

Abstract
To gain initial structure–activity relationships regarding the highly functionalized pentyl side chain attached at C-3 of mithramycin (MTM), we focused on a post-polyketide synthase (post-PKS) tailoring step of the MTM biosynthesis by Streptomyces argillaceus ATCC 12956, which was proposed to be catalyzed by ketoreductase (KR) MtmW. In this last step of the MTM biosynthesis, a keto group of the pentyl side chain is reduced to a secondary alcohol, and we anticipated the generation of an MTM derivative with an additional keto group in the 3-side chain. Insertional inactivation of mtmW, a gene located ca. 8 kb downstream of the mithramycin-PKS genes, yielded an S. argillaceus mutant, which accumulated three new mithramycin analogues, namely mithramycin SA, demycarosyl-mithramycin SK, and mithramycin SK (MTM-SK). The structures of these three compounds confirmed indirectly the proposed role of MtmW in MTM biosynthesis. However, the new mithramycin derivatives bear unexpectedly shorter 3-side chains (ethyl or butyl) than MTM, presumably caused by nonenzymatic rearrangement or cleavage reactions of the initially formed pentyl side chain with a reactive β-dicarbonyl functional group. The major product, MTM-SK, was tested in vitro against a variety of human cancer cell lines, as well as in an in vitro toxicity assay, and showed an improved therapeutic index, in comparison to the parent drug, MTM.
Introduction
Mithramycin (MTM, 1, also known as aureolic acid, mithracin, LA-7017, PA-144, and plicamycin; see Figure 1) is an aureolic acid-type polyketide produced by various soil bacteria of the genus Streptomyces, including Streptomyces argillaceus (ATCC 12956).1–3 The aureolic acid group of anticancer antibiotics includes MTM, chromomycin A3 (CHR), olivomycin A (OLI), UCH9, and the newly discovered durhamycin A.1,4,5 All contain the same tricyclic core moiety with a unique dihydroxy-methoxy-oxo-pentyl side chain attached at C-3 and vary only slightly, with respect to the residue at C-7, which is either a H atom or a small alkyl side chain. However, these naturally occurring aureolic acid antibiotics differ in the nature and linking of their saccharide chains, which consist of various 2,6-dideoxysugar residues. Such structural variations impart subtle differences in the DNA binding and activity profiles among the members of this group.1,6–9 MTM has been used clinically in the United States to treat Paget’s disease and testicular carcinoma.10–13 In addition, MTM’s hypocalcemic effect has been used to manage hypercalcemia in patients with malignancy-associated bone lesions.14 Unfortunately, it exhibits severe side effects, such that the current clinical use of MTM is limited by its gastrointestinal, hepatic, renal, and bone marrow toxicities, which result in nausea, vomiting, and bleeding.11,15 Consequently, it is important to investigate MTM analogues for increased therapeutic indexes.
Figure 1.
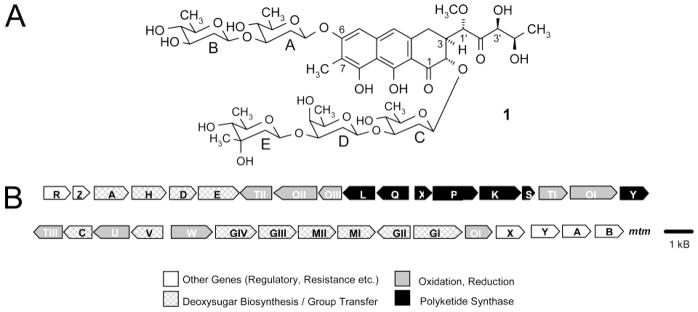
(A) Structure of mithramycin (MTM, 1). (B) Genetic organization of the MTM biosynthetic gene cluster in S. argillaceus ATCC 12956.
MTM interacts with the minor groove of DNA, mainly in GC-rich regions, as a 2:1 MTM:Mg2+ complex, cross-linking the DNA and blocking its template activity for DNA- and RNA-dependent polymerases, thereby inhibiting replication and transcription processes.6 The specific characteristics of the DNA: antibiotic interaction have been intensively investigated by NMR and X-ray crystallographic studies,8,9,16–18 particularly in view of the role played by the oligosaccharide moieties. These and other studies revealed that the C–D units of the trisaccharide chains of MTM, OLI, and CHR stack on the aromatic core of the dimer-partner aglycon, and that the intact C–D–E trisaccharide moiety is essential for dimer formation as well as optimal DNA binding. The role of the C-3 polyoxygenated pentyl side chain is not well characterized, except through the X-ray structure of the MTM–DNA complex,8,9 which reveals that the pentyl side chain interacts with the phosphate backbone of the DNA, leading to the hypothesis that modifications of this alkyl side chain may be profitable. Thus, derivatives, which differ from their parent drug, with respect to the alkyl side chain at C-3, are needed to further investigate structure–activity relationships of the aureolic acid-type antitumor drugs.
Synthetic and semisynthetic approaches toward the production of new aureolic acid-type compounds are complemented by combinatorial biosynthetic procedures, whereby the genes of the biosynthetic pathway of the drugs are altered in the bacteria themselves (e.g., through gene inactivation, expression, or recombination). This method not only allows the generation of new derivatives, the so-called “unnatural natural compounds”, but also simultaneously generates a bacterial strain, which allows the biotechnological mass production of the new derivative. The biosynthetic gene cluster leading to the formation of MTM has been studied in our laboratories over the last years and resulted in the identification of 34 genes and the assignment of various gene product functions for the biosynthesis of MTM (Figure 1B).3,19–28 It has been shown that MTM biosynthesis proceeds through tetracyclic intermediates with glycosylation steps occurring on these tetracyclic biosynthetic intermediates. One of the last steps, the key step in MTM biosynthesis, is the oxidative cleavage of the fourth ring of the fully glycosylated tetracyclic intermediate premithramycin B, which results in the formation of a tricyclic immediate precursor of MTM (see Figure 5).24 This enzymatic reaction is very important, because it causes an alteration of the shaping of the molecule and only tricyclic (and not the tetracyclic) derivatives/analogues of MTM appear to be biologically active. In conjunction with this oxidative cleavage step, a decarboxylation step occurs and the pentyl side chain attached at C-3 is generated, which may play an important role for the biological activity of the aureolic acid class of antitumor agents. To generate the final MTM molecule, the oxidative cleavage step is followed by a ketoreduction step, in which the keto group in the 4′-position of the 3-side chain is reduced to a secondary alcohol. So far, no studies have addressed the altering of this side chain and how this may affect the biological activity of MTM.
Figure 5.
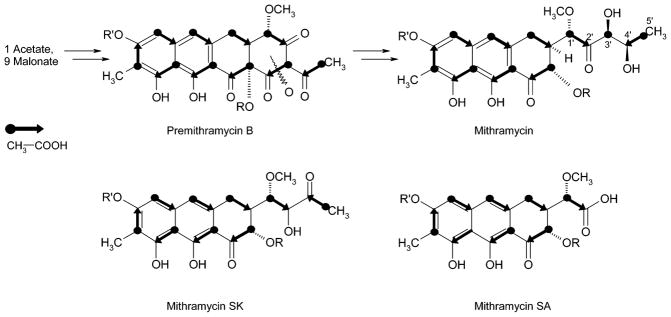
Incorporation experiments with [1-13C]-acetate and [1,2-13C2]-acetate on the MTM-SK and MTM-SA producer S. argillaecus M7W1, compared to the normal incorporation pattern found in MTM (shown above) revealed that the former MTM carbon C-3′ and carbons 3′,4′ and 5′, respectively, were excised during the formation of the new metabolites MTM-SK and MTM-SA, respectively. R and R′ are the deoxysaccharide chains shown in Figure 1.
