure and the localization of variable regions
Determination of the three dimensional structure of B. burgdorferi B31 VlsE (46) revealed one of the most remarkable examples of immune evasion through the optimized localization of variable epitopes. As mentioned previously, VlsE is a surface localized lipoprotein, and is thereby anchored to the outer membrane surface by lipid moieties covalently linked to the N-terminal cysteine residue of the processed protein. Although the unit cell of the crystal structure consists of four monomers with two side-by-side pairs (46), VlsE appears to exist as a monomer rather than a dimer in its native state. The N- and C-terminal sequences and the relatively invariant regions within the cassette form α-helices that constitute the structural framework of VlsE (Fig. 4A). Within this framework, the six VRs constitute random coil regions that form a ‘dome’ on the membrane distal surface of the protein. In the space-filling model (Fig. 4B), these variable regions essentially cover the top of the protein. Therefore, the region of the polypeptide that is most likely to interact with antibodies is precisely the area that undergoes rapid sequence variation during the course of mammalian infection.
Fig. 4.
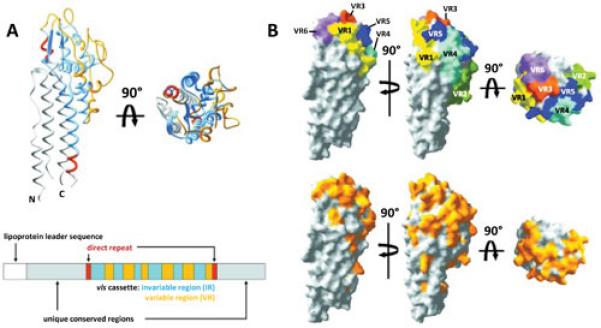
Localization of the variable regions (VRs) in the three-dimensional structure of the B. burgdorferi B31 allele VlsE1. A. Ribbon diagram showing the abundance of alpha helices and the location of the cassette variable regions (VR) (yellow) and invariant regions (blue). Amino acids encoded by the direct repeats at either end of the cassette region are shown in red. The protein is anchored to the outer membrane by lipid moieties associated with the N-terminal cysteine. A schematic of the cassette region and flanking sequences is shown below the 3D structure. B. Space-filling models indicating the locations of variable regions (VRs) 1 through 6. The VRs cover most of the membrane distal surface of the protein. The bottom panel shows the locations of variable amino acid residues in yellow. Modified from Eicken et al. (46) with permission.
It is not known to what degree the relatively invariant lateral surfaces of this elongated protein are exposed to the external fluid phase in the intact organism. The surface lipoproteins may form a contiguous layer during infection that would prevent antibodies from accessing the lateral surfaces. Such a ‘forest’ of outer surface proteins has been documented by cryo electron tomography (cryo ET) in in vitro cultured B. burgdorferi (47), but under these conditions the lipoproteins OspA, OspB, and OspC are highly expressed. During mammalian infection, OspA and OspB are dramatically down-regulated, OspC appears to be transiently expressed, and VlsE expression is thought to increase (29,48,49); however, to date the surface topology of mammalian host-adapted Borrelia has not been examined in detail by cryo ET or other means. It has been shown that antibodies against the invariant regions, particularly the highly immunogenic IR6 region, do not react with VlsE in intact organisms, although they will bind to recombinant VlsE in solution (37)(50). IR6 forms a compact α-helix that is ‘buried’ underneath the variable region random coil structures (46), and thus may lack the surface exposure necessary to permit antibody binding. Recombinant VlsE is difficult to crystallize; it is possible that a significant amount is misfolded and thus may have exposed IR6 available for antibody reactions.
Role in pathogenicity
The earliest indication that the vls system was required for full infectivity was that loss of the encoding plasmid (a 28 kb linear plasmid, lp28-1, in most Lyme Borrelia) resulted in an intermediate infectivity phenotype in experimentally infected mice (15,18,20-23). Needle inoculation of immunocompetent C3H/HeN mice with lp28-1− clones of B. burgdorferi strains B31 or 297 results in a transient infection in which organisms can be cultured from joints and, occasionally, other tissues for up to two weeks post inoculation (but not thereafter). Anti-B. burgdorferi antibody responses occur in the animals infected with the lp28-1− clones, consistent with the establishment of sufficient numbers of spirochetes to induce B cell responses. Surprisingly, lp28-1− clones cause long-term infections in severe combined immunodeficiency (SCID) mouse strains (21,30,51). Trans-complementation with vlsE alone on a shuttle vector does not alter the infectivity phenotype of a lp28-1− clone, indicating that VlsE expression is not sufficient to restore full infectivity (51). Loss of lp28-1 does not affect the colonization of I. scapularis ticks (52). Taken together, these results show that lp28-1 carries virulence determinant(s) that protect Lyme Borrelia against adaptive immune responses in mammalian hosts.
More definitive evidence of the role of the vls system in mammalian infection was provided by elegant studies reported by Bankhead and Chaconas in 2007 (53). Using a telomere resolvase-mediated targeted deletion approach, they were able to delete the entire vls locus from the right end of lp28-1 in B. burgdorferi B31. As a control, they also generated mutants lacking genes (BBF01-BBF19) from the left end of lp28-1, upstream from the region required for plasmid replication. The two Δvls mutant clones tested exhibited the same intermediate infectivity phenotype in C3H/HeN mice as the control lp28-1− strain, whereas the mutants lacking the left end of lp28-1 were fully infectious. Expression of VlsE by itself in the Δvls background was not sufficient to restore full infectivity. These results clearly indicate that an intact vls system is required for persistent infection.
Antibody responses against VlsE
Immune evasion through antigenic variation systems is thought to act through the alteration of surface-exposed epitopes so that antibodies induced against one form of the antigen will not react with subsequent variants. An unexpected finding was that VlsE induces a robust antibody response in human Lyme borreliosis patients and naturally and experimentally infected animals (2,37,54-60). The antibody responses are predominantly against the conserved, non-variable regions of VlsE, particularly the IR6 region (also called C6) within the vls cassette (61,62); serologic reactivity has also been demonstrated against the N-terminal and C-terminal regions outside the central cassette (63). However, McDowell et al. (64) established by VlsE variant cross-absorbtion studies that the variable regions of VlsE also induce antibody responses. Antibodies against conserved regions are typically cross-reactive with other VlsE sequences, and either full-length VlsE or the C6 peptide have been incorporated into many serological diagnostic tests for Lyme borreliosis in humans or dogs (55,56,58,59).
