

Abstract
Mithramycin is an aureolic acid-type antimicrobial and antitumor agent produced by Streptomyces argillaceus. Modifying post-polyketide synthase (PKS) tailoring enzymes involved in the production of mithramycin is an effective way of gaining further information regarding the late steps of its biosynthetic pathway. In addition, new “unnatural” natural products of the aureolic acid-type class are likely to be produced. The role of two such post-PKS tailoring enzymes, encoded by mtmC and mtmTIII, was investigated, and four novel aureolic acid class drugs, two premithramycin-type molecules and two mithramycin derivatives, were isolated from mutant strains constructed by insertional gene inactivation of either of these two genes. From data bank comparisons, the corresponding proteins MtmC and MtmTIII were believed to act as a C-methyltransferase involved in the production of the D-mycarose (sugar E) of mithramycin and as a ketoreductase seemingly involved in the biosynthesis of the mithramycin aglycon, respectively. However, gene inactivation and analysis of the accumulated products revealed that both genes encode enzymes participating in the biosynthesis of the D-mycarose building block. Furthermore, the inactivation of MtmC seems to affect the ketoreductase responsible for 4-ketoreduction of sugar C, a D-olivose. Instead of obtaining premithramycin and mithramycin derivatives with a modified E-sugar upon inactivation of mtmC, compounds were obtained that completely lack the E-sugar moiety and that possess an unexpected 4-ketosugar moiety instead of the D-olivose at the beginning of the lower deoxysaccharide chain. The inactivation of mtmTIII led to the accumulation of 4E-ketomithramycin, showing that this ketoreductase is responsible for the 4-ketoreduction of the D-mycarose moiety. The new compounds of the mutant strains, 4A-ketopremithramycin A2, 4A-keto-9-demethylpremithramycin A2, 4C-keto-demycarosylmithramycin, and 4E-ketomithramycin, indicate surprising substrate flexibility of post-PKS enzymes of the mithramycin biosynthetic pathway. Although the glycosyltransferase responsible for the attachment of D-mycarose cannot transfer the unmethylated sugar to the existing lower disaccharide chain, it can transfer the 4-ketoform of sugar E. In addition, the glycosyltransferase MtmGIV, which is responsible for the linkage of sugar C, is also able to transfer an activated 4-ketosugar. The oxygenase MtmOIV, normally responsible for the oxidative cleavage of the tetracyclic premithramycin B into the tricyclic immediate precursor of mithramycin, can act on a substrate analogue with a modified or even incomplete trisaccharide chain. The same is true for glycosyltransferases MtmGI and MtmGII, both of which partake in the formation and attachment of the A–B disaccharide in mithramycin.
Introduction
Mithramycin (10, also known as plicamycin), a natural product of Streptomyces argillaceus (ATCC 12956) and other Streptomyces spp., along with chromomycin A3, olivomycin A, chromocyclomycin, and UCH9, belongs to the discrete group of aureolic acid anticancer antibiotics.1–19 The aureolic acid antibiotics are effective against a variety of experimental and human tumors.1–4,20–25 Mithramycin has been used clinically to treat testicular carcinoma,1,3,4,19 Paget’s bone disease, and other bone growth disorders.26–29 In particular, its hypocalcemic effect has been used to manage hypercalcemia in patients with malignancy-associated bone lesions.30 Unfortunately, 10 exhibits gastrointestinal, hepatic, renal, and bone marrow toxicity resulting in nausea, vomiting, and bleeding.1,3,29,31 Therefore, its widespread clinical use is limited, making the identification of improved therapeutic mithramycin analogues an important endeavor.
The biosynthetic pathway of 10 has been studied in our laboratories over the past years.4,32–44 On the basis of the knowledge gained thus far, alteration of the biosynthetic pathway provides a mechanism through which modified 10 analogues can be produced “naturally”. We use this approach to target mainly post-polyketide synthase (post-PKS) modifying enzymes, which allow modifications to be made in regions of the molecule identified as crucial for the activity of 10. The mechanism of action of 10 and in particular its DNA binding have been extensively studied. Mithramycin inhibits replication and transcription processes via cross-linking of DNA strands, thereby blocking their template activity for DNA- and RNA-dependent polymerases. It was shown that the deoxysaccharide moieties and the highly functionalized pentyl side chain play major roles2,7–9,19,23,24,28,45 in the DNA binding of 10. In particular, the trisaccharide residue is positioned inside of the DNA minor groove with the D-mycarose moiety sitting in its floor and interacting through H-bonds to both DNA strands. This suggests that changes in the trisaccharide segment may affect binding affinity and sequence selectivity of new 10 analogues. Consequently, the aim of our studies described in this article was to modify the D-mycarose moiety found at the end of the trisaccharide chain.
Past biosynthetic studies revealed to a great extent the sequence of events and role of important post-PKS oxygenases and group transferases in the mithramycin biosynthetic pathway. Inactivation of the four glycosyltransferases (GT) and elucidation of the accumulated products of the resulting mutant strains showed that the GTs MtmGI and MtmGII are involved in the assembly and attachment of the diolivoside fragment A–B,36 while MtmGIII and MtmGIV participate in the assembly of the trisaccharide chain, linking sugar D (a D-oliose) and sugar C (a D-olivose), respectively (see Figures 1 and 2).41 The studies also revealed that the disaccharide A-B can be attached only after the trisaccharide chain has been completed, suggesting the biosynthetic sequence 1 → 3 → 6 → 8 → 9 (Figures 1 and 2). Currently, no GT has been identified for the linkage of D-mycarose, the last sugar of the trisaccharide chain, and so there seemed to be no way to selectively modify the trisaccharide by inhibiting its attachment or to investigate the substrate specificity of this GT.
Figure 1.

Late biosynthetic steps of the mithramycin (10) biosynthetic pathway, including the glycosyltransfer steps, the oxidative cleavage of the fourth ring, and some biosynthetic steps of the deoxysugar units.
Figure 2.