Here, we report the isolation and characterization of three novel MTM derivatives, namely mithramycin SK (MTM-SK, 2), demycarosyl-mithramycin SK (demyc-MTM-SK, 3), and mithramycin SA (MTM-SA, 4), which are all variants, with respect to the 3-side chain. Only compound 3 bears an additional modification in the trisaccharide chain. All these compounds were generated by insertional inactivation of the gene mtmW from the MTM producer Streptomyces argillaceus, which encodes a reductase involved in ketoreduction of the C-3 side chain of MTM.
Results and Discussion
Sequencing and Insertional Inactivation of mtmW
A 1.4 kb region of the MTM gene cluster (later designated as mtmW) located between several sugar biosynthetic genes (mtmV, mtmU, mtmC, and mtmTIII) and glycosyltransferase genes (mtmGIV and mtmGIII) was sequenced (Figure 1B). Analysis of the DNA sequence for coding regions using the CODONPREFERENCE program29 showed the presence of an open reading frame (designated as mtmW). It is comprised of 981 nucleotides with a GTG start codon and a TAG stop codon and codes for a polypeptide of 326 amino acids with an estimated Mr of 35 304. This codon region shows the high GC content and bias for the third codon position, which is characteristic of Streptomyces genes. Comparison of the deduced product of mtmW with other proteins using protein databases revealed similarities with various oxidoreductases. The highest similarities were observed with a putative potassium channel beta subunit of Deinococcus radiodurans30 (40% identical amino acids). It also showed similarity with the EryBII31 (33% identical amino acids) and TylCII32 (33% identical amino acids) proteins. These two proteins are oxidoreductases, which participate in deoxygenation steps during the biosynthesis of the deoxysugars that form part of the macrolide antibiotics erythromycin and tylosin, respectively.
To investigate the possible role of the mtmW gene product in MTM biosynthesis, particularly the 3-side chain ketoreduction step, mtmW was inactivated by gene replacement through the insertion of an apramycin resistance cassette (Figure 2).
Figure 2.

Scheme representing the replacement event in the chromosome of wild-type S. argillaceus strain by a double crossover to construct mutant M7W1.
Upon transformation of the wild-type S. argillaceus ATCC 12956 with pM7W1, transformants were selected for their resistance to apramycin. One of these transformants, named mutant M7W1, was found to be also sensitive to thiostrepton, being the consequence of a double crossover, which results in the replacement of the wild-type gene by the in vitro mutated one. This fact was confirmed by Southern hybridization: the 4.5 kb BamHI fragment of the wild-type strain was replaced by two new BamHI fragments of 3.7 and 2.3 kb, as expected if the replacement occurred. It was also confirmed that the gene replacement only affected mtmW, because expressing this gene in trans, using pAGW, restored MTM production in mutant M7W1.
Isolation and Structure Elucidation of the Novel Compounds
S. argillaceus M7W1 was grown on a R5A liquid medium and, after 5 days of incubation, the culture supernatant was subjected to solid-phase extraction. The material eluted with a methanol:water mixture was analyzed by reverse-phase HPLC. No MTM production was detected. However, two new HPLC peaks were detected. The absorption spectra of the compounds present in these two peaks were characteristic of MTM biosynthetic intermediates in which the opening of the fourth ring had occurred.24 The compounds in these two peaks were purified, with yields of 14.5 mg/L for the major compound (MTM-SK) and 2.2 mg/L for the minor compound (demyc-MTM-SK) (Figure 3). Alternatively, a liquid extraction work-up procedure, followed by conventional chromatography, was used to isolate the new compounds. Following acidification to pH 5.5, the culture was extracted using (i) EtOAc and (ii) n-BuOH. The more-lipophilic compounds, MTM-SK (yield of 9 mg/L) and demyc-MTM-SK (yield of 2 mg/L), were found in the EtOAC extract, whereas the more-hydrophilic MTM-SA (yield of 5 mg/L, Figure 3) was solely found in the n-BuOH extract. Silica gel chromatography was used for both the EtOAc extract and the n-BuOH extract. MTM-SK and demyc-MTM-SK were finally purified using an RP-18 silica gel column, followed by Sephadex-LH 20 chromatography. MTM-SA was finally purified through preparative thin-layer chromatography (TLC), using RP-18 silica gel plates. The exact isolation procedure is described in the discussion regarding the feeding experiments in the Experimental Section.
Figure 3.

Structures of the new metabolites MTM-SK (2), demycarosyl-mithramycin SK (3), and MTM-SA (4) produced by mutant S. argillaceus M7W1.
The structures of the new compounds were elucidated using physicochemical methods. The 1H NMR and 13C NMR spectra (Table 1), in combination with the positive-mode electrospray ionization (ESI) mass spectrum, which revealed a molecular ion of m/z 1077.4 (M + Na+), indicated the molecular formula of the major compound to be C51H74O23 (MW = 1054.4). This was verified by high-resolution positive-mode fast atom bombardment mass spectroscopy (FAB MS) (HR for C51H74O23-Na: calcd, 1077.4519; observed, 1077.4478). The presence of five anomeric carbon signals (δC 97.0, 97.9, 99.9, 100.3, and 100.8) and the analysis of the additional sugar 1H NMR and 13C NMR chemical shifts and coupling constants indicated that the typical MTM glycosylation pattern was upheld. Specifically, a diolivoside is attached to the C-6 position of the aglycon and a trisaccharide composed of an olivose, an oliose, and a mycarose moiety is attached at C-2. All are β-glycosidically linked (10-Hz transaxial coupling between the 1-H and 2-Ha) with the interglycosidic connections, as well as the points of linkage to the aglycon, clearly assigned from the heteronuclear multiple bond connectivity (HMBC) spectra. Also, as in MTM, the aglycon is methylated at the C-7 position (δC 7.9, δH 2.15). The difference between this compound and MTM lies in the side chain at C-3, which is unexpectedly missing one hydroxylated carbon (a difference of 30 amu from MTM). The novel hydroxy-methoxy-oxo-butyl side chain in MTM-SK bears the typical 1′-OCH3 group (δC 60.0, δH 3.55), a hydroxylated carbon at the 2′-position (δC 79.46, δH 4.32), a keto group in the 3′-position (δC 209.9), and terminates with a methyl group (δC 26.3, δH 2.35). This is supported by the coupling pattern associated with the protons of this side chain. In particular, H-1′, which is normally a simple doublet, being situated between H-3 and the 2′-keto group of MTM, is now a doublet of a doublet, as would be expected if the keto function at 2′ had been replaced by a carbon bearing a single proton. Furthermore, the methyl group, which ends the side chain, is now a sharp singlet instead of a doublet and its carbon and proton signals are shifted downfield (δC 26.26, δH 2.35), compared to those found in the normal MTM compound (δC 18.7, δH 1.26), indicative of its proximity to the keto group at C-3′. Important HMBC couplings substantiating this structure include the 4JC–H couplings between C-2′ and H-2 and 1′-OCH3 and the 3JC–H couplings between C-2 and H-1′, C-3 and H-2′, C-2′ and H3-4′, and C-3′ and H-1′. Other typical MTM HMBC couplings were similar to those recently published.28 Thus, the major compound is MTM-SK (2), where SK refers to the shortened side chain with a relocated keto function. The suggested stereochemistry of the center at C-2′ shown in the structure for 2 follows from semiempirical calculations, which propose a hydrogen bond between 2′-OH and 2-O. Only the structure with R-configuration at C-2′ in such an arrangement is in agreement with the NMR data (the dihedral angle between 1′-H and 2′-H is ca. 30°, in agreement with J1′-H/2′-H = 3.4 Hz), whereas for the molecule with the opposite stereochemistry at C-2′, a dihedral angle of ~180° was found.33
Table 1.