In studies conducted to date, immunization with recombinant forms of VlsE or the C6 peptide has not has not resulted in protection of mice against infection with B. burgdorferi expressing the homologous VlsE variant (65) (S.J.N., unpublished data). Also, B. burgdorferi strains with a functional vls system that are experimentally modified to constitutively express an invariant form of VlsE cause persistent infection (51,53). It could be expected that Borrelia expressing invariant VlsE would be eliminated by antibodies against that allele, but that is not the case. Liang et al. (37) have hypothesized that suggested that IR6 may serve as a “decoy” epitope that diverts the immune response away from protective responses to other epitopes on VlsE or other antigens. Although the recombinant B31 VlsE or a single C6 peptide is effective in detecting antibodies induced in humans and dogs infected with a variety of Lyme Borrelia species and strains, Baum et al. (66) found that the white-footed mouse Peromyscus leucopus, a natural host of B. burgdorferi in North America, often produces a weak response to infection with several different B. burgdorferi strains. They suggest that coevolution of the pathogen and host has resulted in the elimination of sequences that induce protective anti-VlsE responses. Overall, much remains to be understood about the nature of the immune response against VlsE and host-pathogen interactions.
VlsE expression
As is the case for many Lyme Borrelia proteins, the expression of VlsE appears to be highly regulated during the organism’s infectious cycle (31,48,67). When B. burgdorferi is acquired by tick larvae, it continues to express VlsE for approximately 96 hours, after which the percentage of VlsE-positive decreases (48). VlsE is expressed at very low levels by the spirochete in the tick in between feedings. Disparate results were obtained regarding VlsE level changes occurring when an infected tick acquires a blood meal from mice. In two studies, the expression increased significantly within 24 hours, in a manner similar to the increase in OspC expression (31,67); in a third report, the percent of VlsE-positive organisms remained <10% during feeding, but increased to ~90% in the skin of the mice at 72 hours after feeding was initiated (48). Piesman et al. (68) found that vlsE transcript levels were higher in B. burgdorferi in the salivary glands as compared to the levels in the midgut of feeding ticks, suggesting that the salivary gland environment is stimulatory toward vlsE expression. Similarly, Koči et al. (67) observed vlsE transcript increases during tick feeding in I. ricinus ticks infected with B. afzelii. In vitro studies are contradictory, with opposite changes in VlsE expression occurring when B. burgdorferi were incubated at pH 7 or pH 8 at 34° C (31,48). However, the reports were in agreement that VlsE expression was high at pH 7.5-8 at 23° C, conditions thought to mimic the tick midgut.
Hudson et al. (29) determined that vlsE is transcribed at higher levels in B. burgdorferi B31 in the presence of endothelial cells than in cell-free liquid culture, suggesting that the presence of these cells or their products triggers an up-regulation of vlsE expression. No vlsE recombination was detected in these studies.
The vlsE promoter region has predicted −35 and −10 sequences typical of RpoD recognition sites (Fig. 5A). The transcriptional initiation site was mapped by Bykowski et al. (48) by primer extension and was found to be located at two adjacent T residues 6 nt downstream of the −10 sequence. One can speculate that the predicted stem loop structure in the B31 has regulatory effects on vlsE expression. However, as mentioned previously, the location and nature of inverted repeats is variable in the few organisms in which the intact sequence of this region is available. Factors that bind to the promoter region have not as yet been reported. Jutras et al. (69) used electrophoretic mobility shift assays (EMSAs) to demonstrate that SpoVG binds to a dsDNA region of the B. burgdorferi B31 vlsE open reading frame; it is thus possible that this factor may affect transcription after initiation (see further discussion of SpoVG below).
Fig. 5.
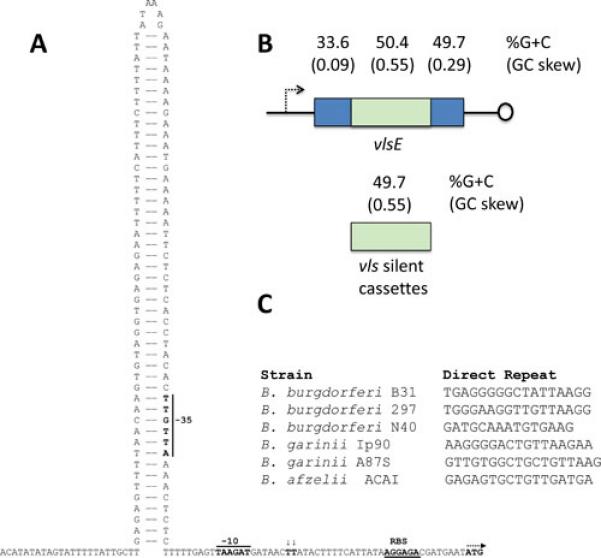
Properties of the vls locus potentially involved in recombination and regulation of transcription. A. Inverted repeat between vlsE and the vls silent cassettes of B. burgdorferi B31 (48);(S.J. Norris and J.K. Howell, unpublished data). The predicted stem loop structure includes the −35 region of the vlsE promoter. B. G+C content and GC skew of vlsE and the vls silent cassettes in B. burgdorferi B31. C. Lack of conservation of the vls direct repeat sequences in Lyme disease Borrelia suggests a limited role of these regions in vlsE recombination (38). Representative sequences are shown.
Crother et al. (49,70) performed novel studies on the expression of B. burgdorferi lipoproteins during mouse or rabbit infection, in which the tissue specimens were extracted by Triton X114 phase partitioning, which preferentially extracts lipoproteins. In the first study (70), SCID mice were inoculated to increase the yield of organisms, and infected joint, heart, ear, and skin tissues were utilized. The extracted proteins were detected by immunoblot analysis with polypeptide-specific or anti-B. burgdorferi polyspecific antisera. In these experiments, VlsE was the most abundant borrelial protein recovered from all tissues except the heart. In the heart, the levels were low throughout the course of infection. In the second study, rabbits were infected by intradermal injection, and rabbits were sacrificed and the skin sites analyzed on days 5, 7, 9, 11, 14, and 21 post inoculation (PI). By this methodology, VlsE protein levels increased dramatically on day 7 PI, and remained high throughout the experiment. Conversely, OspC levels peaked on day 7 PI and decreased rapidly thereafter. When VlsE was analyzed using VlsE-specific antiserum and two dimensional gel electrophoresis, it appeared as a series of spots over a broad pH range; this may reflect the presence of multiple variant VlsE proteins with different isoelectric points (pI’s). In addition, low molecular weight isoforms of VlsE were also present, but it was unclear whether these were degradation products or truncated forms. The results of this study provided evidence that OspC levels are downregulated and VlsE levels are upregulated during the course of experimental rabbit infection.
Further studies by Liang et al. (71,72) examined transcript levels of vlsE, ospC and other lipoprotein genes during the course of B. burgdorferi infection in normal mice, B-cell deficient mice, SCID mice, and SCID mice treated with sera from infected animals. In SCID mice, transcript levels of most lipoprotein genes, including vlsE and ospC, remain at high levels; the same results were obtained with vlsE and ospC in B cell-deficient mice (72). In contrast, ospC transcripts were decreased below detectable levels and vlsE transcript levels are reduced in immunocompetent mice at day 33-40 PI. Treatment of SCID mice with sera from B. burgdorferi infected mice resulted in dramatic decreases (up to 76-fold) in ospC mRNA levels and increases in vlsE transcripts (up to 44-fold) relative to SCID mice treated with prebleed mouse sera (72). Similar results were obtained with a monoclonal antibody against OspC, indicating that anti-OspC antibodies were principally responsible for this effect. Interestingly, the effects varied according to the tissues sampled; for example, neither of the antisera had a significant effect on vlsE transcript levels in joint tissue.