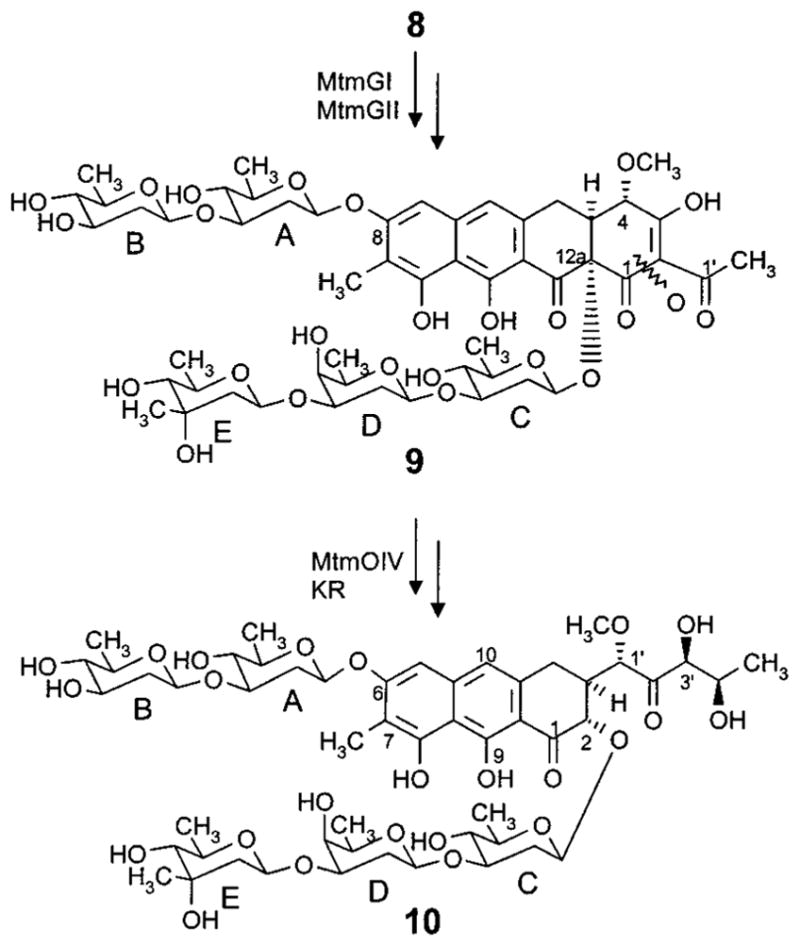
Late biosynthetic steps of the mithramycin (10) biosynthetic pathway, including the glycosyltransfer steps, the oxidative cleavage of the fourth ring, and some biosynthetic steps of the deoxysugar units.
However, three methyltransferases (MT), MtmMI, MtmMII, and MtmC, are present in the mtm gene cluster. Recent investigations on the role of MtmMI and MtmMII identified them as being involved in the biosynthesis of the aglycon. MtmMI is the 4-O-methyltransferase responsible for the conversion of 4-demethylpremithramycinone into premithramycinone (1, Figure 1) prior to the sugar attachments. MtmMII is the C-methyltransferase, which introduces the aromatic methyl group predominantly after completion of the trisaccharide (product 8), but occasionally also after attachment of the first or second sugar of the trisaccharide chain.42 The role for the third potential MT in the mtm gene cluster (MtmC) seemed to be the addition of the one remaining methyl group in 10, specifically that of the mycarose moiety. Thus, it was decided to inactivate MtmC with the hope of revealing a mechanism through which the third sugar could be modified or eliminated. During these studies, the role of the ketoreductase MtmTIII came into question. It seemed that MtmTIII, whose corresponding gene mtmTIII is clustered with the deoxysugar biosynthetic genes and is located immediately upstream of mtmC in the same reading direction, may be responsible for the 4-ketoreduction of sugar C (a D-olivose). To investigate this hypothesis, MtmTIII was also inactivated.
Results
The deduced amino acid sequence of MtmC closely resembles various methyltransferases, most of which are involved in deoxysugar biosynthesis. The highest identity was found with SnoG from the nogalamycin pathway in S. nogalater (62.2%),54 DnrX from the daunorubicin pathway in S. peucetius (37%),55 TylCIII from the tylosin pathway in S. fradiae (32.4%),56,57 and EryBIII from the erythromycin pathway in Sacc. erythraea (31%).58 TylCIII and EryBIII have been proposed to be S-adenosylmethionine-dependent methyltransferases involved in C-3-methylation during the biosynthesis of L-mycarose. On the basis of these similarities, we predicted that mtmC may encode a C-3-methyltransferase involved in the biosynthesis of the D-mycarose moiety of mithramycin. To prove this hypothesis, mtmC was inactivated by gene replacement.44 An apramycin resistance cassette was inserted within the gene in both the same and the reverse orientations with respect to the transcriptional direction of mtmC (M7C1 and M7C1R). As expected, HPLC analysis of cultures of both mutants showed that they did not produce 10. Instead, two identical compounds accumulated in both strains, while a third compound was detected only in M7C1.
The compounds were purified by preparative HPLC, and their structures were determined by NMR and mass spectroscopy. The major compound of both mutant strains exhibits all of the typical NMR signals of a tetracyclic premithramycin-type aglycon including the 9-methyl group, but only has two sugar moieties present. Unexpectedly, one of these sugars contains a hydrated keto group in the 4-position. This is strongly supported by the NMR data (Tables 1 and 2), which reveal a quaternary carbon in the 13C NMR spectrum (δC 90.6, no signal in the HSQC spectrum) and no proton in the 4-position (1H NMR). The H,H-COSY, HSQC, and HMBC data (Table 3) allow the unambiguous assignment of all of the carbons and protons of this 4-ketosugar and also, through the observation of the 3JC–H coupling between C-12a and 1-H of the sugar in the HMBC spectrum, indicate its linkage to the 12a-position of the aglycon. The H,H coupling constants, along with the other 1H and 13C NMR data, prove the other sugar moiety to be an oliose. The presence of only small couplings to the 4B-H indicating the axial position of the 4-OH group is most important. The oliose is linked with its C-1 to the oxygen in the 3-position of the ketosugar, as implied by the 3JC–H couplings between its 1-H and C-3 of the ketosugar as well as between its C-1 and 3-H of the ketosugar moiety visible in the HMBC spectrum (Table 3). The negative FAB mass spectrum shows a molecular ion (M–H− 703, 100%, HR calculated for C34H39O16, 703.2238; found, 703.2206), which corresponds to a mass difference of 16 amu from premithramycin A2 (9-methyl-6)36 and thus supports the structure with the hydrated keto group. Therefore, the main compound is 4A-ketopremithramycin A2 (11, Figure 3).
Table 1.