1H and 13C NMR Analysis of Nonlabeled and Labeled Mithramycin SK (2)
| position | 1H δ (ppm)a multiplicity (JHz) | 13C δ (ppm)a |
1-13Cb
|
1,2-13C2b
|
|
|---|---|---|---|---|---|
| shift (ppm) | relative enrichments | Jcc (Hz) | |||
| 1 | 203.5 | 197.5 | 3.0 | 45 | |
| 2 | 4.70 d (11.5) | 78.2 | 78.1 | 45 | |
| 3 | 2.48 overlap | 43.7 | 45.3 | 3.3 | 44 |
| 4a | 3.15 dd (16, 3) | 28.3 | 30.5 | 44 | |
| 4e | 2.99 overlap | ||||
| 4a | 136.9 | 139.9 | 7.7 | 66 | |
| 5 | 6.87 s | 101.7 | 101.3 | 61 | |
| 6 | 159.9 | 160.3 | 8.1 | 69 | |
| 7 | 111.0 | 111.7 | 69 | ||
| 7-CH3 | 2.15 s | 7.9 | 8.6 | ||
| 8 | 156.2 | 161.9 | 4.7 | 61 | |
| 8a | 108.0 | 108.8 | 61 | ||
| 9 | 165.3 | 177.2 | 2.7 | 69 | |
| 9a | 108.5 | 108.7 | 69 | ||
| 10 | 6.89 s | 117.0 | 113.4 | 66 | |
| 10a | 139.1 | 140.8 | 6.2 | 61 | |
| 1′ | 4.25 dd (3.4, 1.5) | 78.3 | 80.7 | 43 | |
| 1′-OCH3 | 3.55 s | 60.0 | 60.5 | ||
| 2′ | 4.32 d (3.4) | 79.5 | 80.6 | 6.4 | 43 |
| 3′ | 209.9 | 212.6 | 3.2 | 39 | |
| 1A | 5.37 dd (10, 2) | 97.0 | |||
| 2Aa | 1.86 ddd (12, 12, 10) | 37.5 | |||
| 2Ae | 2.48 overlap | 37.5 | |||
| 3A | 3.78 ddd (12, 9, 5) | 81.3 | |||
| 4A | 3.09 dd (9, 9) | 75.4 | |||
| 5A | 3.55 overlap | 72.6 | |||
| 6A (CH3) | 1.34 d (6) | 18.0 | |||
| 1B | 4.75 dd (10, 2) | 99.9 | |||
| 2Ba | 1.59 ddd (12, 12, 10) | 40.0 | |||
| 2Be | 2.20 ddd (12, 5, 2) | 40.0 | |||
| 3B | 3.58 overlap | 71.4 | |||
| 4B | 3.01 dd (9, 9) | 77.6 | |||
| 5B | 3.41 dq (9, 6) | 72.6 | |||
| 6B (CH3) | 1.34 d (6) | 17.7 | |||
| 1C | 5.14 dd (10, 2) | 100.8 | |||
| 2Ca | 1.62 ddd (12, 12, 10) | 37.9 | |||
| 2Ce | 2.51 ddd (12, 5, 2) | 37.9 | |||
| 3C | 3.68 overlap | 81.8 | |||
| 4C | 3.05 dd (9, 9) | 75.7 | |||
| 5C | 3.33 dq (9, 6) | 72.6 | |||
| 6C (CH3) | 1.34 d (6) | 17.9 | |||
| 1D | 4.70 dd (10, 2) | 100.3 | |||
| 2Da | 1.80 ddd (12, 12, 10) | 32.5 | |||
| 2De | 1.95 ddd (12, 5, 2) | 32.5 | |||
| 3D | 3.88 ddd (12, 5, 3) | 77.3 | |||
| 4D | 3.72 bs | 68.9 | |||
| 5D | 3.70 overlap | 71.0 | |||
| 6D (CH3) | 1.34 d (6) | 16.5 | |||
| 1E | 4.98 dd (9.5, 2) | 97.9 | |||
| 2Ea | 1.56 dd (13, 9.5) | 44.3 | |||
| 2Ee | 1.90 dd (13, 2) | 44.3 | |||
| 3E | 70.7 | ||||
| 3E-CH3 | 1.22 s | 27.0 | |||
| 4E | 2.99 d (9) | 76.8 | |||
| 5E | 3.65 overlap | 71.0 | |||
| 6E (CH3) | 1.22 d (6) | 26.3 | |||
| 4′ | 2.35 s | 26.3 | 27.2 | 39 | |
Data collected in acetone-d6 at 500 MHz (1H NMR) and 125.7 MHz (13C NMR).
Data collected in methanol-d4 at 75.4 MHz.
The positive-mode ESI mass spectrum of the second compound revealed a molecular ion of m/z 933.4 (M + Na+), indicating the molecular formula to be C44H62O20 (MW = 910.4), which is seven carbon atoms less than that found for MTM-SK. This was verified by high-resolution positive-mode FAB MS (HR for C44H62O20K: calcd, 949.3472; observed, 949.3482). The presence of only four anomeric carbon signals (δC 97.1, 99.9, 100.4, and 100.8) and, in particular, the nonappearance of the 3E quaternary carbon and the 3E methyl group indicated that the loss of seven carbon atoms was due to the absence of the mycarose moiety normally attached to the 3-position of oliose. Further analysis of the 1H NMR and 13C NMR spectra (see Experimental Section) indicated that the remaining structural features, including the shortened side chain, were identical to MTM-SK; thus, the minor compound was deduced to be demyc-MTM-SK (3). Demycarosyl-mithramycin derivatives have been isolated only once before;28 thus, demyc-MTM-SK provides further evidence for the flexibility of the enzymes, which normally act following the addition of the mycarose moiety, e.g., MtmGI, MtmGII, and MtmOIV.34 The missing mycarose moiety in 3 might also reflect the possibility that the inactivation of mtmW slightly compromised the function of the product of the gene immediately downstream, mtmGIV, which encodes the glycosyltransferase responsible for the attachment of the D-mycarose unit in the mithramycin biosynthesis.28,35
The negative- and positive-mode ESI mass spectrum of the third compound, which was only obtained following a liquid extraction procedure, showed a molecular ion of m/z 1025.4 (M – H)− and m/z 1049.4 (M + Na+), respectively, indicating the molecular formula to be C49H70O23 (MW = 1026.4). The analysis of 1H NMR, 13C NMR, and various two-dimensional (2D) NMR spectra indicated that all structural features of this minor product were identical to those of MTM-SK (2), except the 3-side chain (see Experimental Section). The 1H signals corresponding to the 4′ methyl group and the H-2′of the 3-side chain were missing in the 1H NMR analysis, whereas the 13C NMR analysis confirmed the absence of carbons C-3′ and C-4′. The 13C NMR analysis showed a characteristic new carbonyl carbon at δ 176.8, indicating C-2′ to be a carboxylic acid. Thus, the compound isolated from the n-BuOH extract of the MTM-SK producer possesses a very short 3-side chain ending with a carboxylic acid group at the 2′-position and was consequently named mithramycin SA (MTM-SA (4), for short side chain and acid). It is possible that 4 is (partly) an artifact of the work-up procedure, because its formation from the putative labile compound 5 (see below) is acid-catalyzed, and the culture needed to be adjusted to a slightly acidic pH (5.5) to enable the extraction of 4 into the organic phase. On the other hand, this minor compound may have been missed in the work-up procedure following solid extraction, because it might have gotten absorbed onto the HPLC column.