Overall, studies to date indicate that vlsE transcription is dramatically affected by antibody responses, most likely through the direct or indirect effects of OspC transcript or protein expression levels. It is not known currently if these effects are due to the selection of OspC-underexpressing cells, or a generalized down-regulation of OspC expression in the infecting population. Studies by Anguita et al. (73) suggest that the inflammatory process (in particular, interferon-ϒ expression) may also affect spirochete tissue adaptation and vlsE recombination. Clearly, additional studies are needed to gain a better understanding of this interesting and novel phenomenon.
The vlsE recombination process
Gene conversion clearly represents the major mechanism of vlsE sequence variation, although some have suggested that point mutations may also play a role (74). In terms of the recombination results, vlsE recombination most closely resembles that in the Neisseria gonorrhoeae pilE system and the var system of Plasmodium falciparum, in that the region of DNA replacement is variable in both length and region; in each cases, the sequence changes appear to be restricted to a given area (e.g. the cassette region in vlsE). The vls cassette regions uniformly exhibit high GC content (~50%) and marked GC skew relative to the rest of the B. burgdorferi sensu lato genomes, and the existence of G quadraplex (G4) structure similar to that in neisserial pilE loci has been suggested (75). Beyond these similarities, little is known about the vlsE recombination process. It does not require the extensive array of DNA recombination and repair proteins that are involved in pilE sequence variation (76,77), nor has the involvement of a small RNA (as recently described in N. gonorrhoeae(78)) been implicated. Unlike Neisseria species and most other antigenic variation systems described, vlsE recombination is not detectable during in vitro culture (or during tick infection), indicating that the process activated in some manner by conditions in the mammalian host. However, to date the factor(s) contributing to this activation have not been identified, limiting the study of vlsE recombination to animal infection experiments. Analysis of the recombination process is further hampered by the recalcitrance of the vls locus to genetic manipulation, other than its elimination through upstream insertion of telomere sequences (53).
As a result, the present understanding of vlsE recombination requirements is limited to the aspects listed in Table 2. These points are addressed in greater detail below.
Table 2.
Observations regarding the mechanisms of vlsE recombination
| 1) The cis arrangement of vlsE and the vls silent cassettes appears to be required for recombination to occur. |
| 2) Inverted repeats with the potential of forming stem loop structures are present between vlsE
and the vls cassette arrays in the three strains in which intact vls region sequences have been obtained, but these inverted repeats vary in sequence, location and length. |
| 3) The uniform existence of high GC content and GC skew in vls cassette sequences indicates the involvement of these features in recombination. |
| 4) Among an extensive list of DNA recombination and repair gene products examined to date, only RuvAB (a Holliday junction resolvase) and potentially MutS (77) have a substantial effect on the vlsE recombination process (76,77). |
| 5) The regulatory protein SpoVG binds to a sequence in the vlsE reading frame upstream of the cassette region of B. burgdorferi B31 (69); however, this sequence is not uniformly present in other Lyme disease Borrelia vlsE genes, and the potential effects of SpoVG on vlsE expression and recombination are currently unknown. |
| 6) vlsE expression and recombination are markedly upregulated during infection of mammalian hosts, but the mechanism(s) of these effects are not known. |
Cis arrangement of vlsE and vls silent cassettes
Complete (or nearly complete) vls loci sequences have only been determined for three Borrelia strains: B. burgdorferi B31, B. burgdorferi JD1, and B. garinii Far04. In each of these cases, the reading frames of vlsE and the silent cassettes are arranged in opposite directions, pointing away from one another (Fig. 3). The silent cassette region also begins with a region highly homologous to the 5’ end of the vlsE open reading frame; the promoter, however, is not retained. This region may be a remnant of the initial duplication event that gave rise to the first silent cassette. As described previously, the region at the junction of vlsE and the vlsE cassettes in each strain contains an inverted repeat of 51, 476 and 476 nt respectively, although these differ in location and composition. In the B31 strain, the inverted repeat is in the region between vlsE and the vls silent cassettes, with inclusion of the −35 sequence of the vlsE promoter (Fig. 5A; (29,48); J. K. Howell and S. J. Norris, unpublished data). In the JD1 and Far04 strains, the inverted repeats encompass the 5’ ends of the open reading frames of vlsE and the first silent cassette, ending at the sequence differences within the cassette regions. As mentioned previously, the genes at the far end of lp28-1 in B. burgdorferi B31 are not required for vlsE sequence changes, in that removal of BBF01-BBF19 from lp28-1 did not interfere with vlsE recombination or infectivity of the clone in immunocompetent mice (53).
Based on the limited sampling of intact vls loci, the cis ‘head to head’ arrangement of vlsE and the silent cassettes may be required for vlsE recombination. No sequence changes were detected when mice were infected with B. burgdorferi B31 harboring an intact vlsE gene in a shuttle vector (51), suggesting the importance of the cis arrangement. A possible model is that vlsE “doubles back” onto the silent cassette, promoting close proximity of the donor and recipient regions and thereby strand invasion as part of the gene conversion process. It is conceivable that the inverted repeats could form hairpin loops and in some way facilitate the doubling back. However, one would expect the nearest silent cassettes to recombine more frequently with vlsE in this scenario, but that is not the case (33). If site-directed mutagenesis of the vls locus becomes feasible, then the potential roles of the head to head arrangement and inverted repeat regions in vlsE recombination could be examined more thoroughly.
High G+C content, GC skew, and potential G4 structures
The vlsE cassette region and the vls silent cassettes have a remarkably high G+C content and GC skew compared to the remainder of the B. burgdorferi B31 genome (2,39,75) (Fig. 5B). At 49.7% compared with the 28.2% value for the overall genome, the average percent G+C is 20.5% higher in the silent cassette regions. Although the 3’ end of the vlsE also has a high G+C content, the coding strand GC skew (G-C/G+C) is preserved only in the cassette region. The cassette region GC skew of 0.55 is much higher than that in the leading strand of DNA replication (e.g. 0.18 in the B. burgdorferi B31 chromosome). This pattern is conserved in all of the vls sequences that have been characterized to date, despite significant sequence divergence (as low as 40/49 percent identity/similarity). The preservation of this pattern is therefore likely to be important in the promotion of vlsE recombination.
In N. gonorrhoeae, the segmental recombination of pilS cassette sequences into the pilE pilus protein expression site requires the formation of a G-quadruplex (G4) structure in a region just upstream of the pilE open reading frame (79,80). This conformational change involves the formation of intrachain G-G hydrogen bonds in neighboring G-rich regions to create a complex of four antiparallel segments. It is promoted by the transcription of a small RNA that overlaps with the G4 region (78).