1H NMR Data of 4A-Ketopremithramycin A2 (11), 4A-Keto-9-demethylpremithramycin A2 (12), 4C-Keto-demycarosylmithramycin (13), and 4E-Ketomithramycin (14) in Acetone-d6 at 400 MHz (δ in ppm Relative to Internal TMS)a
|
δ multiplicity (J/Hz)
|
||||
|---|---|---|---|---|
| 11 | 12 | 13 | 14c | |
| 2-H | 4.70 d (12) | |||
| 3-H | 2.78 br t (12) | 4.80 d (12) | ||
| 4-H | 4.24 s br | 4.31 s br | a: 2.92 dd (16)b e: 2.60 dd (16,3) |
2.82 br t (12) a: 3.00 complex |
| 4-OCH3 | 3.6 s | 3.61 d (1) | e: 2.68 dd (16,3) | |
| 4a-H | 3.13 ddd (11,4,3) | 3.13 ddd (11,4,3) | ||
| 5-H | a: 3.95 (16,4,1) e: 3.05 (16,3) |
3.96 d br (16) 3.07 obsc |
6.81 s | 6.86 s |
| 6-H | 6.97 d (1) | 7.03 s br | ||
| 7-H | 6.75 s | 6.69 s | ||
| 7-CH3 | 2.09 s | 2.15 s | ||
| 9-H | 6.45 s | |||
| 9-CH3 | 2.19 s | |||
| 10-H | 6.85 s | 6.84 s | ||
| 1′ | 4.88 d (1.5) | 4.88 d (2) | ||
| 1′-OCH3 | 3.40 s | 3.44 s | ||
| 2′ | 2.65 s | 2.64 s | ||
| 3′ | 4.24 d (3)b | 4.30 d (3)b | ||
| 4′ | 4.23 dq (6.5,3)b | 4.30 dq (6,3)b | ||
| 5′ | 1.22 d (6.5) | 1.27 d (6) | ||
| 1A | 4.94 dd (10,2) | 4.95 dd (10,2) | 5.34 dd (10,2) | 5.33 dd (10,2) |
| 2Aa | 1.86 ddd (12,12,10) | 1.85 ddd (12,12,10) | 1.80 ddd (12,12,10) | 1.85 ddd (12,12,10) |
| 2Ae | 2.45 m br | 2.44 ddd (12,5,2,) | 2.43 ddd (12,5,2)b | 2.40 ddd (12,5,2)b |
| 3A | 3.70 dd (12,5)b | 3.71 dd (12,5)b | 3.81b | 3.70b |
| 4A | 3.06 dd (9,9) | 3.06 dd (9,9) | ||
| 5A | 3.45 q (6) | 3.44 q (6)b | 3.52 dq (9,6) | 3.51 dq (9,6) |
| 5A-CH3 | 1.25 d (6) | 1.25 d (6) | 1.26 d (6) | 1.30 d (6) |
| 1B | 4.68 dd (10,2) | 4.69 dd (10,2) | 4.74 dd (10,2) | 4.72 dd (10,2) |
| 2B | 1.70 ddd (12,12,10) 1.90 ddd (12,5,2) |
1.73 ddd (12,12,10) 1.92 ddd (12,5,2) |
a: 1.55 ddd (12,12,10) e: 2.19 ddd (12,5,2) |
1.58 ddd (12,12,10) 2.20 ddd (12,5,2) |
| 3B | 3.82 ddd (12,5,3) | 3.83 mb | 3.6 ddd (12,9,5) | 3.60 ddd (12,9,5) |
| 4B | 3.52 s br | 3.50 s br | 2.99 dd (9,9) | 3.01 dd (9,9) |
| 5B | 3.70 q (6.5) | 3.70 q (6.5) | 3.40 dq (9,6) | 3.41 dq (9,6) |
| 5B-CH3 | 1.25 d (6.5) | 1.25 d (6.5) | 1.26 d (6) | 1.30 d (6) |
| 1C | 5.09 dd (10,2) | 5.14 dd (10,2) | ||
| 2Ca | 1.88 ddd (12,12,10) | 1.62 ddd (12,12,10) | ||
| 2Ce | 2.42 ddd (12,5,2)b | 2.55 ddd (12,5,2) | ||
| 3C | 3.84 dd (12,5) | 3.70 complex | ||
| 4C | 3.02 dd (9,9) | |||
| 5C | 3.40 q (6) | 3.32 dq (9,6) | ||
| 5C-CH3 | 1.26 d (6) | 1.30 d (6) | ||
| 1D | 4.74 dd (10,2) | 4.7 dd (10,2) | ||
| 2Da | 1.80 ddd (12,12,10) | 1.76 ddd (12,12,10) | ||
| 2De | 1.98 ddd (12,5,2) | 1.93 ddd (12,5,2) | ||
| 3D | 3.81 ddd (12,5,3)b | 3.82 complex | ||
| 4D | 3.56 s br | 3.54 s br | ||
| 5D | 3.70 q br (6.5) | 3.74 q (6) | ||
| 5D-CH3 | 1.26 d (6.5) | 1.24 d (6) | ||
br = broad; obsc = obscured by solvent or water; OH signals not visible due to 5% water in the samples.
Complex, partially overlapped by other signal(s).
Sugar E signals: 1-E, 5.42 dd (10,2); 2-Ea, 1.98 dd (14,10); 2-Ee, 2.30 dd (14,2); 3-E-CH3, 1.31 s; 5-E, 4.66 q (6); 5E-CH3, 1.30 d (6).
Table 2.
13C NMR Data of 4A-Ketopremithramycin A2 (11), 4A-Keto-9-demethylpremithramycin A2 (12), 4C-Keto-demycarosylmithramycin (13), and 4E-Ketomithramycin (14) in Acetone-d6 at 125.7 MHz (δ in ppm Relative to Internal TMS, Assignments Were Made with the Help of Couplings Determined in the HSQC and HMBC Spectra)a
|
δ
|
δ
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| 11 | 12 | 13 | 14 | 11 | 12 | 13 | 14 | ||
| 1 | 196.1 | n.o. | 203.7 | 204.0 | 1A | 97.9 | 97.9 | 96.8 | 96.9 |
| 2 | 112.6 | 112.6 | 77.6 | 76.8 | 2A | 36.9 | 36.9 | 37.2 | 37.5 |
| 3 | 193.1 | n.o. | 42.4 | 42.8 | 3A | 82.0 | 82.1 | 80.4 | 81.4 |
| 4 | 77.6 | 77.6 | 27.1 | 27.3 | 4A | 90.6 | 91.0 | 75.2 | 75.4 |
| 4-OCH3 | 61.2 | 61.2 | 5A | 70.5 | 70.5 | 72.3 | 72.6 | ||
| 4a | 42.7 | 42.5 | 136.4 | 136.5 | 6A | 12.7 | 12.7 | 17.9 | 17.9 |
| 5 | 26.5 | 27.0 | 101.7 | 101.7 | 1B | 100.5 | 100.5 | 99.4 | 100.0 |
| 5a | 134.3 | 133.0 | 2B | 34.8 | 34.8 | 39.7 | 40.0 | ||
| 6 | 117.6 | 117.8 | 159.6 | 159.2 | 3B | 68.8 | 68.9 | 70.9 | 71.3 |
| 7 | 102.3 | 103.0 | 111.1 | 111.1 | 4B | 70.3 | 70.6 | 77.0 | 77.5 |
| 7-CH3 | 7.97 | 7.9 | 5B | 71.5 | 71.5 | 72.6 | 72.6 | ||
| 8 | 161.7 | 163.0 | 155.7 | 156.2 | 6B | 16.4 | 16.7 | 17.6 | 16.5 |
| 8a | 107.8 | 108.4 | 1C | 101.3 | 100.9 | ||||
| 9 | 110.4 | 102.0 | 164.0 | 165.3 | 2C | 36.3 | 38.0 | ||
| 9a | 108.7 | 108.6 | 3C | 81.4 | 81.8 | ||||
| 9-CH3 | 7.7 | 4C | 91.3 | 75.7 | |||||
| 10 | 157.1 | 160.6 | 117.3 | 117.2 | 5C | 73.8 | 72.6 | ||
| 10a | 106.8 | 107.1 | 138.9 | 139.0 | 6C | 12.7 | 17.6 | ||
| 1′ | 202.9 | 202.9 | 81.9 | 82.1 | 1D | 100.1 | 100.4 | ||
| 1′-OCH3 | 58.7 | 58.8 | 2D | 34.4 | 36.0 | ||||
| 2′ | 27.9 | 27.3 | 212.1 | 211.3 | 3D | 68.7 | 68.9 | ||
| 3′ | 79.1 | 79.2 | 4D | 69.9 | 70.2 | ||||
| 4′ | 68.3 | 68.4 | 5D | 71.5 | 71.1 | ||||
| 5′ | 19.3 | 19.6 | 6D | 16.4 | 14.7 | ||||
| 1E | 97.5 | ||||||||
| 2E | 48.0 | ||||||||
| 3E | 69.2 | ||||||||
| 3E-CH3 | 23.8 | ||||||||
| 4E | 206.0 | ||||||||
| 5E | 71.7 | ||||||||
| 6E | 18.0 | ||||||||
n.o. = not observed.