Rearrangement Reactions and Incorporation Experiments
The structures of MTM-SK (2) and its demycarosyl derivative 3 were intriguing, because we expected compound 5 to be accumulated from the inactivation of ketoreductase MtmW. However, compound 5 has a highly functional and, thus, reactive 3-side chain, which contains two keto functions in the β-position, relative to each other, separated by a carbon atom bearing another oxygen atom. This β-dicarbonyl constellation might trigger a Favorskii-like rearrangement (Figure 4A), for which an 1,2-acyl shift induced by deprotonation of the central alcohol can be envisaged, followed by the addition of water on the resulting aldehyde and a consequent departure of formic acid. Moore and co-workers recently discussed a similar Favorskii-type rearrangement, in regard to the enterocin biosynthesis; however, there, the reaction occurs after an oxidative cleavage of a C–C bond and therefore is more likely to be enzymatically catalyzed.36,37 MTM-SA (4) also gives indirect evidence for the labile structure 5, because its formation from 5 is possible through the attack of water at the carbonyl adjacent to the methoxy group, followed by retro-aldol cleavage to yield MTM-SA (4) and hydroxyacetone (Figure 4B).
Figure 4.
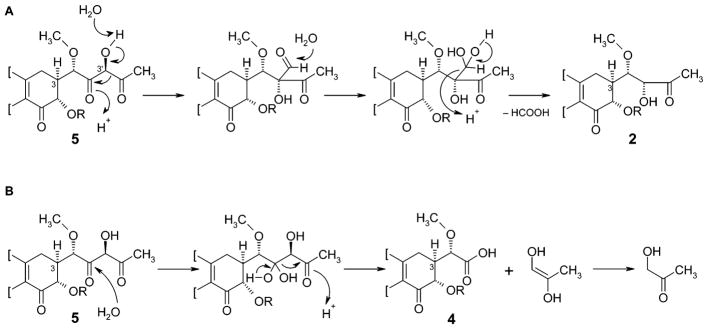
Formation of MTM-SK (2) and MTM-SA (4) through a Favorskii-like rearrangement, and retro-aldol-type cleavage of the reactive product 5, proposed to be generated by the inactivation of the ketoreductase encoding gene mtmW.
To prove the excision of carbon 3′, two feeding experiments using [1-13C]-acetate and [1,2-13C2]-acetate were performed (Table 1). The oxidative rearrangement during the biosynthesis of MTM leads to an acetate incorporation pattern, shown in Figure 5, in which carbons 4′ and 5′ of the 3-side chain stem from the starter unit, whereas carbons 1′, 2′, and 3′ were once the end of the polyketide chain.3,38 If C-3′ is lost, two intact acetate units “facing each other” from opposite directions should result. The results, also shown in Figure 5, are consistent with these expectations and, thus, prove the loss of carbon 3′.
Although we favor the nonenzymatic rearrangement reaction previously shown for the formation of MTM-SK (2), a participation of an enzyme, especially an oxygenase, as discussed for the enterocin biosynthesis,37 cannot be excluded. Therefore, alternative oxidative rearrangement mechanisms are also possible, for which either one of the oxygenases of the mtm gene cluster3 or any other S. argillaceus oxygenase might be responsible.
Biological Activity
The antitumor activity of the two compounds was tested against a variety of tumor cell lines, including the 60 human tumor cell line assay of the NCI (National Cancer Institute, Bethesda, MD).39–41 Compilation of the average log(GI50) values showed that both compounds were active, with MTM-SK (activity up to 9 times higher than that of MTM) being much more active than demyc-MTM-SK (ca. 25 times less active than MTM) (Table 2a). MTM-SK was particularly active against melanoma, leukemia, and CNS cancer cells (log(GI50) values of −7.64, −7.59, and −7.61, respectively). Given the increased activity observed for 2, a neutral red uptake analysis of squamous, melanoma, lung, and breast carcinomas was performed (Frevert42), which not only confirmed the increased activity of mithramycin SK as compared to MTM, but also showed an even more pronounced improvement of activity (up to ca. 90 times better) (Table 2b). In addition, toxicity assays using this same process and mouse 3T3 fibroblast (nontumor) cells showed that 2, with an IC50 value of 1.96 × 10−5 M, is more than 1500-fold less toxic than MTM (IC50 values ranging from 1.29 × 10−8 to 3.45 × 10−9 M). (From Frevert,42 Ginsburg et al.,43 and Givens et al.44) Thus, MTM-SK (2) displays a significantly improved therapeutic index, up to 4 orders of magnitude better, compared to its parent drug, MTM.
Table 2.
Antitumor Analysis Comparing Mithramycin (1), Mithramycin SK (2), and Demycarosylmithramycin SK (3)a
| 1 | comparison with 2
|
comparison with 3
|
|||||
|---|---|---|---|---|---|---|---|
| 2 | Δ1–2 | activity improvement factore | 3 | Δ1–3 | activity improvement factore | ||
| (a) Average log(GI50) Valuesb | |||||||
| leukemia (5)c | −6.65 | −7.59 | 0.94 | 8.7 | −5.55 | −1.10 | 0.08 |
| NSCLC (8)c | −6.73 | −7.37 | 0.64 | 4.4 | −5.30 | −1.43 | 0.04 |
| colon (7)c | −6.65 | −7.32 | 0.67 | 4.7 | −5.35 | −1.30 | 0.05 |
| CNS (5)c | −6.78 | −7.61 | 0.83 | 6.8 | −5.30 | −1.48 | 0.03 |
| melanoma (8)c | −6.72 | −7.64 | 0.92 | 8.3 | −5.37 | −1.35 | 0.04 |
| ovarian (6)c | −6.60 | −7.53 | 0.93 | 8.5 | −5.23 | −1.37 | 0.04 |
| renal (8)c | −6.73 | −7.29 | 0.56 | 3.6 | −5.14 | −1.59 | 0.03 |
| prostate (2)c | −6.90 | −7.48 | 0.58 | 3.8 | −5.25 | −1.65 | 0.02 |
| breast (8)c | −6.59 | −5.89 | −0.70 | 0.2 | −5.15 | −1.44 | 0.04 |
| (b) Average log(IC50) Valuesd | |||||||
| squamous carcinoma | −5.04 | −5.99 | 0.95 | 8.9 | |||
| melanoma | −5.05 | −6.25 | 1.20 | 15.8 | |||
| lung carcinoma (A549) | −4.92 | −6.88 | 1.96 | 91.2 | |||
| breast carcinoma (MCF-7) | −4.95 | −6.74 | 1.79 | 61.6 | |||
Data show that 2 exhibits an activity that is up to 90 times higher than that of 1, whereas 3 is ~25 times less active than 1.
