

Abstract
Mechanotransduction in bone is fundamental to proper skeletal development. Deficiencies in signaling mechanisms that transduce physical forces to effector cells can have severe consequences for skeletal integrity. Therefore, a solid understanding of the cellular and molecular components of mechanotransduction is crucial for correcting skeletal modeling and remodeling errors and designing effective therapies. In recent years, progress has been made on many fronts regarding our understanding of bone cell mechanotransduction, including subcellular localization of mechanosensitive components in bone cells, the discovery of mechanosensitive G-protein-coupled receptors, identification of new ion channels and larger pores (eg, hemichannels) involved in physical signal transduction, and cell adhesion proteins, among others. These and other recent mechanisms are reviewed to provide a synthesis of recent experimental findings, in the larger context of whole bone adaptation.
Keywords: Mechanical loading, Stress, Strain, Osteocytes, Osteoblasts, Mechanotransduction, Biological factors, Mechanical signals, Bone adaptation
Introduction
Mechanical loading of the skeleton through exercise and/or physical activity presents the resident bone cell populations with a potent stimulus for improving bone health and fracture susceptibility. Improvements in skeletal health associated with mechanical loading come largely from load-induced increases in bone formation, reductions in bone resorption, and changes in the physical properties of the bone matrix itself. Mechanical loading of bone through voluntary exercise is perhaps the most cost-effective method for improving fracture susceptibility, with the least degree of unwanted side effects. Despite the numerous and well-known positive side effects that accompany vigorous exercise (improvements in cardiovascular health, immune function, glucose metabolism, to name a few), isolation of the specific cellular and molecular mechanisms of mechano-transduction in bone remains a heavily pursued area of investigation in order to someday selectively mimic the effects of loading on bone pharmacologically, while leaving other physiologic systems undisturbed.
The ability to harness the anabolic and anti-catabolic potential of the mechanotransduction pathways in bone requires a detailed understanding of signaling cascades that are activated, the receptors that activate them, and the downstream changes in gene expression and protein translation/degradation that are manifest. These processes are not completely understood, but significant progress is being made in unraveling the complexity of mechanotransduction in bone. The goal of this review is to provide some discussion of recent findings related to the cellular mechanisms of bone cell mechanotransduction. These recent discoveries warrant particular discussion and synthesis because they have already begun to reshape the type of questions and hypotheses in current bone mechanobiology experiments. The scope of this review is therefore limited, in most areas of research, to developments that have been communicated in the past several years. For a more general discussion of mechanotransduction pathways and processes that include a more historical and fundamental treatment of the subject, the reader is referred to several excellent reviews [1–3].
The Osteocyte as the Mechanosensor: The Long Arm(s) of the Law
Many decades ago, it was postulated that the osteocytes were the best candidate for a sensor cell type in bone [4]. They are ideally situated to sense small changes in the mechanical environment of all the skeletal elements, owing to their sheer number, pervasive presence in all parts of the skeletal elements, and their connectivity and ability to communicate with one another (Fig. 1). Several recent studies have supported, with convincing data, this view of the osteocyte in mechanotransduction. While earlier studies demonstrated that osteocytes are required for mechanotransduction to occur [5], more recent studies have focused on how osteocytes might differ from their precursor cells—the osteoblasts—in terms of sensitivity to mechanical stimulation. For example, Kamel et al. [6] investigated whether the osteocytic cell line MLO-Y4 is more sensitive to fluid flow than two other osteoblast models—2T3 osteoblastic cells and primary neonatal calvarial osteoblasts. The osteocytic cultures released significant amounts of prostaglandin E2 (PGE2) into the media during fluid flow stimulation at very low levels of shear stress (~2–8 dyn/cm2), whereas the PGE2 response in the 2T3 cells was undetectable at similar levels of shear stress. Increasing the shear stress to a level that did elicit a response from the osteoblastic models (16–24 dyn/cm2) revealed an eightfold greater response in the osteocytes. Moreover, translocation of β-catenin to the nucleus, a marker of Wnt signaling and mechanotransduction in bone cells [7], was observed after low levels of shear stress in the osteocyte line but not in the osteoblast lines. A similar conclusion regarding the sensitivity of osteocytic versus osteoblastic cell culture models was reached recently by Lu et al. [8]. They cultured osteoblastic MC3T3 and osteocytic MLO-Y4 cells on a very cleverly engineered substrate that permitted cell adherence and only in specified locations. These micropatterned substrates allowed for networks of cells to be developed, with cell bodies separate from one another. When exposed to low-velocity fluid flow (generating 5 dyn/cm2), the MLO-Y4 cells exhibited significantly greater Ca2+ responses, as measured using the Fura-2 system, than was recorded for the MC3T3 cells. As shear stress was increased, the two cell types exhibited less difference in responsiveness. Interestingly, the MC3T3 cells exhibited an enhanced ATP response compared to MLO-Y4 cells in response to fluid shear, so it appears that the osteocyte lines might not be more sensitive to shear for all outcome measures.