The high GC content and skew in the vls cassette region suggests that similar secondary structure changes may be involved in vlsE recombination. In 2013, Walia and Chaconas (75) explored the possible occurrence of G4 structure in vls sequences. First, they noted that PCR amplification of B. burgdorferi B31 vlsE resulted in an unexpected lower molecular weight band in which the variable region and one of the 17 bp direct repeats were missing. This phenomenon is apparently due to aberrant base pairing between the two identical direct repeats during the amplification process. Thus it is not thought to occur during vlsE recombinations; rather, it indicates unusual base-pairing properties of the 17 bp repeat, which includes a stretch of five G residues. In solution, the 17 bp region of B. burgdorferi B31 (or a shorter 14 bp oligonucleotide that also encompasses the G5 stretch) forms a stable, higher order complex in the presence of high concentrations of KCl, conditions that favor G4 formation (75). The occurrence of hydrogen bonding between the G-rich regions was indicated by protection against methylation by methylsulfoxide. It should be noted, however, that direct repeats at the ends of vlsE cassette regions are variable both between and within strains; their lack of conservation argues against a sequence-specific role of these regions (e.g. recognition by a protein involved in the recombination process). Walia and Chaconas (75) further reported that stretches of 3 or more G’s are found at very high frequencies in the coding strand of the vlsE cassette regions of B. burgdorferi strains B31, N40, and JD1; indeed, this is a common property of all known vls sequences. Whereas all of these findings support the potential formation of G4 or other intrastrand secondary structures in vls sequences, to date no G4 structures have been demonstrated either conceptually (based on sequence) or biophysically using native vls regions. This represents a promising area of future research.
DNA repair and recombination proteins
Dresser et al. (76) and Lin et al. (77) examined a comprehensive list of B. burgdorferi B31 genes encoding predicted DNA repair and recombination proteins for their potential role in vlsE recombination. Mutations in these genes were introduced by either site-directed mutagenesis (76) or random transposon mutagenesis (77). Surprisingly, only a few genes were found to have a marked effect on vlsE sequence changes during mouse infection (and hence also on survival of the B. burgdorferi strains in immunocompetent mice). Both studies identified ruvA and ruvB as important genes in the recombination process, in that the rate of accumulation and diversity of vlsE sequence changes were dramatically reduced in clones in which these genes were mutated (76,77). RuvA and RuvB form a Holliday junction branch migrase. Holliday junctions are mobile junctions between two homologous DNA duplexes, in which strand exchange results in base pairing between homologous regions and formation of a structure with four double-stranded branches. RuvAB branch migrases promote the release and reformation of the base pairs, resulting in migration of the branch point in a zipper-like manner. This branch migration process may be important in the random ‘selection’ of regions of the silent cassette sequences for exchange with the parental vlsE sequences. In most bacterial systems, the endonuclease RuvC is required to resolve the Holliday junction and complete homologous recombination. Borrelia lack an obvious ruvC homologue; it possible that putative prophage nucleases on the multiple cp32 plasmids could fulfill this function (76). In the Lin et al. study (77), mutation of mutS also appeared to have a relatively small effect on the rate of vlsE sequence variation during infection. The other genes examined had no obvious effect on vlsE recombination; this list includes genes encoding the recombination proteins RecA, RecG and RecJ, repair proteins MutL and MutS2, general DNA replication/processing proteins NucA, Mag, Mfd, Nth, SbcC, SbcD, and PriA, plasmid-encoded putative recombinases BBD20 and BBG32, predicted DNA methyltransferase BBE29, and Rep helicase. The finding that the single strand DNA-binding protein RecA is not necessary for vlsE sequence changes was also determined previously by Liveris et al. (81). The apparent lack of involvement of this long list of genes differs from the N. gonorrhoeae pilE system, in which many proteins of the common recombination pathways (including RecA) are required (see (75,80)). This pattern, coupled with the induction of vlsE recombination during mammalian infection, suggests that this process may utilize as yet undiscovered recombinase(s) or other proteins (or RNAs) that become active in the mammalian environment and are specific for the vls sequences.
Potential role of SpoVG
It is likely that proteins or small RNAs that specifically recognize vls sequences are involved in the recombination process. SpoVG was initially described as a cytoplasmic protein that is required for normal sporulation, vegetative cell structure, and cell division in Bacillus subtilis. However, homologs of this protein are found in most eubacteria. In a search for proteins that bind to vlsE DNA sequences, Jutras et al. (69) identified a B. burgdorferi homologue of SpoVG that binds to an 18-bp sequence within the 5’ region of the B31 vlsE gene, just upstream of the cassette region. The predicted SpoVG sequence is highly conserved in all Borrelia species, and also has high similarity with SpoVG sequences in other bacteria. However, the B31 vlsE sequence to which SpoVG binds is not well conserved in other Lyme disease Borrelia outside the B31 ‘clade’. Further study is thus necessary to determine if the observed binding activity is related to a conserved mechanism among B. burgdorferi sensu lato for regulation of vlsE expression or recombination.
Induction of vlsE expression and recombination
As mentioned previously, vlsE recombination has not been detected consistently in in vitro cultures or in infected ticks, yet can be detected as early as three days after the initiation of mammalian infection (30,33,34). The fact that the recombination can be detected as soon as organisms can be cultured, combined with its occurrence in SCID animals, argues against these results being due to immune selection in the infected animals, although it is clear that the immune response accelerates the clearance of the parental clone used for inoculation (33). Thus recombination appears to be induced in the mammalian environment by unknown mechanisms that appear to be unrelated to temperature, CO2 concentration, presence of serum components, or other simple explanations. This situation differs from that of relapsing fever organisms and N. gonorrhoeae, in which recombination of variable major protein genes and pilE occur readily during in vitro culture. vlsE sequence changes have been detected by incubation of B. burgdorferi with mouse or rabbit tissue explants using sensitive PCR procedures, but it is as yet unclear whether these exceedingly rare events are occurring at rates higher than in standard in vitro cultures. As described previously, vlsE transcription and VlsE protein expression is increased during mammalian infection, and it is possible that gene expression facilitates the recombination process. Attempts to mutate vlsE in its native lp28-1 location have thus far been unsuccessful, but would be useful in determining whether transcription and/or translation contribute to the occurrence of cassette region sequence changes, as well as in identifying important cis elements.
Evolution of the vls system
Lyme borreliosis (LB) and relapsing fever (RF) Borrelia are closely related, with nearly complete synteny across the chromosomes and a high degree of homology among the protein products. Plasmid structure and content in these groups differ considerably and likely account in part for the biological differences observed, including the colonization and transmission by Ixodes vs. Ornithodoros ticks and distinct patterns of clinical manifestations. However, recent characterizations of B. miyamotoi and strain LB-2001 genome sequences indicate a blurring of these phenotypic and genotypic lines, in that both organisms are firmly positioned within the RF group genetically but are transmitted by Ixodes ticks (82). Overall, the phylogeny of Borrelia species clearly indicate that they are monophyletic, i.e. arose from a common primordial ancestor distinct from those that gave rise to other major groups of spirochetes, including the Spirochaeta/Treponema/Sphaerochaeta group, the Leptospira/Leptonema/Turneriella group, and Brachyspira.