Number in parentheses is the number of cell lines tested in each family.
Average log(IC50) values resulting from the neutral red assay.42
The activity improvement factor is equal to 10Δ1–x, where x is the identifying value for compound 2 or 3. An activity improvement factor of 1.0 corresponds to no difference in activity.
Experimental Section
Microorganisms, Culture Conditions, and Plasmids
Streptomyces argillaceus ATCC 12956 was used as the source of chromosomal DNA. For sporulation on a solid medium, it was grown at 30 °C on plates containing medium A.47 For protoplast transformation, it was grown in a YEME medium containing 17% sucrose. For growth in a liquid medium, the organism was grown in a TSB medium (trypticase soya broth, Oxoid). Escherichia coli XL1blue (Stratagene, Germany) was used as the host for plasmid propagation. When plasmid-containing clones were grown, the medium was supplemented with the appropriate antibiotics: thiostrepton, 25 μg/mL; tobramycin, 20 μg/mL; ampicillin, 100 μg/mL; or apramycin, 25 μg/mL. Plasmids pBSKT,21 pIJ2921,48 pIAGO,49 and pEFBA (a pBSK derivative containing an apramycin resistance cassette; Ernestina Fernández, unpublished) were used for subcloning experiments.
DNA Manipulation Techniques
Plasmid DNA preparations, restriction endonuclease digestions, alkaline phosphatase treatments, ligations, Southern hybridization, and other DNA manipulations were performed according to standard procedures for E. coli50 and Streptomyces.51
DNA Sequencing
Sequencing was performed on double-stranded DNA templates using the dideoxynucleotide chain-termination method.52 Both DNA strands were sequenced with primers supplied in the kits or with internal oligoprimers (17-mer) using an ALF-express automatic DNA sequencer (Pharmacia Biotech). Computer-aided database searching and sequence analyses were conducted using the University of Wisconsin Genetics Computer Group programs package (UWGCG)29 and the BLAST program.53–55
Insertional Inactivation
For inactivation of the mtmW gene, a 4.5 kb BamHI fragment containing mtmW, mtmGIV, and portions of adjacent genes was subcloned into the BamHI site of pBSKT, generating pM7W0. Then, an apramycin resistance cassette containing the aac(3)-IV gene was subcloned as a 1.5 kb SmaI-EcoRV fragment into the unique BglII site (blunt-ended) located within the coding region for mtmW and orientated in the direction of transcription of mtmW, thus generating pM7W1 (Figure 2). This construct was used to transform protoplasts of S. argillaceus. Transformants, in which the wild-type region of the chromosome was replaced by the in vitro mutated one through a double crossover at both sides of the apramycin cassette, were recognized by their resistance to apramycin and sensitivity to thiostrepton. The replacement was also verified by Southern analysis.
Complementation of the mtmW-minus Mutant
A 1.1 kb XhoI-FspI fragment comprising the mtmW gene was subcloned into the SalI–SmaI sites of pIJ2921, generating pAGW0. The gene was then rescued with SphI (using the 5′-end SphI site of the polylinker) and subcloned into the unique SphI site of the bifuncional (Streptomyces–E. coli) plasmid pIAGO, immediately downstream of the erythromycin resistance promoter, ermEp, generating pAGW. This construct was used for complementing the M7W1 mutant.
Isolation of Compounds
A seed culture was prepared using TSB inoculated with spores of S. argillaceus M7W1 and incubated in an orbital shaker for 24 h at 30°C and 250 rpm. This seed culture was used to inoculate (at 2.5% v/v) eight 2-liter Erlenmeyer flasks, each containing 400 mL of R5A medium.47 The flasks were incubated for 5 days under the previously described conditions. The entire culture obtained was centrifuged (12 000 rpm, 30 min), the pellets were discarded, and the supernatant was filtered (using membrane filters with a pore size of 0.45 μm). The filtrate was applied to a solid-phase extraction cartridge (Supelclean LC-18, 10 g, Supelco), and the retained material was eluted with a mixture of methanol and water. A linear gradient from 0% to 100% methanol in 60 min, at 10 mL/min, was used. Fractions were taken every 5 min and, after HPLC analysis, the new compounds were found in fractions eluted between 40 and 55 min. These were evaporated under vacuum, redissolved in a mixture of dimethyl sulfoxide and methanol (50:50), and chromatographed using a μBondapak C18 preparative column (PrepPak Cartridge, 25 mm × 100 mm, Waters), with acetonitrile (ACN) and water as solvents, at a flow rate of 10 mL/min. In the first step, a linear gradient from 30% to 50% ACN in 30 min was used. The material in the two peaks collected in this step was further purified under isocratic conditions with 37.5% ACN in water as a solvent. The isolated products were finally dried in vacuo and weighed.
An alternative work-up procedure via liquid extraction and conventional chromatography is described below (in the Feeding Experiments section).
Feeding Experiments
A seed culture was prepared using TSB inoculated with spores of S. argillaceus M7W1 and incubated in an orbital shaker for 24 h at 30°C and 250 rpm. This seed culture was used to inoculate (at 2.5% v/v) sixteen 250-mL Erlenmeyer flasks, each containing 100 mL of modified R5 medium. Thirty-two hours after the inoculation, the pulse feeding of sodium acetate was started and continued for 36 h at 12-h intervals (four feedings for a total of 1 g of sodium acetate per liter of culture). For the single- and double-labeled experiments, 1-13C and 1,2-13C sodium acetate compounds, respectively, were used. The culture was then grown for an additional 52 h, for a total of 120 h before extraction. Following acidification with HCl to pH 5.5, the culture was extracted first with EtOAc and then with BuOH. The 13C-labeled MTM-SK and demyc-MTM-SK were isolated from the EtOAc extract (0.21 g) using several chromatographic steps. The first step was a silica gel column (25 cm × 3 cm, eluted with CHCl3 and MeOH, 9:1). The second column, an RP-18 silica gel column (11 cm × 1.75 cm), was eluted with water and ACN (65:35) being used to apply pressured air. Finally, MTM-SK and demyc-MTM-SK were purified using a Sephadex LH-20 column (100 cm × 2.5 cm, eluted with MeOH). Yields: MTM-SK, 13.7 mg; demyc-MTM-SK, 3.2 mg (both obtained as amorphous solids). MTM-SA was isolated from the BuOH extract (4.8 g), using (i) a silica gel column (25 cm × 3 cm, CHCl3:MeOH = 8.5:1.5), followed by (ii) a final purification via preparative TLC (20 cm × 20 cm RP-18 silica gel plates, Merck RP-18 F254, solvent system, water/ACN (65:35)). The yellow zone containing MTM-SA (Rf = 0.43) was cut out, and the pure compound was obtained as an amorphous yellow solid by extraction with MeOH and evaporation to dryness.