Fig. 1.
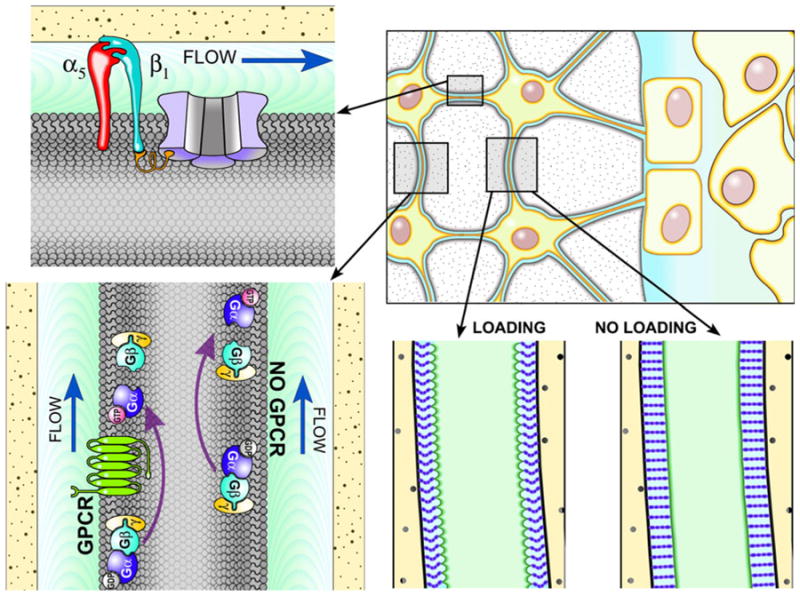
Top right, Osteocytes are likely the mechanosensory cell type in bone. Osteocytes reside in a mineralized matrix, and have long cytoplasmic processes that allow communication with one another and with cells on the bone surfaces. Bottom right, As bone is loaded in bending, extracellular fluid (light blue) movement occurs between the osteocyte cell body/processes (light green) and the lacuno-canalicular walls (pale yellow). The fluid movement in those spaces creates drag forces that pull on tethering structures (purple bands extending from the cell process to the canalicular wall) of the glyco-calyx, which suspend the osteocyte from the bony walls. Those drag forces create radial strains on the cell processes, and induce mechano-sensory proteins to signal. Bottom left, A potential candidate for a mechanosensor in bone is G-protein signaling, which can become activated (GTP-bound) by ligand-independent mechanical perturbation of the G-protein-coupled receptor (GPCR, shown in green), or by GPCR-independent mechanical perturbation of the membrane. Top left, The α5β1 integrin was recently shown to directly promote opening of Cx43 hemichannels (shown in light purple) in the membrane, which allow mechanical signaling molecules to act on key receptors
Beyond the recent studies aimed at determining whether osteocytes might be more sensitive to mechanical stimulation than osteoblasts, an intriguing study looked at whether the osteocyte’s cell processes or the cell body is primarily responsible for mechanical signal reception. Burra et al. [9] developed a transwell filter system for culturing MLO-Y4 and primary osteocytes that allowed them to mechanically stimulate the osteocyte cell body (which resided on one side of the membrane) or the cell processes (which extended through to the opposing side of the membrane) via dropping a bolus of liquid from a specified height. When they stimulated the cell-body side of the membrane, hemichannels on the cell body opened (as monitored microscopically via entrance of an extracellular dye into the cell) but those on the cell processes did not. When they flipped the membranes over and dropped media onto the cell process side of the membrane, the same result was found—opening of hemichannels on the cell body but not the cell processes. Adachi et al. [10] took a different approach to probe the osteocyte for local differences in sensitivity to mechanical stimulation. They cultured primary chick osteocytes with Phex antibody-coated microparticles, which facilitated adherence of the particles to the osteocyte membranes. The microparticles could then be used to apply mechanical stimulation locally by manipulating them with a glass microneedle, and the calcium response was measured. Generation of a Ca2+ transient required more than twice as much deformation when applied at the cell body versus application to the cell process. These results, in conjunction with recent computer modeling simulations [11], suggest that the osteocyte cell process might be the site of activity for mechanical signal reception in the osteocyte.