All Lyme disease organisms characterized to date have vls antigenic variation systems, and all relapsing fever organisms have variable major protein (VMP) antigenic variation systems, which feature a single expression site in which either variable large protein (vlp) or variable small protein (vsp) open reading frames can be inserted. The two systems differ fundamentally, in that vlsE sequence changes arise from replacement with random segments from silent cassettes, whereas most recombinations in the VMP system involve replacement of the expression site gene with a nearly complete open reading frame from one of ~ 40 vlp and vsp promoterless gene copies (83). These recombination events predominantly utilize two sites termed Upstream Homology Sequences (UHS) and Downstream Homology Sequences (DHS) that are shared among nearly all of the ‘donor’ vlp and vsp copies. This feature is remarkable, in that the vlp and vsp sequences are otherwise essentially unrelated to each other.
Despite this diversity in the LB and RF recombination mechanisms, the vlp and vsp RF lipoprotein gene families each have relatives in LB organisms. vlsE has a moderate level of sequence homology with the vlp genes; for example, comparison of the originally described variant vlsE1 with B. hermsii vlp17 reveals nucleotide sequence identity of 58.8% and deduced amino acid sequence identity and similarity of 37.4% and 58.8%, respectively (2). In addition to the multiple vsp copies, RF organisms also contain a vsp-related ‘variable tick protein’ (vtp) gene that does not participate in vlp/vsp recombination but is preferentially expressed in the tick environment. Interestingly, LB organisms have a prominent vsp homolog encoding outer surface protein C (ospC), which is predominantly expressed during the transmission of Borrelia from the tick to the mammal and in the mammal during the early stages of infection. Therefore the vlp and vsp gene families each must have evolved from single primordial vlp and vsp genes in the common Borrelia ancestor and then undergone duplication and remarkable sequence and functional diversification during the development of the separate LB and RF Borrelia lineages. This scenario is shown diagrammatically in Fig. 6.
Fig. 6.
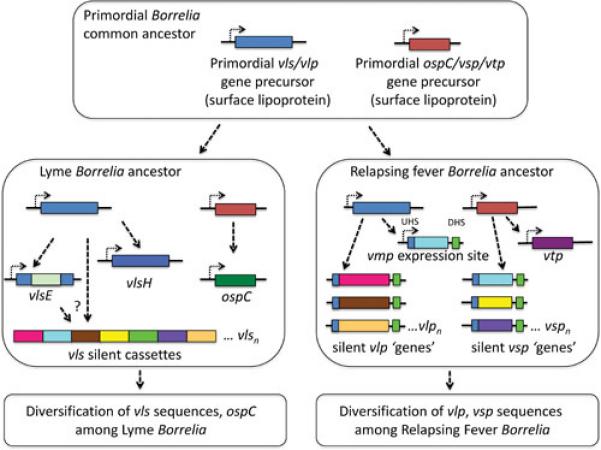
Model for the evolution of vlp- and vsp-related genes in Borrelia. It is hypothesized that a common ancestor of LB and RF organisms contained single copies of primordial vlp and vsp homologues. Following the divergence of LB and RF groups, these precursor genes duplicated and developed into the vls system and ospC in a primordial LB organism, and into the VMP system and vtp in a primordial RF ancestor. Each of these antigenic variation systems and related surface proteins continued to evolve and diverge under the pressure of immune selection.
Is there any evidence of a common vlp precursor? Surprisingly, there is a paralogous gene in LB Borrelia that may be a remnant of this ancient ancestral gene. In B. burgdorferi B31, it is a fragmented pseudogene. However, in several strains, including B. burgdorferi JD1, the reading frame is intact; some strains, including B. burgdorferi WI91-23 and B. garinii PBr, have two intact copies. These genes, located on 28 kb, 38 kb, or 36 kb plasmids, often have been mistakenly annotated as vlsE in genomic sequences; it is suggested that they be referred to as vls homologues or vlsH. In JD1, the lp28-6 encoded gene BbuJD1_Z01 encodes a 40.6 kDa lipoprotein (e.g. BbuJD1_Z01) that is 28% identical and 43% similar to B31 VlsE1. One conserved region is contained in Invariant Region 6 (IR6, also called C6), a 25 aa region that forms an alpha helix embedded in the membrane distal region of VlsE (46). This region of VlsE is also highly immunogenic and is used as a diagnostic antigen in immunologic tests for Lyme disease. Several strains, including B. burgdorferi B31, N40, 29805, and 64b, have an identical frameshift following the 56th codon; this occurrence may indicate ancestral inheritance of this damaged gene from a common progenitor. The vlsH genes and their products have not been studied; it would be of interest to examine their expression and other properties.
Both OspC and VlsE are surface-localized lipoproteins that are thought to be expressed sequentially during mammalian infection, with OspC being the predominant surface protein in the first days or weeks of infection followed by replacement by VlsE (84). Likely due to the constant assault of the host antibody response, these proteins are the most heterogeneous of all LB Borrelia polypeptides. The natural hosts of LB Borrelia (such as the white-footed mouse Peromyscus leucopus) are continually exposed to different strains of Borrelia delivered through multiple tick bites. Thus antibodies against OspC and the ‘framework’ portions of VlsE from prior infections would select for organisms that expressed different epitopes, promoting the fixation of mutations that change the antigenic structure without destroying the proteins’ structural integrity. This constant immune pressure thus resulted in strains expressing increasingly heterogeneous versions of this surface proteins. In the case of VlsE, this selection not only drove the development of the vls antigenic variation system, but also accelerated the evolution of diverse framework regions of this protein.
Conclusions
The vls system represents one of the most elaborate and elegant antigenic variation systems in bacterial pathogens. Its consistent presence in LB organisms indicates its importance in the tenuous survival of these spirochetes during their tightrope act of continual transmission between mammalian and arthropod hosts. The intense immune selection not only drove the parallel but divergent evolution of two different antigenic variation systems in RF and LB Borrelia, but also has promoted VlsE framework divergence within different LB species and strains. In addition, the patterns of expression of OspC in LB spirochetes and Vtp in RF organisms could be considered forms of phase variation to evade the immune response; OspC heterogeneity provides yet another example of the power of antibody reactions in the selection of antigenic variants. Together, these elaborate adaptations promote survival of Lyme borreliosis organisms for months to years in mammalian hosts, thus assuring the passage of future generations to arthropods and back again to sylvan mammals and, as accidental hosts, humans.