Table 3.
Long-Range Couplings Observed in the HMBC Spectra of the New Premithramycin (11, 12) and Mithramycin Derivatives (13, 14) (Most Important Couplings Are Highlighted in Bold)
| H-position | C-position
|
|||
|---|---|---|---|---|
| 11 | 12 | 13 | 14 | |
| 2 | 1,3,1′,1C | 3,1′,1C | ||
| 3 | 4 | |||
| 4 | 1,4-OCH3,4a,5 | |||
| α | 4a | 4a,10 | ||
| β | 2,3,4a,9a,10 | 2,4a,9a,10 | ||
| 4-OCH3 | 4 | 4 | ||
| 4a | ||||
| 5 | 6,7,8,8a,9,9a,10 | 6,7,10 | ||
| α | 4a,5a | |||
| β | ||||
| 6 | 5,6a,7,10a,11a | 5,7,11a | ||
| 7 | 6,8,9,10a | 6,8,9,10a | ||
| 7-CH3 | 5,6,7,8,8a,9a | 6,7,8,10a | ||
| 9 | 7,8,10,10a | |||
| 9-CH3 | 8,9,10 | |||
| 10 | 4,5,8a,9a,10a | 4,5,8,8a,9,9a,10a | ||
| 14 | 2,13 | 13 | ||
| 1′ | 2,3,4,1′-OCH3,2′ | 3,4,1′-OCH3,2′ | ||
| 1′-OCH3 | 1′,3′ | 2,1′ | ||
| 3′ | 2′,4′,5′ | 2′ | ||
| 4′ | 2′,5′ | |||
| 5′ | 2′,3′,4′ | 3′,4′ | ||
| 1A | 12a | 6 | 6,2A | |
| 2Aa | 1A,3A | 1A,3A | 1A,3A | 1A,3A |
| 2Ae | 3A,4A | 3A,4A | 1A,3A,4A | 1A,3A,4A |
| 3A | 1B | 1B | 2A,4A,1B | 1B |
| 4A | 3A,5A,6A | 3A,5A | ||
| 5A | 1A,6A | 1A,6A | 1A,3A,4A,6A | 1A,4A |
| 6A | 4A,5A | 4A,5A | 4A,5A | 5A |
| 1B | 3A | 3A | 3A | 3A,2B |
| 2Ba | 1B,3B | 1B,3B | 1B,3B,4B | 1B,3B |
| 2Be | 1B,3B,4B | 1B,3B,4B | ||
| 3B | 2B,4B,5B | 4B | ||
| 4B | 5B | 5B | 3B,5B,6B | 3B,5B |
| 5B | 4B,6B | 4B,6B | 1B,3B,4B,6B | 1B,3B,4B |
| 6B | 5B | 5B | 3B,4B,5B | 2B,5B |
| 1C | 2,2C | |||
| 2Ca | 1C,3C | 1C,3C | ||
| 2Ce | 1C,3C,4C | 1C,3C,4C | ||
| 3C | 2C,1D | 4C,1D | ||
| 4C | 3C,5C | |||
| 5C | 1C,3C,4C,6C | 1C,4C | ||
| 6C | 4C,5C | 2C,4C,5C | ||
| 1D | 3C | 3C | ||
| 2Da | 1D,3D | 1D,3D | ||
| 2De | 4D | |||
| 3D | 2D,4D | |||
| 4D | 2D,3D,5D | 2D,3D | ||
| 5D | 1D,4D,6D | 3D,4D | ||
| 6D | 4D,5D | |||
| 1E | 2E | |||
| 2Ea | 1E | |||
| 2Ee | 1E,4E | |||
| 3E-CH3 | 1E,2E,3E,4E | |||
| 5E | 1E,4E | |||
| 6E | 4E,5E | |||
Figure 3.

New structures resulting from the mtmC- and mtmTIII-inactivation experiments: 4A-ketopremithramycin A2 (11), 4A-keto-9-demethylpremithramycin A2 (12), 4C-keto-demycarosylmithramycin (13), and 4E-ketomithramycin (14).
Compared to this compound, the minor compound found in both mutant strains differs only in that it is missing the aromatic 9-methyl group. Instead, 9-H can be observed in the 1H NMR spectrum at δ 6.45. The chemical shifts and coupling constants of all other 1H NMR signals are almost identical with the major compound allowing us to suggest 4A-keto-9-demethylpremith-ramycin A2 (12) as the structure for this minor compound.
The third compound, which is only observed as a product of the M7C1 mutant, exhibits the typical UV light (366 nm)-induced yellow fluorescence of the tricyclic mithramycin chromophore. Indeed, the NMR data reveal a compound that possesses the tricyclic mithramycinone skeleton, but only four deoxysugar building blocks. Comparison with the mithramycin NMR data11,37,39 indicates that the A-B disaccharide attached at C-6 is unchanged, while the lower chain attached at C-2 is identical to the disaccharide containing the hydrated 4-ketosugar found in 11 and 12. Thus, 4C-keto-demycarosylmithramycin (13) was deduced as the structure of this third compound. Long-range couplings observed in the HMBC spectrum (Table 3) as well as the negative FAB mass spectrum (M− 956, 100%) confirm these conclusions.
Interestingly, all three novel compounds discovered by inactivation of mtmC are chemically in a keto/hydrate equilibrium. The keto form of the olivose attached at C-12a and C-2, respectively, is the minor form (10–20%), visible in the 1H NMR spectrum (shifted 1-, 3-, and 5-positions, data not shown). The keto form is not detectable in the 13C NMR. Changing the solvent from acetone to DMSO increases the amount of the keto form (data not shown), which becomes dominant after being dissolved in DMSO for 72 h.
While the absence of the D-mycarose moiety in all three new compounds is in agreement with the proposed function for MtmC as the C-3-methyltransferase necessary for the formation of the D-mycarose unit (see Discussion), the appearance of a hydrated keto group at the 4-position of the first sugar of the lower saccharide chain (normally a D-olivose, as in 10) was unexpected and intriguing. This suggested that in both mutants another enzymatic function, a sugar 4-ketoreductase, was also affected. Our initial interpretation was that the inactivation of mtmC was causing a polar effect on genes transcribed in the same direction and following mtmC.34,36,38–42 Since a potential ketoreductase gene, mtmTIII, is located adjacent to mtmC and immediately upstream in the same reading direction, this gene was consequently thought to be most likely affected.