Mechanical Signal Transduction: Connexing the Dots
One of the hallmarks of the osteocyte network, which makes it such an attractive candidate for the mechanosensory apparatus in bone, is its ability to rapidly communicate and transmit cell-to-cell information. That communication is facilitated by the presence of gap junctions where osteocyte processes from neighboring cells touch one another. Gap junctions in bone are formed by opposing connexons, and each connexon is formed by an arrangement of six connexin molecules. In bone cells, connexin 43 (Cx43) is highly expressed compared to the other known connexins. Because efficient communication among osteocytes is a prerequisite for mechanotransduction to work, a number of investigators have looked into the role that Cx43 might play in mechano-transduction. It should be pointed out that connexons can exist unopposed from a neighboring cell, in which case the connexon is considered to be a hemichannel. In fact, hemi-channels have been proposed as the mechanism by which bone cells release PGE2 upon mechanical stimulation (Fig. 1). Siller-Jackson et al. [12] showed that PGE2 release from fluid-sheared MLO-Y4 cells could be nearly completely inhibited when the cells were pretreated with a blocking antibody specific for Cx43 hemichannels. Further, mechanically induced opening of the Cx43 hemichannel appears to be controlled by physical perturbation of the α5β1 integrin through a direct mechanical linkage [13••]. This view is not without controversy, as Li et al. [14] have reported that another pore-forming protein—the ATP-gated P2X7 receptor—is likely the mechanism by which PGE2 might be released from the cell in response to mechanical stimulation.
In vivo models of mechanotransduction have shed additional light on the role of Cx43 in bone mechanobiology. Three independent studies have recently been completed that employed the same floxed loss-of-function Cx43 mouse model in conjunction with skeletal loading. The loxP sites were cut using different Cre drivers, including Dermo1/Twist2-Cre [15], Osteocalcin-Cre [16•], and Dmp1-Cre [17], but all three studies revealed a gain of mechano-responsiveness on the periosteal surface when Cx43 is deleted from the bone. It is difficult to rationalize why disruption of an important communication apparatus among osteocytes would have positive effects on mechanotransduction. Nevertheless, while the function of Cx43 in load-induced periosteal bone gain appears to be consistent across these three studies, the role of Cx43 more broadly in mechanotransduction is still unclear as two studies have shown that Dermo1/Twist2-Cre [15] and Col2.3-Cre [18] -mediated deletion of the allele results in a loss of mechano-responsiveness on the endocortical surface. Moreover, although disruption of Cx43 resulted in reduced load-induced bone gain on the endocortex, it also resulted in osteoprotective effects from disuse on the same surface [19]. Clearly, the complexity of these outcomes highlights the realization that there is a great deal yet to learn about how Cx43 regulates mechanobiology in bone.