Acknowledgment
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01 AI037277. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.van der Woude MW, Bäumler AJ. Phase and antigenic variation in bacteria. Clin Microbiol Rev. 2004;17:581–611. doi: 10.1128/CMR.17.3.581-611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang JR, Hardham JM, Barbour AG, Norris SJ. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell. 1997;89:275–285. doi: 10.1016/s0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 3.Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanek G, Strle F. Lyme disease--European perspective. Infect Dis Clinics North Amer. 2008;22:327–339. doi: 10.1016/j.idc.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Müllegger RR, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355–368. doi: 10.2165/0128071-200809060-00002. [DOI] [PubMed] [Google Scholar]
- 6.Stanek G, Reiter M. The expanding Lyme Borrelia complex—clinical significance of genomic species? Clin Microbiol Infect. 2011;17:487–493. doi: 10.1111/j.1469-0691.2011.03492.x. [DOI] [PubMed] [Google Scholar]
- 7.Norris SJ, Coburn J, Leong JM, Hu LT, Höök M. Pathobiology of Lyme disease Borrelia. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular and Cellular Biology. Caister Academic Press; Hethersett, Norwich, UK: pp. 299–331. [Google Scholar]
- 8.Skare JT, Carroll JA, Yang XF, Samuels DS, Akins DR. Gene regulation, transcriptomics, and proteomics. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular and Cellular Biology. Caister Academic Press; Hethersett, Norwich, UK: pp. 67–102. [Google Scholar]
- 9.Radolf JD, Lukehart SA. Pathogenic Treponema: Molecular and Cellular Biology. Caister Academic Press; Hethersett, Norwich, UK: 2006. [Google Scholar]
- 10.Kraiczy P, Stevenson B. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: Structure, function and regulation of gene expression. Ticks and Tick-borne Diseases. 2013;4:26–34. doi: 10.1016/j.ttbdis.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Taeye SW, Kreuk L, van Dam AP, Hovius JW, Schuijt TJ. Complement evasion by Borrelia burgdorferi: it takes three to tango. Trends Parasitol. 2013;29:119–128. doi: 10.1016/j.pt.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Tilly K, Bestor A, Dulebohn DP, Rosa PA. OspC-independent infection and dissemination by host-adapted Borrelia burgdorferi. Infect Immun. 2009;77:2672–2682. doi: 10.1128/IAI.01193-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, Byram R, Dorward D, Vanraden MJ, Stewart P, Rosa P. Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect Immun. 2006;74:3554–3564. doi: 10.1128/IAI.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease, a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- 15.Labandeira-Rey M, Baker E, Skare J. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infect Immun. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbour AG. Plasmid analysis of Borrelia burgdorferi, the Lyme disease agent. J Clin Microbiol. 1988;26:475–478. doi: 10.1128/jcm.26.3.475-478.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson RC, Marek N, Kodner C. Infection of Syrian hamsters with Lyme disease spirochetes. J Clin Microbiol. 1984;20:1099–1101. doi: 10.1128/jcm.20.6.1099-1101.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norris SJ, Howell JK, Garza SA, Ferdows MS, Barbour AG. High- and low-infectivity phenotypes of clonal populations of in vitro-cultured Borrelia burgdorferi. Infect Immun. 1995;63:2206–2212. doi: 10.1128/iai.63.6.2206-2212.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Johnson RC. Analysis and comparison of plasmid profiles of Borrelia burgdorferi sensu lato strains. J Clin Microbiol. 1995;33:2679–2685. doi: 10.1128/jcm.33.10.2679-2685.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Kodner C, Coleman L, Johnson RC. Correlation of plasmids with infectivity of Borrelia burgdorferi sensu stricto type strain B31. Infect Immun. 1996;64:3870–3876. doi: 10.1128/iai.64.9.3870-3876.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Labandeira-Rey M, Seshu J, Skare JT. The absence of linear plasmid 25 or 28-1 of Borrelia burgdorferi dramatically alters the kinetics of experimental infection via distinct mechanisms. Infect Immun. 2003;71:4608–4613. doi: 10.1128/IAI.71.8.4608-4613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grimm D, Eggers CH, Caimano MJ, Tilly K, Stewart PE, Elias AF, Radolf JD, Rosa PA. Experimental assessment of the roles of linear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infectious cycle. Infect Immun. 2004;72:5938–5946. doi: 10.1128/IAI.72.10.5938-5946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purser JE, Norris SJ. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc Natl Acad Sci USA. 2000;97:13865–13870. doi: 10.1073/pnas.97.25.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- 25.Casjens S, Palmer N, van Vugt R, Huang WM, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, Haft D, Hickey E, Gwinn M, White O, Fraser CM. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol Microbiol. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- 26.Hinnebusch J, Bergstrom S, Barbour AG. Cloning and sequence analysis of linear plasmid telomeres of the bacterium Borrelia burgdorferi. Mol Microbiol. 1990;4:811–820. doi: 10.1111/j.1365-2958.1990.tb00651.x. [DOI] [PubMed] [Google Scholar]
- 27.Hinnebusch J, Barbour AG. Linear plasmids of Borrelia burgdorferi have a telomeric structure and sequence similar to those of a eukaryotic virus. J Bacteriol. 1991;173:7233–7239. doi: 10.1128/jb.173.22.7233-7239.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobryn K, Chaconas G. ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol Cell. 2002;9:195–201. doi: 10.1016/s1097-2765(01)00433-6. [DOI] [PubMed] [Google Scholar]
- 29.Hudson CR, Frye JG, Quinn FD, Gherardini FC. Increased expression of Borrelia burgdorferi vlsE in response to human endothelial cell membranes. Mol Microbiol. 2001;41:229–239. doi: 10.1046/j.1365-2958.2001.02511.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang JR, Norris SJ. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun. 1998;66:3689–3697. doi: 10.1128/iai.66.8.3689-3697.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Indest KJ, Howell JK, Jacobs MB, Scholl-Meeker D, Norris SJ, Philipp MT. Analysis of Borrelia burgdorferi vlsE gene expression and recombination in the tick vector. Infect Immun. 2001;69:7083–7090. doi: 10.1128/IAI.69.11.7083-7090.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nosbisch LK, de Silva AM. Lack of detectable variation at Borrelia burgdorferi vlsE locus in ticks. J Med Entomol. 2007;44:168–170. doi: 10.1603/0022-2585(2007)44[168:lodvab]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 33.Coutte L, Botkin DJ, Gao L, Norris SJ. Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Pathog. 2009;5:e1000293. doi: 10.1371/journal.ppat.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Embers ME, Liang FT, Howell JK, Jacobs MB, Purcell JE, Norris SJ, Johnson BJ, Philipp MT. Antigenicity and recombination of VlsE, the antigenic variation protein of Borrelia burgdorferi, in rabbits, a host putatively resistant to long-term infection with this spirochete. FEMS Immunol Med Microbiol. 2007;50:421–429. doi: 10.1111/j.1574-695X.2007.00276.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhang JR, Norris SJ. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun. 1998;66:3698–3704. doi: 10.1128/iai.66.8.3698-3704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawabata H, Myouga F, Inagaki Y, Murai N, Watanabe H. Genetic and immunological analyses of Vls (VMP-like sequences) of Borrelia burgdorferi. Microb Pathog. 1998;24:155–166. doi: 10.1006/mpat.1997.0183. [DOI] [PubMed] [Google Scholar]