To further clarify this situation, two experiments were carried out: (i) complementation experiments with mtmC cloned in a multicopy plasmid using the M7C1 and M7C1R mutants as transformation hosts, and (ii) inactivation of mtmTIII. In the first experiment, overexpression of mtmC restored the production of mithramycin in both the M7C1R and the M7C1 mutants.44 The anticipated 4C-ketomithramycin was not produced. Thus, it is unlikely that the presence of the 4-keto group of sugar A in 11 and 12 and sugar C in 13 is caused by a polar effect. For the second experiment, the mtmTIII-minus mutant was constructed by inserting an apramycin resistance cassette within the mtmTIII gene (Figure 4). This mutant (M7T3) yielded only one compound, a mithramycin derivative, which was subsequently identified as 4E-ketomithramycin (14). The negative APCI-MS reveals an (M–H)− peak at m/z 1081.3 (31%), which is 2 amu smaller than the corresponding peak for mithramycin (10) and in agreement with a calculated molecular formula of C52H74O24, which lacks two protons as compared to the mithramycin molecular formula. While most of the NMR signals of this novel compound 14 are very similar to those of mithramycin,11,37,39 significant differences can be seen for the signals of sugar E, which is a 4-keto-D-mycarose in 14 and D-mycarose in 10. Most obvious, the signal for C-4E (δ 76.1 in 10) is shifted downfield to δ 206.0 in 14, typical for a keto group. This keto group signal shows 3JC–H couplings to the 3E-and 5E-methyl group protons (see Table 3), which proves its location at the 4-position. Other significant shift differences for C-1E, C-3E, and C-5E as well as for the protons attached to these carbons were also observed, all in agreement with a 4E-carbonyl group. In contrast to the keto group in the 4C-position mentioned in context with structure 13, the 4E-keto group in 14 clearly appears as a carbonyl, not as a hydrate (keto:hydrate ≈ 95:5), as the 13C NMR spectrum shows no signal in the δ 90 region.
Figure 4.
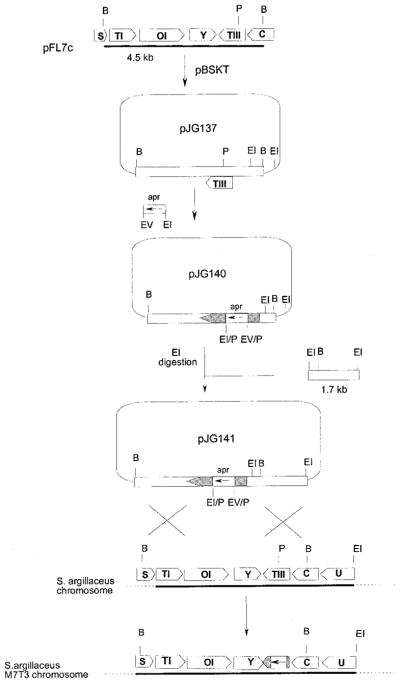
Scheme representing the insertional inactivation of mtmTIII and the replacement in the chromosome of the wild-type mtmTIII allele by the in vitro mutated one. B, BamHI; EI, EcoRI; EV, EcoRV; P, PmlI; apr, apramycin resistance gene.
Discussion
The results of the above-described experiments clearly point out that MtmC is the 3-C-methyltransferase necessary for the biosynthesis of the D-mycarose moiety of mithramycin. This is supported by its sequence similarity to TylC3 (TylCIII) and EryBIII, other proteins that act as methyltransferases involved in L-mycarose building block biosyntheses. Particular proof came from the inactivation of the mtmC gene leading to the accumulation of compounds lacking the D-mycarose moiety of the trisaccharide chain. It seems that the glycosyltransferase responsible for the attachment of D-mycarose to the preceding D-oliose moiety, possibly MtmGIV (see below), is unable to recognize an unmethylated sugar donor substrate for this particular glycosyl transfer step and, therefore, seems to be inhibited when the methyl group is missing in the sugar cosubstrate.
The accumulation of products showing an unreduced C-4 carbonyl group at the first sugar moiety of the lower disaccharide chain was also proven to be the consequence of the inactivation of mtmC, since mithramycin biosynthesis was restored in the corresponding mutants by expressing in trans the mtmC gene. Since a polar effect on the neighbor gene mtmTIII could be ruled out, this result suggests that inactivation of mtmC is affecting a ketoreductase other than MtmTIII. Possible candidates are MtmTI or MtmTII, whose functions within the biosynthetic pathway of 10 have yet to be determined. Type-II PKSs are likely to be assembled in a highly structured multifunctional array resulting in a 3-dimensional structure, which might be crucial to assist folding of the growing polyketide chain as well as for the activity of certain biosynthetic enzymes. Similar unpredicted results as found here led already to the conclusion that a complex association of many proteins is sometimes necessary in type-II PKS systems and that removal of one component can have unpredictable indirect effects on the behavior of the remaining activities.59 Thus, the modification of the spatial structure of the protein cluster by inactivation of mtmC may in this case be the underlying cause by which the ketoreductase responsible for the 4-reduction of the first sugar building block of the trisaccharide chain, although expressed, is rendered inactive.
Inactivation of MtmTIII showed that this enzyme is responsible for the 4-ketoreduction step of the mycarose building block biosynthesis, considering the product isolated from the MtmTIII mutant (14) contains a 4-ketomycarose instead of a mycarose unit at the end of the trisaccharide chain. That MtmTIII is responsible for a ketoreduction in a sugar building block biosynthesis seemed at first somewhat surprising, since the deduced amino acid sequence of MtmTIII resembles mostly ketoreductases involved in polyketide aglycon biosyntheses. The closest resemblance was found for DnrE and DauE,40 which catalyze an early step of the biosynthesis of anthracyclinones, namely the reduction of the tetracyclic aklaviketone to aklavi-none. Only weak resemblance could be found with ketoreductases involved in deoxysugar biosyntheses. A possible explanation for this is that the reduction of the 4-keto group of this sugar moiety happens after its attachment. Thus, MtmTIII has to act on a large molecule, composed of a tetracyclic polyketide-derived aglycon (like aklaviketone) and an attached trisaccharide, instead of on a single NDP-deoxysugar. Transfer of biosynthetic intermediates of deoxysugars that are still unreduced at their 4-position is possible; however, it is very unusual and rarely found in antibiotic biosynthetic pathways. For example, in a recent publication, Liu and co-workers generated new methymycin/pikromycin analogues bearing modified sugars, revealing a highly relaxed sugar cosubstrate specificity of the glycosyltransferase DesVII. Despite its unusual substrate flexibility, DesVII failed to transfer 4-ketosugars.60 In contrast, an EryBIV mutant from the erythromycin producer, Saccharopolyspora erythraea, has been previously reported to transfer a 4-keto-L-mycarose unit to the erythronolide B aglycon.61 Also, the GT responsible for the glycosyltransfer step, through which the D-mycarose moiety is linked during the biosynthesis of 10, as well as the GT, which attaches the first sugar unit of the trisaccharide chain, are capable of transferring 4-ketosugars, as was shown in products 11–13 and 14, respectively. Since such a capability is rarely found among GTs, it seems reasonable to assume that a single GT may be responsible for both of these transfer steps and that, as suggested by the results presented above, the corresponding sugars are reduced after attachment as ketosugars. A dual role for one of the mtm GTs is also necessary because of the presence of five sugar moieties, but only four GT encoding genes in the mtm gene cluster. MtmGIV has previously been shown to be responsible for the attachment of the first sugar of the trisaccharide chain, a D-olivose, and thus is the best candidate for such a double role. As with all GTs, MtmGIV is believed to have two binding sites, one for the alcohol substrate or the acceptor substrate (here 1) and a second for the sugar cosubstrate, the donor substrate. It seems that MtmGIV may have a unique sugar binding site, which recognizes and selectively attaches two different sugars, a 4-ketoolivose as the first sugar and a 4-ketomycarose as the last sugar of the trisaccharide chain, respectively. The change of the alcohol acceptor substrate, here in particular the absence or presence of a D-olivose-D-oliose disaccharide chain, might also influence the sugar donor binding site, thereby changing its specificity and thus accounting for the differentiation between NDP-ketoolivose and NDP-ketomycarose as sugar cosubstrates.