Developments in Physical Signal Reception: Making Sense of the Sensors
The mechanoreceptor in bone has long eluded skeletal biologists. The protein (or proteins) that senses the mechanical perturbation in the local environment, and transforms that physical signal into a biochemical cascade, has been a mystery for many years. Mechanosensors have historically fallen into one of three main categories: ion channels, G-protein-coupled receptors (GPCRs), and cytoskeletal/integrin complexes. In the realm of ion channels, several new reports have expanded our view of mechanoreception. The first is from the transient receptor potential (Trp) family of channels. Specifically, TrpV4, a receptor known to be sensitive to mechanical perturbation (most notably, cell swelling) and osmolarity in other tissues, was recently shown to modulate the response to mechanical disuse in mice. Mizoguchi et al. [20] subjected TrpV4 knockout and wild-type (WT) mice to tail suspension for 2 weeks and measured changes in bone mass and formation rates. Tail-suspended TrpV4 knockout mice failed to lose bone and did not exhibit reduced bone formation rates as was observed in tail suspended WT mice. The TrpV4 channel has been shown to be sensitive to shear stress in other cell types [21], thus its role in bone as a mechanosensor is worth further investigation.
Another Trp channel that has received significant attention in bone cell mechanotransduction is TrpP1, also known as Pkd1. Several years ago, it was convincingly demonstrated that that Pkd1 and Pkd2 regulate mechanotransduction in kidney epithelial cells. Pkd1 and Pkd2 reside largely on the primary cilium—a nonmotile ~250-nm thick “antenna” that extends into the tubular lumen. Deflection of the cilium from fluid movement in the renal tubules activates Pkd1, which causes the Pkd2 channel to open, kicking off a Ca2+ cascade that has multiple downstream effects. This mechanism was recently investigated in bone by Xiao et al. [22•]. In this experiment, the Dmp1-Cre transgene was used to inactivate floxed Pkd1 alleles in osteocytes. Mutant mice subjected to in vivo ulnar loading exhibited a ~70 % reduction in load-induced apposition rates compared to control mice, indicating that osteocytic polycystin-1 is an important protein in the anabolic response to skeletal loading. Similar effects have been found in vitro, where cilium disruption via chemical treatment or gene silencing inhibits the response to fluid flow [23, 24]. The primary cilium, and its compliment of associated proteins, is an attractive candidate for mechanosensing in the osteocytes, in light of the importance of fluid movement in the canaliculolacunar network. But it is difficult to envisage how the cilium would physically fit in the extracellular space. The distance between the canalicular wall and the osteocyte cell process/body is typically 50 to 80 nm [25] and could be much smaller if measured on sections processed with newly refined fixation techniques that reduce cell shrinkage [26]. In culture, MC3T3 and MLO-Y4 cells exhibit primary cilia that are ~1–3 μm in length [27]. If this structure is acting as a flow sensor, as it appears to do in other cell types that have an adjacent lumen to project into, it would have very little room to do so.
A channel protein that can be removed from the list of candidates is the α1 subunit of the L-type voltage-sensitive calcium channel (VSCC) (cav1.3). The L-type VSCC has been shown previously in cell culture models to regulate shear-induced nitric oxide and PGE2 release [28], but deletion of the α1 pore-forming subunit of this channel in mice did not alter the anabolic response to mechanical loading, despite having a significant effect on skeletal development [29]. This result does not exclude a role for the L-type VSCC in mechanotransduction, but if the L-type VSCC plays a prominent role in the process, it is likely that other pore-forming subunits can substitute for cav1.3.
G-protein signaling mechanisms in mechanotransduction are beginning to become clearer. Earlier, in a cleverly designed set of experiments, Gudi et al. [30] showed that when purified G-proteins are reconstituted into otherwise empty phospholipid vesicles, they could be activated (GTP hydrolysis) almost immediately upon fluid shear. This flow-induced activation was independent of a GPCR presence, but rather, was modulated by membrane stiffness. Living cell membranes are peppered with GPCRs, and so this group next asked whether the receptors themselves are responsive to ligand-independent mechanically induced activation. Here, they focused on the parathyroid hormone receptor 1 (PTHR1), as Chow et al. [31] showed earlier that parathyroidectomized rodents have impaired bone mechanotransduction. Using GPCR conformation-sensitive fluorescence resonance energy transfer in MC3T3 cells, Zhang et al. [32•] found that fluid shear stress leads to ligand-independent response (conformational change) of the PTHR1, similar to that found when the cells were treated with PTH 3–34 (Fig. 1). This response occurred within seconds and was modulated by membrane fluidity/stiffness. Similar conformational changes in the endothelial B2 bradykinin GPCR were reported earlier by the same group [33], thus the mechanosensitive nature of many GPCRs might be fairly widespread.