HPLC Analysis
Analyses were performed with a reversed-phase column (Symmetry C18, 4.6 cm × 250 mm, Waters), with ACN and 0.1% trifluoroacetic acid in water as solvents. A linear gradient from 10% to 100% ACN in 30 min, at a flow rate of 1 mL/min, was used. Detection and spectral characterization of peaks were made with a photodiode array detector and Millennium software, extracting bi-dimensional chromatograms at 280 nm.
Structure Elucidation and Characterization
The structures of the new MTM derivatives were elucidated by NMR spectroscopy and mass spectrometry. The electrospray ionization mass spectra (ESI-MS) were acquired using a Finnigan MAT LCQ mass spectrometer. The high-resolution positive-mode fast atom bombardment mass spectrometry (FAB) spectra were acquired using a model VG70SQ double focusing magnetic sector MS instrument. The UV spectra were recorded on a Varian model CARY50 spectrophotometer, and IR spectra were obtained from a pure sample of KBr disks in a Bio-Rad model FTS3000MX FT-IR spectrometer. All NMR data, if not stated otherwise, were recorded in acetone-d6, using either 300 or 400 MHz Varian Inova equipment or a 500 MHz Bruker model DMX instrument. The NMR data for 2 are listed in Table 1; the NMR data for 3 and 4 are given below. All NMR assignments are based on H,H–COSY, HSQC, and HMBC spectra, allowing an unambiguous assignment of all NMR signals. For the semiempirical calculations, the generation of the two possible diastereoisomeric three-dimensional structures of MTM-SK was performed, using the builder module of the InsightII software (Accelrys, 2000), on the basis of the known structure of MTM.2 Energy minimizations were performed within the discovery module of the InsightII software, using force field CFF91 running on a Silicon Graphics computer (model O2).33
NMR and MS Analysis Data for Mithramycin SK, C51H74O23 (2)
ESI-MS m/z (relative intensity) [M + Na+]: 1077.4 (100). HR-FAB m/z [M + Na+]: calcd, 1077.4519; found, 1077.4478. UV (MeOH) λmax (ε): 421.5 (10 600), 315.5 (7400), 285.0 (54 500) nm. FT-IR (KBr) ν: 3423 (OH), 2974 (CH), 2931 (CH), 2883 (CH), 1712 (C=O), 1600 (C=O), 1521 (C=C), 1371, 1164, 1068 cm−1. 1H NMR δ and 13C NMR δ data are given in Table 1.
NMR and MS Analysis Data for Demycarosyl-mithramycin SK, C44H62O20 (3)
ESI-MS m/z (relative intensity) [M + Na+]: 933.4 (100). HR-FAB m/z [M + K+]: calcd, 949.3472; found, 949.3482. UV (MeOH) λmax (ε): 421.5 (13 600), 315.9 (10 300), 285.0 (61 900) nm. FT-IR (KBr) ν: 3413 (OH), 2974 (CH), 2931 (CH), 2884 (CH), 1710 (C=O), 1629 (C=O), 1580 (C=C), 1369, 1166, 1068 cm−1. 1H NMR (500 MHz, acetone-d6, δ): 1.33 (d, 12H, J = 6 Hz, 6A–H3, 6B–H3, 6C–H3, and 6D–H3), 1.58 (ddd, 1H, J = 12, 12, 10 Hz, 2B–Ha), 1.62 (ddd, 1H, J = 12, 12, 10 Hz, 2C–Ha), 1.76 (ddd, 1H, J = 12, 12, 10 Hz, 2D–Ha), 1.90 (ddd, 1H, J = 12, 12, 10 Hz, 2A–Ha), 1.95 (ddd, 1H, J = 12, 5, 2 Hz, 2D–He), 2.17 (s, 3H, 7-CH3), 2.21 (ddd, 1H, J= 12, 5, 2 Hz, 2B–He), 2.34 (s, 3H, 4′-H3), 2.47 (overlap, 1H, 3-H), 2.49 (overlap, 1H, 2A–He), 2.51 (overlap, 1H, 2C–He), 2.99 (dd, 1H, J = 9, 9 Hz, 4B–H), 3.01 (overlap, 2H, 4-He), 3.01 (dd, 1H, J = 9, 9 Hz, 4C–H), 3.08 (dd, 1H, J = 9, 9 Hz, 4A–H), 3.19 (dd, 1H, J = 16, 3 Hz, 4-Ha), 3.35 (dq, 1H, J = 9, 6 Hz, 5C–H), 3.38 (dq, 1H, J = 9, 6 Hz, 5B–H), 3.54 (bs, 1H, 4D–H), 3.56 (s, 3H, 1′-OCH3), 3.56 (overlap, 1H, 5A–H), 3.58 (overlap, 1H, 3B–H), 3.69 (overlap, 1H, 3C–H), 3.71 (bq, 1H, J = 6 Hz, 5D–H), 3.78 (ddd, 1H, J = 12, 9, 5 Hz, 3A–H), 3.80 (ddd, 1H, J = 12, 5, 3 Hz, 3D–H), 4.24 (dd, 1H, J = 3.4, 1.5 Hz, 1′-H), 4.31 (d, 1H, J = 3.4 Hz, 2′-H), 4.69 (dd, 1H, J = 10, 2 Hz, 1D–H), 4.77 (d, 1H, J = 11.5 Hz, 2-H), 4.77 (dd, 1H, J = 10, 2 Hz, 1B–H), 5.14 (dd, 1H, J = 10, 2 Hz, 1C–H), 5.43 (dd, 1H, J = 10, 2 Hz, 1A–H), 6.94 (s, 2H, 5-H, and 10–H). 13C NMR (125.7 MHz, acetone-d6, δ): 7.9 (7-CH3), 16.5 (C-6D), 17.6 (C-6B), 17.9 (C-6C and C-6A), 26.2 (C-4′), 28.3 (C-4), 35.2 (C-2D), 37.5 (C-2A), 37.9 (C-2C), 40.0 (C-2B), 43.8 (C-3), 60.0 (1′-OCH3), 68.9 (C-3D), 70.2 (C-4D), 71.3 (C-5D), 72.7 (C-5A and C-5C), 75.4 (C-4A), 75.7 (C-4C), 77.5 (C-4B), 78.1 (C-2), 79.3 (C-1′), 79.5 (C-2′), 81.3 (C-3A), 81.7 (C-3C), 97.1 (C-1A), 99.9 (C-1B), 100.4 (C-1D), 100.8 (C-1C), 101.7 (C-5), 108.0 (C-8a), 108.6 (C-9a), 111.1 (C-7), 117.1 (C-10), 137.0 (C-4a), 139.1 (C-10a), 156.1 (C-8), 160.0 (C-6), 165.4 (C-9), 203.6 (C-1), and 209.8 (C-3′).