The experiments described here also raise important issues regarding the flexibility of other enzymes involved in the biosynthesis of 10. For example, MtmGIII, which normally attaches D-oliose to the (already 4-reduced!) D-olivose of premithramycin A1 (3), shows a unique flexibility toward its acceptor substrate by attaching D-oliose also to a 4-ketosugar. To our knowledge, this is the first example in a deoxysaccharide biosynthesis in which a GT was able to link a deoxysugar to another genetically modified deoxysugar that includes a keto group neighboring the linkage position. The hydrate formation, which changes 4-keto-D-olivose to 4-hydroxy-D-olivose, may be indeed helpful for this process, since 4-hydroxy-olivose is stereoelectronically more similar to olivose than the keto form. In addition, compound 13 is the first case in which MtmGI and MtmGII have been shown to attach the disaccharide chain even though the trisaccharide chain is incomplete. It is also worth pointing out that oxygenase MtmOIV was capable of oxidatively opening the fourth ring of the 13 precursor, although this lacks the mycarose moiety and has the (hydrated) keto function in the 4C-position as compared to the natural substrate premithramycin B (9). Furthermore, MtmOIV was able to process the precursor of 4E-ketomithramycin (14) despite its keto function in the 4E-position. Along with the previously published 10 derivatives, 7-demethylmithramycin and premithramycin A4, 13 and 14 are further examples demonstrating the relatively high degree of substrate flexibility of MtmOIV. This is an important finding, since MtmOIV plays a key role in the generation of novel mithramycin derivatives by combinatorial biosynthesis due to its ability to convert biologically inactive tetracyclic premithramycins into active tricyclic mithramycins.39
In conclusion, the work described here on the two neighboring genes mtmC and mtmTIII reveals that the corresponding enzymes, MtmC and MtmTIII, are responsible for the 3-methyltransfer and the 4-ketoreduction, respectively, occurring during the generation of the D-mycarose moiety of 10. Glycosyltransferase MtmGIV presumably displays a double role, likely being responsible for the linkage of both the first and the third sugar of the trisaccharide chain. These sugars appear to be attached as 4-ketosugars, and the 4-ketoreduction happens after the glycosyltransfer step. Finally, four new compounds, two of which are potentially active 10 analogues, were created as a result of the inactivation experiments. All these results also provide important additional information regarding the substrate specificity of several of the post-PKS enzymes involved in mithramycin biosynthesis.
Experimental Section
Microorganisms, Culture Conditions, and Vectors
Streptomyces argillaceus ATCC 12956, a mithramycin producer, was used as the source of chromosomal DNA. For sporulation it was grown for 7 days at 30 °C on plates containing A medium.36 For protoplast regeneration, the organism was grown on R5 solid medium,46 and R5A medium was used as the liquid medium for production.36 Escherichia coli XL1-Blue47 was used as the host for subcloning and was grown at 37 °C in TSB medium (tryptic soy broth, Oxoid). When plasmid-containing clones were grown, the medium was supplemented with the appropriate antibiotics: 5 or 50 μg/mL of thiostrepton for liquid or solid cultures, respectively, 100 μg/mL of ampicillin, 25 μg/mL of apramycin, or 20 μg/mL of tobramycin. Plasmid pBSKT38 was the vector used for insertional inactivation experiments.
DNA Manipulation
Plasmid DNA preparations, restriction endonuclease digestions, alkaline phosphatase treatments, ligations, and other DNA manipulations were performed according to standard procedures for E. coli 48 and for Streptomyces.46
Insertional Inactivation of mtmC
A 5.5 kb PstI fragment containing the mtmC gene was subcloned as a XbaI–HindIII fragment (using these sites from the polylinker) into the same sites of pBSKT, generating pM7C0. A BamHI–BglII fragment from pUO9090, containing an apramycin resistance cassette, was then subcloned in both orientations into the unique BamHI site in pM7C0 located within the mtmC gene, generating two different constructs: pM7C1, with the resistance gene oriented in the same direction of transcription as mtmC, and pM7C1R, with the resistance gene inserted in the opposite direction. These constructs were used to transform protoplasts of the wild-type strain S. argillaceus ATCC 12956. Transformants were selected on the basis of resistance to 25 μg/mL of apramycin. To select disruptants in which the wild-type region of the chromosome was replaced by the in vitro mutated one through a double crossover, transformant colonies were grown in TSB medium without antibiotics. After 72 h, protoplasts were obtained and plated onto a regeneration medium. Growing colonies were tested for resistance to apramycin and sensitivity to thiostrepton (50 μg/mL). The replacement was also confirmed by Southern analysis.
Complementation of the mtmC-Minus Mutants
The 2 kb SnaBI–PmlI fragment from pLP7 was subcloned into the SmaI site of pUK21, generating PAGC0. Then the SpeI fragment using the sites from the flanking polylinker was subcloned into the XbaI site of pIAGO, in the correct orientation and downstream of the erythromycin resistance promoter, generating the multicopy plasmid pAGC1. Protoplasts of mutants M7C1 and M7C1R were transformed with pAGC1.
Insertional Inactivation of mtmTIII
A 4.5 kb BamHI fragment containing the mtmTIII gene was subcloned into the BamHI site of pBSKT. In this construct, an apramycin resistance cassette was subcloned as an EcoRI–EcoRV (this site blunt-ended) fragment into the unique PmlI site which is located within the mtmTIII coding region. This construct (pJG140) was digested with EcoRI, and a 1.7 kb EcoRI fragment was inserted to produce pJG141, in which the right-hand side of the insert was enlarged with additional chromosomal DNA from that region to facilitate the occurrence of a crossover. This final construct (pJG141) was used to transform protoplasts of the wild-type strain S. argillaceus ATCC 12956. Transformants in which the replacement of the wild-type region of the chromosome was replaced by the in vitro mutated one through a double crossover at both sides of the apramycin cassette were recognized by resistance to apramycin and sensitivity to thiostrepton (Figures 4 and 5). The replacement was also verified by Southern analysis.
Figure 5.
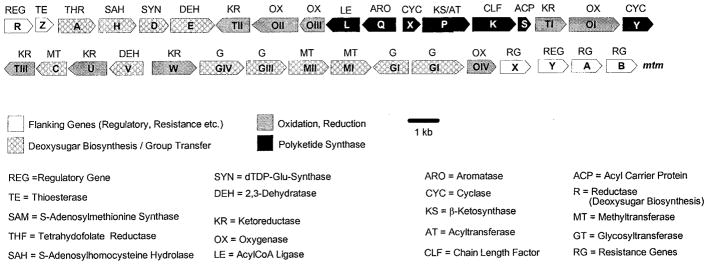
Genetic organization of the mithramycin gene cluster.