Our understanding of integrin/cytoskeletal-mediated mechanotransduction has also expanded in the past several years. Most notably, the β1 integrin subunit appears to be emerging as a crucial mechanical signaling/sensing molecule (Fig. 1). Litzenberger et al. [34] showed that MLO-Y4 cells stably transfected with a dominant-negative fragment of the β1 subunit exhibited reduced PGE2 release, deficient cyclooxygenase-2 upregulation, and blunted receptor activator of NF-κB ligand/osteoprotegerin responses to oscillatory fluid flow, compared to control-transfected cells. Similar results were found by Watabe et al. [35], who subjected primary mandibular, long bone, and calvarial osteoblasts to pulsed ultrasound stimulation. They reported that antibody-mediated blockade of the β1 subunit (in conjunction with the α5 subunit) inhibited β-catenin, Akt, Bcl-2, and mTOR responses to mechanical stimulation. In vivo, Col2.3-Cre-mediated deletion of floxed β1 integrin alleles had little to no effect on the skeletal phenotype. However, a bizarre phenotypic result was reported when the mice were subjected to disuse. Phillips et al. [36] reported that osteoblast/osteocyte deletion of β1 not only protected mice from the normal (as was observed in the WT) tail suspension-induced deterioration of bone mechanical properties, but mechanical properties were actually improved by tail suspension in the mutants.
Conclusions
The past several years have witnessed a multitude of studies aimed at sorting out many of the complexities of mechanobiology in bone. Some of these studies have provided new experimental support for very old ideas (e.g., that the osteocyte is the mechanosensory cell type in bone), further clarification of more recently discovered phenomena (e.g., integrin-mediated and ion channel-mediated mechanotransduction), and the introduction of novel mechanisms for bone mechanobiology (e.g., ligand-independent GPCR mechanical signaling, primary cilium effects). These advances in our understanding of mechanotransduction have opened up new avenues of investigation, and provided a better understanding of how mechanotransduction might integrate into general skeletal development and maintenance. Moreover, elucidation of the normal mechanisms of mechanotransduction can help us understand disease processes at the cellular level. The ultimate goal of these studies—a detailed understanding of the precise cellular and molecular mechanisms involved in mechanical signal processing—moves closer to a reality every year.
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Riddle RC, Donahue HJ. From streaming-potentials to shear stress: 25 years of bone cell mechanotransduction. J Orthop Res. 2009;27:143–9. doi: 10.1002/jor.20723. [DOI] [PubMed] [Google Scholar]
- 2.Iqbal J, Zaidi M. Molecular regulation of mechanotransduction. Biochem Biophys Res Commun. 2005;328:751–5. doi: 10.1016/j.bbrc.2004.12.087. [DOI] [PubMed] [Google Scholar]
- 3.Turner CH, Pavalko FM. Mechanotransduction and functional response of the skeleton to physical stress: the mechanisms and mechanics of bone adaptation. J Orthop Sci. 1998;3:346–55. doi: 10.1007/s007760050064. [DOI] [PubMed] [Google Scholar]
- 4.Hert J, Pribylova E, Liskova M. Reaction of bone to mechanical stimuli. 3. Microstructure of compact bone of rabbit tibia after intermittent loading. Acta Anat (Basel) 1972;82:218–30. [PubMed] [Google Scholar]
- 5.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Kamel MA, Picconi JL, Lara-Castillo N, Johnson ML. Activation of beta-catenin signaling in MLO-Y4 osteocytic cells versus 2T3 osteoblastic cells by fluid flow shear stress and PGE2: implications for the study of mechanosensation in bone. Bone. 2010;47:872–81. doi: 10.1016/j.bone.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norvell SM, Alvarez M, Bidwell JP, Pavalko FM. Fluid shear stress induces beta-catenin signaling in osteoblasts. Calcif Tissue Int. 2004;75:396–404. doi: 10.1007/s00223-004-0213-y. [DOI] [PubMed] [Google Scholar]