NMR and MS Analysis Data for Mithramycin SA, C49H70O23 (4)
Positive-mode ESI-MS m/z (relative intensity) [M + Na+], 1049.4 (100); negative-mode ESI-MS m/z (relative intensity) [M – H]−, 1025.4 (100). UV (MeOH) λmax (ε): 421.5 (10 300), 316.9 (26 800), 284.5 (50 600) nm. FT-IR (KBr) ν: 3443 (OH), 2924, (CH), 2853 (CH), 1739 (C=O), 1646 (C=O), 1457 (C=C), 1369, 1026, 823 cm−1. 1H NMR (400 MHz, pyridine-d5, δ): 1.50 (s, 3H, 3E–CH3), 1.52 (d, 3H, J = 6.5 Hz, 6E–H3), 1.62 (d, 9H, J = 6.0 Hz, 6A–H3, 6C–H3, and 6D–H3), 1.68 (d, 3H, J = 6.0 Hz, 6B–H3), 1.77 (bt, 2H, J = 10.0 Hz, 2E–Ha and 2B–Ha), 1.92 (bdd, 1H, J = 12, 9 Hz, 2C–Ha), 2.02 (bdd, 1H, J = 12, 11 Hz, 2D–Ha), 2.08–2.22 (overlap, 2H, 2A–Ha, and 2D–He), 2.28 (dd, 1H, J = 9, 2 Hz, 2E–He), 2.38 (bd, 1H, J = 10 Hz, 2A–He), 2.47 (s, 3H, 7-CH3), 2.54 (m, 1H, 2C–He), 2.79 (m, 1H, 2B–He), 2.93 (m, 1H, 4-He), 3.11 (bt, 1H, J = 15.2 Hz, 4-Ha) 3.14 (bt, 1H, J = 11 Hz, 3-H), 3.36 (d, 1H, J = 9 Hz, 4E–H), 3.49–3.72 (overlap, 4H, 3A–H, 3B–H, 3C–H, and 5A–H), 3.55 (dd, 1H, 9, 8.5 Hz, 4C–H), 3.62 (s, 3H, 1′-OCH3), 3.84 (bdd, 1H, J = 12.0, 4.5 Hz, 3D–H), 3.93–4.02 (overlap, 4H, 4A–H, 4B–H, 4D–H, 5C–H, and 5D–H), 3.98 (dq, 1H, J = 10.0, 6.0 Hz, 5E–H), 4.29 (dq, 1H, J = 10.0, 6.0 Hz, 5B–H), 4.76 (bd, 1H, J = 10 Hz, 1D–H), 4.86 (d, 1H, J = 1.5 Hz, 1′-H), 4.92 (d, 1H, J = 11 Hz, 2-H), 5.00 (dd, 1H, J = 10, 2 Hz, 1B–H), 5.41 (dd, 1H, J = 10, 2 Hz, 1C–H), 5.53 (dd, 1H, J = 10, 2 Hz, 1E–H), 5.61 (dd, J = 10, 2 Hz, 1A–H), 6.61 (s, 1H, 10-H), and 7.01 (s, 1H, 5-H). 13C NMR (75.4 MHz, methanol-d4, δ): 7.1 (7-CH3), 15.9 (C-6D), 17.1 (C-6B), 17.3 (C-6C), 17.4 (C-6A), 18.0 (C-6E), 26.3 (3E-CH3), 29.7 (C-4), 32.3 (C-2D), 37.1 (C-2A), 37.5 (C-2C), 39.7 (C-2B), 44.6 (C-2E and C-3), 59.6 (1′-OCH3), 68.7 (C-4D), 70.7 (C-3E and C-5E), 70.9 (C-3B and C-5D), 72.5 (C-5C and C-5B), 72.9 (C-5A), 75.1 (C-4A), 75.7 (C-4C), 76.2 (C-3D), 76.8 (C-4E), 77.0 (C-2 and C-4B), 79.5 (C-3A), 81.5 (C-3C), 82.2 (C-1′), 97.5 (C-1A), 97.6 (C-1E), 98.8 (C-1B and C-1D), 100.0 (C-1C), 100.1 (C-5), 108.0 (C-8a), 108.6 (C-9a), 111.7 (C-7), 117.2 (C-10), 138.5 (C-4a), 138.7 (C-10a), 159.2 (C-6), 160.5 (C-8), 165.0 (C-9), 176.8 (C-2′), and 198.4 (C-1).
Acknowledgments
This work was supported by grants of the National Institutes of Health (No. CA091901, to J.R.) and the Spanish Ministry of Science and Technology (BIO97-0771, to J.A.S.) as well as the Plan Regional de Investigación del Principado de Asturias (GE-ME01-05, to J.A.S.). We thank the National Cancer Institute (Bethesda, MD) and Dr. J. Frevert (BioteCon AG, Berlin, Germany) for the antitumor assays, and the referees for helpful discussions.
JA034162H
Contributor Information
José A. Salas, Email: jasf@sauron.quimica.uniovi.es.
Jürgen Rohr, Email: jrohr2@uky.edu.
References
- 1.Skarbek JD, Speedie MK. In: Antitumor Compounds of Natural Origin: Chemistry and Biochemistry. Aszalos A, editor. Vol. 1. CRC Press; Boca Raton, FL: 1981. pp. 191–235. [Google Scholar]
- 2.Wohlert SE, Künzel E, Machinek R, Méndez C, Salas JA, Rohr J. J Nat Prod. 1999;62:119–121. doi: 10.1021/np980355k. [DOI] [PubMed] [Google Scholar]
- 3.Rohr J, Méndez C, Salas JA. Bioorg Chem. 1999;27:41–54. [Google Scholar]
- 4.Katahira R, Uosaki Y, Ogawa H, Yamashita Y, Nakano H, Yoshida M. J Antibiot. 1998;51:267–274. doi: 10.7164/antibiotics.51.267. [DOI] [PubMed] [Google Scholar]
- 5.Jayasuriya H, Lingham RB, Graham P, Quamina D, Herranz L, Genilloud O, Gagliardi M, Danzeisen R, Tomassini JE, Zink DL, Guan ZQ, Singh SB. J Nat Prod. 2002;65:1091–1095. doi: 10.1021/np010642f. [DOI] [PubMed] [Google Scholar]
- 6.Gause GF. In: Antibiotics III–Mechanism of Action of Antimicrobial Antitumor Agents. Corcoran JW, Hahn FE, editors. Vol. 3. Springer-Verlag; Berlin, New York: 1975. pp. 197–202. [Google Scholar]
- 7.Fox KR, Howarth NR. Nucleic Acids Res. 1985;13:8695–8714. doi: 10.1093/nar/13.24.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sastry M, Patel DJ. Biochemistry. 1993;32:6588–6604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- 9.Sastry M, Fiala R, Patel DJ. J Mol Biol. 1995;251:674–689. doi: 10.1006/jmbi.1995.0464. [DOI] [PubMed] [Google Scholar]
- 10.Majee S, Dasgupta D, Chakrabarti A. Eur J Biochem. 1999;260:619–626. doi: 10.1046/j.1432-1327.1999.00159.x. [DOI] [PubMed] [Google Scholar]
- 11.Elias EG, Evans JT. J Bone Jt Surg, Am Vol. 1972;54-A:1730–1736. [PubMed] [Google Scholar]
- 12.Brown JH, Kennedy BJ. New Engl J Med. 1965;272:111–118. doi: 10.1056/NEJM196501212720301. [DOI] [PubMed] [Google Scholar]
- 13.Ryan WG, Schwartz TB, Northrop G. J Am Med Assoc. 1970;213:1153–1157. [PubMed] [Google Scholar]
- 14.Robins PR, Jowsey J. J Lab Clin Med. 1973;82:576–586. [PubMed] [Google Scholar]
- 15.Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Goodman Gilman A, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; New York, St. Louis, MO: 1996. pp. 1267–1268. [Google Scholar]
- 16.Silva DJ, Kahne DE. J Am Chem Soc. 1993;115:7962–7970. [Google Scholar]
- 17.Silva DJ, Goodnow R, Jr, Kahne D. Biochemistry. 1993;32:463–471. doi: 10.1021/bi00053a010. [DOI] [PubMed] [Google Scholar]
- 18.Gao XL, Mirau P, Patel DJ. J Mol Biol. 1992;223:259–279. doi: 10.1016/0022-2836(92)90730-8. [DOI] [PubMed] [Google Scholar]
- 19.Lombo F, Blanco G, Fernandez E, Méndez C, Salas JA. Gene. 1996;172:87–91. doi: 10.1016/0378-1119(96)00029-7. [DOI] [PubMed] [Google Scholar]