Isolation, Purification, and HPLC of the New Compounds
Detection of mithramycin-related compounds was performed on a reversed phase column (Symmetry C18, 4.6 × 250 mm, Waters), with acetonitrile and water supplemented with 0.1% trifluoroacetic acid as solvents (linear gradient from 10 to 100% acetonitrile in 30 min, at a flow rate of 1 mL/min). Detection and spectral characterization of peaks were accomplished with a photodiode array detector and Millennium software (Waters), extracting bidimensional chromatograms at 280 nm. To isolate the new compounds, spores of the mutants were initially grown in TSB medium during 24 h at 30 °C and 250 rpm. This seed culture was used to inoculate (at 2.5%, v/v) eight 2 L Erlenmeyer flasks containing 400 mL of R5A medium. After incubation for 4 days at the above conditions, the cultures were centrifuged, filtered, and extracted as described.36 Mithramycin-related compounds (identified according to their spectral characteristics) were purified by preparative HPLC on a μBondapak C18 radial compression cartridge (PrepPak Cartridge, 25 × 100 mm, Waters). Short gradients using 0.1% trifluoroacetic acid in water and either methanol or acetonitrile, at 10 mL/min, were optimized for resolution of individual peaks. The purified material collected in each case was diluted 4-fold with water, applied to a solid-phase extraction column (Lichrolut RP-18, Merck), washed with water to eliminate trifluoroacetic acid, eluted with methanol, and dried in vacuo. Yields: 11, 12 mg/L; 12, 10 mg/L; 13, 3 mg/L; 14, 12 mg/L (for comparison reasons; the production of 10 by the wild-type strain of S. argillaceus is 10–12 mg/L).
Structure Elucidation of 4A-Ketopremithramycin A2, 4A-Keto-9-demethylpremithramycin A2, 4C-Keto-demycarosylmithramycin, and 4E-Ketomithramycin
The structures of new premithramycin and mithramycin derivatives were elucidated by NMR and mass spectroscopy. The positive fast atom bombardment (FAB) and high-resolution (HR-FAB) mass spectra were acquired at the University of South Carolina, Department of Biochemistry and Chemistry facilities in Columbia, SC, using a VG70SQ double focusing magnetic sector MS instrument. The atmospheric pressure chemical ionization mass spectra (APCI-MS) were acquired at the Medical University of South Carolina, Regional Mass Spectroscopy Center, using a Finnigan MAT LCQ. All the NMR data were performed in d6-acetone/D2O (95:5), either on a Varian Inova 400 or a Bruker DMX 500 instrument. The NMR data are listed in Tables 1 and 2; the important long-range C–H couplings observed in the HMBC spectra are shown in Table 3. All NMR assignments are based on DEPT, H,H-COSY, HSQC, TOCSY, and HMBC spectra,37,49–53 allowing an unambiguous assignment of all NMR signals.
Acknowledgments
This work was supported by grants of the South Carolina Commission of Higher Education (2000–2001), the U.S. Department of Defense, and the National Institutes of Health (R01CA91901) to J.R., and by the Spanish Ministry of Education and Science to J.A.S. through the “Plan Nacional en Biotecnologia” (BIO97-0771). Drs. William Cotham and Michael Walla, Department of Chemistry and Biochemistry, University of South Carolina, are acknowledged for recording the FAB mass spectra.
References
- 1.Skarbek JD, Speedie MK. Antitumor Antibiotics of the Aureolic Acid Group: Chromomycin A3, Mithramycin A, and Olivomycin A. In: Aszalos A, editor. Antitumor Compounds of Natural Origin. Vol. 1. CRC Press; Boca Raton, FL: 1981. pp. 191–235. [Google Scholar]
- 2.Van Dyke MW, Dervan PB. Biochemistry. 1983;22:2373–2377. doi: 10.1021/bi00279a011. [DOI] [PubMed] [Google Scholar]
- 3.Remers WA. The Chemistry of Antitumor Antibiotics. Vol. 1. Wiley-Inter-science; New York: 1979. pp. 133–175. [Google Scholar]
- 4.Rohr J, Méndez C, Salas JA. Bioorg Chem. 1999;27:41–54. [Google Scholar]
- 5.Katahira R, Katahira M, Yamashita Y, Ogawa H, Kyogoku Y, Yoshida M. Nucleic Acids Res. 1998;26:744–755. doi: 10.1093/nar/26.3.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katahira R, Uosaki Y, Ogawa H, Yamashita Y, Nakano H, Yoshida M. J Antibiot. 1998;51:267–274. doi: 10.7164/antibiotics.51.267. [DOI] [PubMed] [Google Scholar]
- 7.Sastry M, Patel DJ. Biochemistry. 1993;32:6588–6604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- 8.Sastry M, Fiala R, Patel DJ. J Mol Biol. 1995;251:674–689. doi: 10.1006/jmbi.1995.0464. [DOI] [PubMed] [Google Scholar]
- 9.Rama Krishna N, Miller DM, Sakai TT. J Antibiot. 1990;43:1543–1552. doi: 10.7164/antibiotics.43.1543. [DOI] [PubMed] [Google Scholar]
- 10.Thiem J, Schneider G, Sinnwell V. Liebigs Ann Chem. 1986:814–824. [Google Scholar]
- 11.Thiem J, Meyer B. Tetrahedron. 1981;37:551–558. [Google Scholar]
- 12.Roush WR, Lin XF. J Am Chem Soc. 1995;117:2236–2250. [Google Scholar]
- 13.Thiem J, Schneider G. Angew Chem. 1983;95:54–55. [Google Scholar]
- 14.Thiem J, Meyer B. J Chem Soc, Perkin Trans 2. 1979:1331–1336. [Google Scholar]
- 15.Thiem J, Schöttmer B. Angew Chem. 1987;99:591–592. [Google Scholar]
- 16.Roush WR, Lin XF. Tetrahedron Lett. 1993;34:6829–6832. [Google Scholar]
- 17.Franck RW, Marzabadi CH. J Org Chem. 1998;63:2197–2208. [Google Scholar]
- 18.Roush WR, Hartz RA, Gustin DJ. J Am Chem Soc. 1999;121:1990–1991. [Google Scholar]
- 19.Majee S, Dasgupta D, Chakrabarti A. Eur J Biochem. 1999;260:619–626. doi: 10.1046/j.1432-1327.1999.00159.x. [DOI] [PubMed] [Google Scholar]
- 20.Jones DE, Jr, Cui DM, Miller DM. Oncogene. 1995;10:2323–2330. [PubMed] [Google Scholar]