- 37.Liang FT, Alvarez AL, Gu Y, Nowling JM, Ramamoorthy R, Philipp MT. An immunodominant conserved region within the variable domain of VlsE, the variable surface antigen of Borrelia burgdorferi. J Immunol. 1999;163:5566–5573. [PubMed] [Google Scholar]
- 38.Wang G, van Dam AP, Dankert J. Analysis of a VMP-like sequence (vls) locus in Borrelia garinii and Vls homologues among four Borrelia burgdorferi sensu lato species. FEMS Microbiol Lett. 2001;199:39–45. doi: 10.1111/j.1574-6968.2001.tb10648.x. [DOI] [PubMed] [Google Scholar]
- 39.Wang D, Botkin DJ, Norris SJ. Characterization of the vls antigenic variation loci of the Lyme disease spirochaetes Borrelia garinii Ip90 and Borrelia afzelii ACAI. Mol Microbiol. 2003;47:1407–1417. doi: 10.1046/j.1365-2958.2003.03386.x. [DOI] [PubMed] [Google Scholar]
- 40.Casjens SR, Fraser-Liggett CM, Mongodin EF, Qiu WG, Dunn JJ, Luft BJ, Schutzer SE. Whole genome sequence of an unusual Borrelia burgdorferi sensu lato isolate. J Bacteriol. 2011;193:1489–1490. doi: 10.1128/JB.01521-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casjens SR, Mongodin EF, Qiu WG, Dunn JJ, Luft BJ, Fraser-Liggett CM, Schutzer SE. Whole-genome sequences of two Borrelia afzelii and two Borrelia garinii Lyme disease agent isolates. J Bacteriol. 2011;193:6995–6996. doi: 10.1128/JB.05951-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casjens SR, Mongodin EF, Qiu WG, Luft BJ, Schutzer SE, Gilcrease EB, Huang WM, Vujadinovic M, Aron JK, Vargas LC, Freeman S, Radune D, Weidman JF, Dimitrov GI, Khouri HM, Sosa JE, Halpin RA, Dunn JJ, Fraser CM. Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS One. 2012;7:e33280. doi: 10.1371/journal.pone.0033280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mongodin EF, Casjens SR, Bruno JF, Xu Y, Drabek EF, Riley DR, Cantarel BL, Pagan PE, Hernandez YA, Vargas LC, Dunn JJ, Schutzer SE, Fraser CM, Qiu WG, Luft BJ. Inter- and intra-specific pan-genomes of Borrelia burgdorferi sensu lato: genome stability and adaptive radiation. BMC Genomics. 2013;14:693. doi: 10.1186/1471-2164-14-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schutzer SE, Fraser-Liggett CM, Casjens SR, Qiu WG, Dunn JJ, Mongodin EF, Luft BJ. Whole-genome sequences of thirteen isolates of Borrelia burgdorferi. J Bacteriol. 2011;193:1018–1020. doi: 10.1128/JB.01158-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schutzer SE, Fraser-Liggett CM, Qiu WG, Kraiczy P, Mongodin EF, Dunn JJ, Luft BJ, Casjens SR. Whole-genome sequences of Borrelia bissettii, Borrelia valaisiana, and Borrelia spielmanii. J Bacteriol. 2012;194:545–546. doi: 10.1128/JB.06263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eicken C, Sharma V, Klabunde T, Lawrenz MB, Hardham JM, Norris SJ, Sacchettini JC. Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia burgdorferi. J Biol Chem. 2002;277:21691–21696. doi: 10.1074/jbc.M201547200. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Lin T, Botkin DJ, McCrum E, Winkler H, Norris SJ. Intact flagellar motor of Borrelia burgdorferi revealed by cryo-electron tomography: evidence for stator ring curvature and rotor/C-ring assembly flexion. J Bacteriol. 2009;191:5026–5036. doi: 10.1128/JB.00340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bykowski T, Babb K, von Lackum K, Riley SP, Norris SJ, Stevenson B. Transcriptional regulation of the Borrelia burgdorferi antigenically variable VlsE surface protein. J Bacteriol. 2006;188:4879–4889. doi: 10.1128/JB.00229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crother TR, Champion CI, Whitelegge JP, Aguilera R, Wu XY, Blanco DR, Miller JN, Lovett MA. Temporal analysis of the antigenic composition of Borrelia burgdorferi during infection in rabbit skin. Infect Immun. 2004;72:5063–5072. doi: 10.1128/IAI.72.9.5063-5072.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang FT, Nowling JM, Philipp MT. Cryptic and exposed invariable regions of VlsE, the variable surface antigen of Borrelia burgdorferi sl. J Bacteriol. 2000;182:3597–3601. doi: 10.1128/jb.182.12.3597-3601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lawrenz MB, Wooten RM, Norris SJ. Effects of vlsE complementation on the infectivity of Borrelia burgdorferi lacking the linear plasmid lp28-1. Infect Immun. 2004;72:6577–6585. doi: 10.1128/IAI.72.11.6577-6585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grimm D, Tilly K, Bueschel DM, Fisher MA, Policastro PF, Gherardini FC, Schwan TG, Rosa PA. Defining plasmids required by Borrelia burgdorferi for colonization of tick vector Ixodes scapularis (Acari: Ixodidae) J Med Entomol. 2005;42:676–684. doi: 10.1093/jmedent/42.4.676. [DOI] [PubMed] [Google Scholar]
- 53.Bankhead T, Chaconas G. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol Microbiol. 2007;65:1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x. [DOI] [PubMed] [Google Scholar]
- 54.Lawrenz MB, Hardham JM, Owens RT, Nowakowski J, Steere AC, Wormser GP, Norris SJ. Human antibody responses to VlsE antigenic variation protein of Borrelia burgdorferi. J Clin Microbiol. 1999;37:3997–4004. doi: 10.1128/jcm.37.12.3997-4004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bacon RM, Biggerstaff BJ, Schriefer ME, Gilmore RD, Jr., Philipp MT, Steere AC, Wormser GP, Marques AR, Johnson BJ. Serodiagnosis of Lyme disease by kinetic enzyme-linked immunosorbent assay using recombinant VlsE1 or peptide antigens of Borrelia burgdorferi compared with 2-tiered testing using whole-cell lysates. J Infect Dis. 2003;187:1187–1199. doi: 10.1086/374395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marangoni A, Moroni A, Accardo S, Cevenini R. Borrelia burgdorferi VlsE antigen for the serological diagnosis of Lyme borreliosis. Eur J Clin Microbiol Infect Dis. 2008;27:349–354. doi: 10.1007/s10096-007-0445-7. [DOI] [PubMed] [Google Scholar]