- 8.Lu XL, Huo B, Chiang V, Guo XE. Osteocytic network is more responsive in calcium signaling than osteoblastic network under fluid flow. J Bone Miner Res. 2011;27:563–574. doi: 10.1002/jbmr.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burra S, Nicolella DP, Francis WL, Freitas CJ, Mueschke NJ, Poole K, et al. Dendritic processes of osteocytes are mechano-transducers that induce the opening of hemichannels. Proc Natl Acad Sci U S A. 2010;107:13648–53. doi: 10.1073/pnas.1009382107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adachi T, Aonuma Y, Tanaka M, Hojo M, Takano-Yamamoto T, Kamioka H. Calcium response in single osteocytes to locally applied mechanical stimulus: differences in cell process and cell body. J Biomech. 2009;42:1989–95. doi: 10.1016/j.jbiomech.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 11.Wu D, Ganatos P, Spray DC, Weinbaum S. On the electrophysiological response of bone cells using a Stokesian fluid stimulus probe for delivery of quantifiable localized picoNewton level forces. J Biomech. 2011;44:1702–8. doi: 10.1016/j.jbiomech.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, Sprague E, et al. Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J Biol Chem. 2008;283:26374–82. doi: 10.1074/jbc.M803136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13••.Batra N, Burra S, Siller-Jackson AJ, Gu S, Xia X, Weber GF, Desimone D, Bonewald LF, Lafer EM, Sprague E, Schwartz MA, Jiang JX. Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc Natl Acad Sci U S A. 2012;109:3359–64. doi: 10.1073/pnas.1115967109. This article describes the role of the α5β1 integrin in directly modulating Cx43 hemichannel opening in bone cells, and implicates a role for phosphatidylinositol 3-kinase in the process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Liu D, Ke HZ, Duncan RL, Turner CH. The P2X7 nucleotide receptor mediates skeletal mechanotransduction. J Biol Chem. 2005;280:42952–9. doi: 10.1074/jbc.M506415200. [DOI] [PubMed] [Google Scholar]
- 15.Grimston S, Watkins M, Brodt M, Silva M, Civitelli R. Variable bone formation response to skeletal axial load in mice with a conditional deletion of the Connexin43 (Cx43) gene (Gja1) J Bone Miner Res. 2011;26(Suppl 1):S75. [Google Scholar]
- 16•.Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, Srinivasan S, Gross TS, Donahue HJ. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. This article presents data from skeletally loaded mice in which Cx43 has been deleted from osteoblasts/osteocytes; the authors found an enhancement of mechanotransduction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bivi N, Farlow N, Brun L, Benson J, Condon K, Robling AG, et al. Unexpected enhanced response to mechanical loading of mice lacking Cx43 exclusively in osteocytes. J Bone Miner Res. 2011;26(Suppl 1):S11. [Google Scholar]
- 18.Grimston SK, Screen J, Haskell JH, Chung DJ, Brodt MD, Silva MJ, et al. Role of connexin43 in osteoblast response to physical load. Ann N Y Acad Sci. 2006;1068:214–24. doi: 10.1196/annals.1346.023. [DOI] [PubMed] [Google Scholar]
- 19.Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res. 2011;26:2151–60. doi: 10.1002/jbmr.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizoguchi F, Mizuno A, Hayata T, Nakashima K, Heller S, Ushida T, et al. Transient receptor potential vanilloid 4 deficiency suppresses unloading-induced bone loss. J Cell Physiol. 2008;216:47–53. doi: 10.1002/jcp.21374. [DOI] [PubMed] [Google Scholar]
- 21.Gao X, Wu L, O’Neil RG. Temperature-modulated diversity of TRPV4 channel gating: activation by physical stresses and phorbol ester derivatives through protein kinase C-dependent and -independent pathways. J Biol Chem. 2003;278:27129–37. doi: 10.1074/jbc.M302517200. [DOI] [PubMed] [Google Scholar]