- 21.Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. J Clin Invest. 1991;88:1613–1621. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray R, Thomas S, Miller DM. Am J Med. 1990;300:203–208. doi: 10.1097/00000441-199010000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Ray R, Snyder RC, Thomas S, Koller CA, Miller DM. J Clin Invest. 1989;83:2003–2007. doi: 10.1172/JCI114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder RC, Ray R, Blume S, Miller DM. Biochemistry. 1991;30:4290–4297. doi: 10.1021/bi00231a027. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy BJ, Yarbro JW, Kickertz V, Sandberg-Wollheim M. Cancer Res. 1968;28:91–97. [PubMed] [Google Scholar]
- 26.Reddy SV, Menaa C, Singer FR, Demulder A, Roodman GD. J Bone Miner Res. 1999;14:3–8. doi: 10.1002/jbmr.5650140203. [DOI] [PubMed] [Google Scholar]
- 27.Cortes EP, Holland JF, Moskowitz R, Depoli E. Cancer Res. 1972;32:74–76. [PubMed] [Google Scholar]
- 28.Hall TJ, Schaeublin M, Chambers TJ. Biochem Biophys Res Commun. 1993;195:1245–1253. doi: 10.1006/bbrc.1993.2178. [DOI] [PubMed] [Google Scholar]
- 29.Elias EG, Evans JT. J Bone Jt Surg. 1972;54A:1730–1736. [PubMed] [Google Scholar]
- 30.Robins P, Jowsey J. J Lab Clin Med. 1973;82:576–586. [PubMed] [Google Scholar]
- 31.Hardmann JG, Limbird LE, Molinoff PB, Ruddon RW, Goodman Gilman A, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; New York, St. Louis: 1996. pp. 1267–1268. [Google Scholar]
- 32.Lombó F, Blanco G, Fernández E, Méndez C, Salas JA. Gene. 1996;172:87–91. doi: 10.1016/0378-1119(96)00029-7. [DOI] [PubMed] [Google Scholar]
- 33.Fernández E, Lombó F, Méndez C, Salas JA. Mol Gen Genet. 1996;251:692–698. doi: 10.1007/BF02174118. [DOI] [PubMed] [Google Scholar]
- 34.Lombó F, Siems K, Braña AF, Méndez C, Bindseil K, Salas JA. J Bacteriol. 1997;179:3354–3357. doi: 10.1128/jb.179.10.3354-3357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohr J, Weissbach U, Beninga C, Kunzel E, Siems K, Bindseil KU, Lombó F, Prado L, Braña AF, Méndez C, Salas JA. Chem Commun. 1998:437–438. [Google Scholar]
- 36.Fernández E, Weissbach U, Sánchez Reillo C, Braña AF, Méndez C, Rohr J, Salas JA. J Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wohlert SE, Künzel E, Machinek R, Méndez C, Salas JA, Rohr J. J Nat Prod. 1999;62:119–121. doi: 10.1021/np980355k. [DOI] [PubMed] [Google Scholar]
- 38.Lombó F, Braña AF, Méndez C, Salas JA. J Bacteriol. 1999;181:642–647. doi: 10.1128/jb.181.2.642-647.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prado L, Fernández E, Weissbach U, Blanco G, Quirós LM, Braña AF, Méndez C, Rohr J, Salas JA. Chem Biol. 1999;6:19–30. doi: 10.1016/s1074-5521(99)80017-9. [DOI] [PubMed] [Google Scholar]
- 40.Prado L, Lombó F, Braña AF, Méndez C, Rohr J, Salas JA. Mol Gen Genet. 1999;261:216–225. doi: 10.1007/s004380050960. [DOI] [PubMed] [Google Scholar]
- 41.Blanco G, Fernández E, Fernández MJ, Braña AF, Weissbach U, Künzel E, Rohr J, Méndez C, Salas JA. Mol Gen Genet. 2000;262:991–1000. doi: 10.1007/pl00008667. [DOI] [PubMed] [Google Scholar]
- 42.Fernández Lozano MJ, Remsing LL, Quiros LM, Braña AF, Fernández E, Sánchez C, Méndez C, Rohr J, Salas JA. J Biol Chem. 2000;275:3065–3074. doi: 10.1074/jbc.275.5.3065. [DOI] [PubMed] [Google Scholar]
- 43.Lombó F, Künzel E, Prado L, Braña AF, Bindseil KU, Frevert J, Bearden D, Méndez C, Salas JA, Rohr J. Angew Chem, Int Ed. 2000;112:808–811. doi: 10.1002/(sici)1521-3773(20000218)39:4<796::aid-anie796>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 44.González A, Remsing LL, Lombó F, Fernández MJ, Prado L, Braña AF, Künzel E, Rohr J, Méndez C, Salas JA. Mol Gen Genet. 2001;263:827–835. doi: 10.1007/s004380000372. [DOI] [PubMed] [Google Scholar]
- 45.Aich P, Dasgupta D. Biochemistry. 1995;34:1376–1385. doi: 10.1021/bi00004a032. [DOI] [PubMed] [Google Scholar]
- 46.Hopwood DA, Bibb MJ, Chater KF, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Ward JM, Schrempf H. Genetic Manipulation of Streptomyces. A Laboratory Manual. The John Innes Foundation; Norwich, U.K: 1985. [Google Scholar]
- 47.Bullock WO, Fernández JM, Short JN. BioTechniques. 1987;5:376. [Google Scholar]
- 48.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual. 1–3 Cold Spring Harbor Laboratory Press; Plainview, NY: 1989. [Google Scholar]
- 49.Norwood TJ, Boyd J, Heritage JE, Soffe N, Campbell ID. J Magn Reson. 1990;87:488–501. [Google Scholar]
- 50.Bax A, Summers MF. J Am Chem Soc. 1986;108:2093–2094. [Google Scholar]
- 51.Norwood TJ, Boyd J, Heritage JE, Soffe N, Campbell ID. J Magn Reson. 1990;87:488–501. [Google Scholar]
- 52.Bax A, Ikura M, Kay LE, Torchia DA, Tschudin R. J Magn Reson. 1990;86:304–318. [Google Scholar]
- 53.Sklenar V, Poitto M, Leppik R, Staudek V. J Magn Reson, Ser A. 1993;102:241–245. [Google Scholar]
- 54.Torkkell S, Ylihonko K, Hakala J, Skurnik M, Mäntsälä P. Mol Gen Genet. 1997;256:203–209. doi: 10.1007/s004380050562. [DOI] [PubMed] [Google Scholar]
- 55.Lomovskaya N, Doi-Katayama Y, Filippini S, Nastro C, Fonstein L, Gallo M, Colombo AL, Hutchinson CR. J Bacteriol. 1998;180:2379–2386. doi: 10.1128/jb.180.9.2379-2386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bate N, Butler AR, Smith IP, Cundliffe E. Microbiology. 2000;146:139–146. doi: 10.1099/00221287-146-1-139. [DOI] [PubMed] [Google Scholar]
- 57.Chen H, Zhao Z, Hallis TM, Guo Z, Liu H-w. Angew Chem Int Ed. in press. [Google Scholar]
- 58.Gaisser S, Böhm GA, Doumith M, Raynal M-C, Dhillon N, Cortés J, Leadlay PF. Mol Gen Genet. 1998;258:78–88. doi: 10.1007/s004380050709. [DOI] [PubMed] [Google Scholar]
- 59.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 60.Yamase H, Zhao L, Liu H-w. J Am Chem Soc. 2001;123 in press. [Google Scholar]
- 61.Salah-Bey K, Doumith M, Michel JM, Haydock S, Cortes J, Leadlay PF, Raynal MC. Mol Gen Genet. 1998;257:542–553. doi: 10.1007/s004380050680. [DOI] [PubMed] [Google Scholar]