- 57.Goettner G, Schulte-Spechtel U, Hillermann R, Liegl G, Wilske B, Fingerle V. Improvement of Lyme borreliosis serodiagnosis by a newly developed recombinant immunoglobulin G (IgG) and IgM line immunoblot assay and addition of VlsE and DbpA homologues. J Clin Microbiol. 2005;43:3602–3609. doi: 10.1128/JCM.43.8.3602-3609.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ledue TB, Collins MF, Young J, Schriefer ME. Evaluation of the LIAISON(R) Borrelia burgdorferi Test, a Recombinant VlsE-based Chemiluminescence Immunoassay for the Diagnosis of Lyme Disease. Clin Vaccine Immunol CVI.00195-00108. 2008 doi: 10.1128/CVI.00195-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy SA, O'Connor TP, Hanscom JL, Shields P. Evaluation of a canine C6 ELISA Lyme disease test for the determination of the infection status of cats naturally exposed to Borrelia burgdorferi. Vet Ther. 2003;4:172–177. [PubMed] [Google Scholar]
- 60.Magnarelli LA, Stafford KC, 3rd, Ijdo JW, Fikrig E. Antibodies to whole-cell or recombinant antigens of Borrelia burgdorferi, Anaplasma phagocytophilum, and Babesia microti in white-footed mice. J Wildl Dis. 2006;42:732–738. doi: 10.7589/0090-3558-42.4.732. [DOI] [PubMed] [Google Scholar]
- 61.Liang FT, Philipp MT. Analysis of antibody response to invariable regions of VlsE, the variable surface antigen of Borrelia burgdorferi. Infect Immun. 1999;67:6702–6706. doi: 10.1128/iai.67.12.6702-6706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liang FT, Philipp MT. Epitope mapping of the immunodominant invariable region of Borrelia burgdorferi VlsE in three host species. Infect Immun. 2000;68:2349–2352. doi: 10.1128/iai.68.4.2349-2352.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang FT, Bowers LC, Philipp MT. C-terminal invariable domain of VlsE is immunodominant but its antigenicity is scarcely conserved among strains of Lyme disease spirochetes. Infect Immun. 2001;69:3224–3231. doi: 10.1128/IAI.69.5.3224-3231.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McDowell JV, Sung SY, Hu LT, Marconi RT. Evidence that the variable regions of the central domain of VlsE are antigenic during infection with Lyme disease spirochetes. Infect Immun. 2002;70:4196–4203. doi: 10.1128/IAI.70.8.4196-4203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang F, Jacobs M, Philipp M. C-terminal invariable domain of VlsE may not serve as target for protective immune response against Borrelia burgdorferi. Infect Immun. 2001;69:1337–1343. doi: 10.1128/IAI.69.3.1337-1343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baum E, Hue F, Barbour AG. Experimental infections of the reservoir species Peromyscus leucopus with diverse strains of Borrelia burgdorferi, a Lyme disease agent. MBio. 2012;3:e00434–00412. doi: 10.1128/mBio.00434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koči J, Derdakova M, Peterkova K, Kazimirova M, Selyemova D, Labuda M. Borrelia afzelii gene expression in Ixodes ricinus (Acari: Ixodidae) ticks. Vector Borne Zoonotic Dis. 2006;6:296–304. doi: 10.1089/vbz.2006.6.296. [DOI] [PubMed] [Google Scholar]
- 68.Piesman J, Zeidner NS, Schneider BS. Dynamic changes in Borrelia burgdorferi populations in Ixodes scapularis (Acari: Ixodidae) during transmission: studies at the mRNA level. Vector Borne Zoonotic Dis. 2003;3:125–132. doi: 10.1089/153036603768395825. [DOI] [PubMed] [Google Scholar]
- 69.Jutras BL, Chenail AM, Rowland CL, Carroll D, Miller MC, Bykowski T, Stevenson B. Eubacterial SpoVG homologs constitute a new family of site-specific DNA-binding proteins. PLoS ONE. 2013;8:e66683. doi: 10.1371/journal.pone.0066683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crother TR, Champion CI, Wu XY, Blanco DR, Miller JN, Lovett MA. Antigenic composition of Borrelia burgdorferi during infection of SCID mice. Infect Immun. 2003;71:3419–3428. doi: 10.1128/IAI.71.6.3419-3428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liang FT, Nelson FK, Fikrig E. Molecular adaptation of Borrelia burgdorferi in the murine host. J Exp Med. 2002;196:275–280. doi: 10.1084/jem.20020770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liang FT, Yan J, Mbow ML, Sviat SL, Gilmore RD, Mamula M, Fikrig E. Borrelia burgdorferi changes its surface antigenic expression in response to host immune responses. Infect Immun. 2004;72:5759–5767. doi: 10.1128/IAI.72.10.5759-5767.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anguita J, Thomas V, Samanta S, Persinski R, Hernanz C, Barthold SW, Fikrig E. Borrelia burgdorferi-induced inflammation facilitates spirochete adaptation and variable major protein-like sequence locus recombination. J Immunol. 2001;167:3383–3390. doi: 10.4049/jimmunol.167.6.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sung SY, McDowell JV, Marconi RT. Evidence for the contribution of point mutations to vlsE variation and for apparent constraints on the net accumulation of sequence changes in vlsE during infection with Lyme disease spirochetes. J Bacteriol. 2001;183:5855–5861. doi: 10.1128/JB.183.20.5855-5861.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walia R, Chaconas G. Suggested role for G4 DNA in recombinational switching at the antigenic variation locus of the Lyme disease spirochete. PLoS One. 2013;8:e57792. doi: 10.1371/journal.pone.0057792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dresser AR, Hardy PO, Chaconas G. Investigation of the genes involved in antigenic switching at the vlsE locus in Borrelia burgdorferi: an essential role for the RuvAB branch migrase. PLoS Pathog. 2009;5:e1000680. doi: 10.1371/journal.ppat.1000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lin T, Gao L, Edmondson DG, Jacobs MB, Philipp MT, Norris SJ. Central role of the Holliday junction helicase RuvAB in vlsE recombination and infectivity of Borrelia burgdorferi. PLoS Pathog. 2009;5:e1000679. doi: 10.1371/journal.ppat.1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cahoon LA, Seifert HS. Transcription of a cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog. 2013;9:e1003074. doi: 10.1371/journal.ppat.1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cahoon LA, Seifert HS. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science. 2009;325:764–767. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cahoon LA, Seifert HS. Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Mol Microbiol. 2011;81:1136–1143. doi: 10.1111/j.1365-2958.2011.07773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liveris D, Mulay V, Sandigursky S, Schwartz I. Borrelia burgdorferi vlsE antigenic variation is not mediated by RecA. Infect Immun. 2008;76:4009–4018. doi: 10.1128/IAI.00027-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barbour AG. Phylogeny of a relapsing fever Borrelia species transmitted by the hard tick Ixodes scapularis. Infect Genet Evol. 2014 doi: 10.1016/j.meegid.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dai Q, Restrepo BI, Porcella SF, Raffel SJ, Schwan TG, Barbour AG. Antigenic variation by Borrelia hermsii occurs through recombination between extragenic repetitive elements on linear plasmids. Mol Microbiol. 2006;60:1329–1343. doi: 10.1111/j.1365-2958.2006.05177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tilly K, Bestor A, Rosa PA. Lipoprotein succession in Borrelia burgdorferi: similar but distinct roles for OspC and VlsE at different stages of mammalian infection. Mol Microbiol. 2013;89:216–227. doi: 10.1111/mmi.12271. [DOI] [PMC free article] [PubMed] [Google Scholar]