- 20.Lombo F, Siems K, Braña AF, Méndez C, Bindseil K, Salas JA. J Bacteriol. 1997;179:3354–3357. doi: 10.1128/jb.179.10.3354-3357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lombo F, Braña AF, Méndez C, Salas JA. J Bacteriol. 1999;181:642–647. doi: 10.1128/jb.181.2.642-647.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez E, Weissbach U, Reillo CS, Braña AF, Méndez C, Rohr J, Salas JA. J Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohr J, Weissbach U, Beninga C, Künzel E, Siems K, Bindseil K, Prado L, Lombo F, Braña AF, Méndez C, Salas JA. Chem Commun. 1998:437–438. [Google Scholar]
- 24.Prado L, Fernandez E, Weissbach U, Blanco G, Quiros LM, Braña AF, Méndez C, Rohr J, Salas JA. Chem Biol. 1999;6:19–30. doi: 10.1016/s1074-5521(99)80017-9. [DOI] [PubMed] [Google Scholar]
- 25.Blanco G, Fernandez E, Fernandez MJ, Braña AF, Weissbach U, Künzel E, Rohr J, Méndez C, Salas JA. Mol Gen Genet. 2000;262:991–1000. doi: 10.1007/pl00008667. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Lonzano MJ, Remsing LL, Quiros LM, Braña AF, Fernandez E, Sanchez C, Méndez C, Rohr J, Salas JA. J Biol Chem. 2000;275:3065–3074. doi: 10.1074/jbc.275.5.3065. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez A, Remsing LL, Lombo F, Fernandez MJ, Prado L, Braña AF, Künzel E, Rohr J, Méndez C, Salas JA. Mol Gen Genet. 2001;264:827–835. doi: 10.1007/s004380000372. [DOI] [PubMed] [Google Scholar]
- 28.Remsing LL, Garcia-Bernardo J, Gonzalez A, Künzel E, Rix U, Braña AF, Bearden DW, Méndez C, Salas JA, Rohr J. J Am Chem Soc. 2002;124:1606–1614. doi: 10.1021/ja0105156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devereux J, Haeberli P, Smithies O. Nucleic Acid Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, Haft DH, Gwinn ML, Nelson WC, Richardson DL, Moffat KS, Qin H, Jiang L, Pamphile W, Crosby M, Shen M, Vamathevan JJ, Lam P, McDonald L, Utterback T, Zalewski C, Makarova KS, Aravind L, Daly MJ, Fraser CM. Science. 1999;286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Summers RG, Donadio S, Staver MJ, Wendt-Pienkowski E, Hutchinson CR, Katz L. Microbiology (Reading, UK) 1997;143:3251–3262. doi: 10.1099/00221287-143-10-3251. [DOI] [PubMed] [Google Scholar]
- 32.Bate N, Butler AR, Smith IP, Cundliffe E. Microbiology (Reading, UK) 2000;146:139–146. doi: 10.1099/00221287-146-1-139. [DOI] [PubMed] [Google Scholar]
- 33.Dinur U, Hagler AT. In: Reviews in Computational Chemistry. Lipkowitz KB, Boyd DB, editors. Vol. 2. VCH Publishers; New York: 1991. pp. 99–164. [Google Scholar]
- 34.Rix U, Fischer C, Remsing LL, Rohr J. Nat Prod Rep. 2002;19:542–580. doi: 10.1039/b103920m. [DOI] [PubMed] [Google Scholar]
- 35.Glycosyltransferase MtmGIV is proposed to be responsible for the attachment of both the first and the last sugar of the trisaccharide unit in MTM. Only the latter activity seems somewhat compromised by the insertional inactivation of MtmW. This might be possible, if the tailoring proteins MtmW (KR) and MtmGIV (GT) arrange themselves close to each other (similar to that in the genetic arrangement), and if only the docking of one set of substrates (alcohol acceptor and NDP-sugar donor) of MtmGIV is slightly affected by the inactivated enzyme MtmW. Note that both proposed substrates for the second glycosyltransfer step are considerably larger than the corresponding substrates of the first glycosyltransfer step.28
- 36.Piel J, Hertweck C, Shipley PR, Hunt DM, Newman MS, Moore BS. Chem Biol. 2000;7:943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 37.Xiang L, Kalaitzis JA, Nilsen G, Chen L, Moore BS. Org Lett. 2002;4:957–960. doi: 10.1021/ol0255155. [DOI] [PubMed] [Google Scholar]
- 38.Montanari A, Rosazza JPN. J Antibiot. 1990;43:883–889. doi: 10.7164/antibiotics.43.883. [DOI] [PubMed] [Google Scholar]
- 39.Boyd MR. Princ Pract Oncol. 1989;3:1–12. [Google Scholar]
- 40.Boyd MR. In: Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval. Teicher B, editor. Humana Press, Inc; Totowa, NJ: 1997. pp. 23–42. [Google Scholar]
- 41.Rabow AA, Shoemaker RH, Sausville EA, Covell DG. J Med Chem. 2002;45:818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]
- 42.Frevert J. personal communication.
- 43.Ginsburg H, Nissani E, Krugliak M, Williamson DH. Mol Biochem Parasitol. 1993;58:7–16. doi: 10.1016/0166-6851(93)90085-c. [DOI] [PubMed] [Google Scholar]
- 44.Givens KT, Kitada S, Chen AK, Rothschiller J, Lee DA. Invest Ophthalmol Visual Sci. 1990;31:1856–1862. [PubMed] [Google Scholar]
- 45.Boyd MR. Drug Dev Res. 1995;34:91–109. [Google Scholar]
- 46.NCI-MTM data acquired from the following website. http://dtp.nci.nih.gov/docs/static_pages/compounds/24559.htm.
- 47.Fernandez E, Weissbach U, Sanchez Reillo C, Braña AF, Méndez C, Rohr J, Salas JA. J Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janssen G, Bibb MJ. Gene. 1993;124:133–134. doi: 10.1016/0378-1119(93)90774-w. [DOI] [PubMed] [Google Scholar]
- 49.Aguirrezabalaga I, Olano C, Allende N, Rodriguez L, Braña AF, Méndez C, Salas JA. Antimicrob Agents Chemother. 2000;44:1266–1275. doi: 10.1128/aac.44.5.1266-1275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 51.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. The John Innes Foundation; Norwich, U.K: 2000. [Google Scholar]
- 52.Sanger F, Nicklen S, Coulson AR. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 54.Altschul SF, Lipman DJ. Proc Natl Acad Sci USA. 1990;87:5509–5513. doi: 10.1073/pnas.87.14.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Nucleic Acid Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]