- 22•.Xiao Z, Dallas M, Qiu N, Nicolella D, Cao L, Johnson M, Bonewald L, Quarles LD. Conditional deletion of Pkd1 in osteo-cytes disrupts skeletal mechanosensing in mice. FASEB J. 2011;25:2418–32. doi: 10.1096/fj.10-180299. This article reports on the effect of deleting the primary cilium-associated protein, Pkd1, from osteocytes, in the skeletal response to mechanical loading. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon RY, Temiyasathit S, Tummala P, Quah CC, Jacobs CR. Primary cilium-dependent mechanosensing is mediated by adenylyl cyclase 6 and cyclic AMP in bone cells. FASEB J. 2010;24:2859–68. doi: 10.1096/fj.09-148007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, et al. Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proc Natl Acad Sci U S A. 2007;104:13325–30. doi: 10.1073/pnas.0700636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.You LD, Weinbaum S, Cowin SC, Schaffler MB. Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec A Discov Mol Cell Evol Biol. 2004;278:505–13. doi: 10.1002/ar.a.20050. [DOI] [PubMed] [Google Scholar]
- 26.McNamara LM, Majeska RJ, Weinbaum S, Friedrich V, Schaffler MB. Attachment of osteocyte cell processes to the bone matrix. Anat Rec (Hoboken) 2009;292:355–63. doi: 10.1002/ar.20869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao ZS, Quarles LD. Role of the polycytin-primary cilia complex in bone development and mechanosensing. Ann N Y Acad Sci. 2010;1192:410–21. doi: 10.1111/j.1749-6632.2009.05239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rawlinson SC, Pitsillides AA, Lanyon LE. Involvement of different ion channels in osteoblasts’ and osteocytes’ early responses to mechanical strain. Bone. 1996;19:609–14. doi: 10.1016/s8756-3282(96)00260-8. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Zhao L, Ferries IK, Jiang L, Desta MZ, Yu X, et al. Skeletal phenotype of mice with a null mutation in Cav 1.3 L-type calcium channel. J Musculoskelet Neuronal Interact. 2010;10:180–7. [PubMed] [Google Scholar]
- 30.Gudi S, Nolan JP, Frangos JA. Modulation of GTPase activity of G proteins by fluid shear stress and phospholipid composition. Proc Natl Acad Sci U S A. 1998;95:2515–9. doi: 10.1073/pnas.95.5.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chow JW, Fox S, Jagger CJ, Chambers TJ. Role for parathyroid hormone in mechanical responsiveness of rat bone. Am J Physiol. 1998;274:E146–54. doi: 10.1152/ajpendo.1998.274.1.E146. [DOI] [PubMed] [Google Scholar]
- 32•.Zhang YL, Frangos JA, Chachisvilis M. Mechanical stimulus alters conformation of type 1 parathyroid hormone receptor in bone cells. Am J Physiol Cell Physiol. 2009;296:C1391–1399. doi: 10.1152/ajpcell.00549.2008. This article reports that mechanical perturbation of the PTHR1 induces conformational changes in the receptor that are ligand independent, implicating a role for G-protein-coupled receptors in mechanotransduction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci U S A. 2006;103:15463–8. doi: 10.1073/pnas.0607224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Litzenberger JB, Kim JB, Tummala P, Jacobs CR. Beta1 integrins mediate mechanosensitive signaling pathways in osteocytes. Calcif Tissue Int. 2010;86:325–32. doi: 10.1007/s00223-010-9343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watabe H, Furuhama T, Tani-Ishii N, Mikuni-Takagaki Y. Mechanotransduction activates alphabeta integrin and PI3K/Akt signaling pathways in mandibular osteoblasts. Exp Cell Res. 2011;317:2642–9. doi: 10.1016/j.yexcr.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 36.Phillips JA, Almeida EA, Hill EL, Aguirre JI, Rivera MF, Nachbandi I, et al. Role for beta1 integrins in cortical osteocytes during acute musculoskeletal disuse. Matrix Biol. 2008;27:609–18. doi: 10.1016/j.matbio.2008.05.003. [DOI] [PubMed] [Google Scholar]