

Abstract
Disruption of the MECP2 gene leads to Rett syndrome (RTT), a severe neurological disorder with features of autism1. MECP2 encodes a methyl-DNA-binding protein2 that has been proposed to function as a transcriptional repressor, but despite numerous studies examining neuronal gene expression in Mecp2 mutants, no clear model has emerged for how MeCP2 regulates transcription3–9. Here we identify a genome-wide length-dependent increase in gene expression in MeCP2 mutant mouse models and human RTT brains. We present evidence that MeCP2 represses gene expression by binding to methylated CA sites within long genes, and that in neurons lacking MeCP2, decreasing the expression of long genes attenuates RTT-associated cellular deficits. In addition, we find that long genes as a population are enriched for neuronal functions and selectively expressed in the brain. These findings suggest that mutations in MeCP2 may cause neurological dysfunction by specifically disrupting long gene expression in the brain.
To identify common features of genes whose expression is misregulated in RTT, we surveyed gene expression datasets from studies of Mecp2 mutant mice, asking if genes that are misregulated when MeCP2 function is disrupted have anything in common with respect to histone modifications, mRNA expression, sequence composition, or gene length. No common features were identified for genes that are down-regulated when MeCP2 function is disrupted; however, we found that genes that are up-regulated in the Mecp2 knockout (MeCP2 KO) brains are significantly longer than the genome-wide average (Fig. 1a). The extreme length of the genes up-regulated in MeCP2 KO brains is apparent in multiple studies performed by different laboratories5–9 (Supplementary Table 1). The misexpression of long genes is a specific feature of the RTT brain, as genesets identified as misregulated in sixteen different mouse models of neurological dysfunction and disease did not display similarly long length (Extended Data Fig. 1).
Figure 1. Length-dependent gene misregulation in Mecp2 mutant mice and human RTT brain.

a, Boxplots summarizing lengths of genes (Refseq transcription start site to termination site) detected as misregulated in independent studies of Mecp2 mutant mice. HYP, hypothalamus5; CB, cerebellum6; AMG, amygdala7; HC, hippocampus8; STR, striatum9; LVR, liver9. MeCP2-induced (blue), genes down-regulated in MeCP2 knockout (MeCP2 KO) and up-regulated in MeCP2 overexpression (MeCP2 OE) mice. MeCP2-repressed (red), genes up-regulated in MeCP2 KO and down-regulated in MeCP2 OE (see Methods). b, Mean expression changes across brain regions and liver of Mecp2 mutant mice for genes ≤100 kb (gray) and >100 kb (red). c–d, Genome-wide changes in gene expression assessed by RNA-seq analysis of mouse cortical tissue from MeCP2 KO compared to wild type (c) or microarray analysis of human RTT brain samples compared to age-matched controls16 (d). In c, d lines represent mean fold-change in expression for genes binned according to gene length (200 gene bins, 40 gene step; see Methods); the ribbon is S.E.M. of each bin. *, p < 0.05; **, p < 0.01; ***, p < 1×10−10, n.s. p ≥ 0.05; one-sample (a) or two sample (b) t-test, Bonferroni correction. Error bars represent S.E.M.
To determine if the extent of gene misregulation in Mecp2 mutant mice is directly correlated with gene length, we interrogated published microarray datasets of gene expression and plotted mRNA fold-change (MeCP2 KO compared to wild type) versus gene length10. We found widespread length-dependent misregulation of gene expression in MeCP2 KO brains, with the longest genes in the genome displaying the highest level of up-regulation relative to shorter genes, which show a reduction or no change in gene expression (Fig. 1b, c and Extended Data Fig. 1). Consistent with previous studies, the magnitude of the length-dependent gene misregulation in the absence of MeCP2 is small, but is widespread (affecting genes across the continuum of gene lengths) and reproducibly detected (Fig. 1b and Extended Data Fig. 1). Importantly, length-dependent gene misregulation in the MeCP2 KO is not an artifact of the method of gene expression analysis used, as this effect was detected using a variety of methodologies including microarrays, total RNA-seq, quantitative PCR, and non-amplification-based nCounter analysis (Fig. 1b, c, Extended Data Fig. 1 and Supplementary Discussion). Furthermore, these observations are corroborated by the recent finding of Nelson and colleagues that long genes are up-regulated in specific neuronal cell types when MeCP2 function is disrupted11.
Additional copies of MECP2 cause neurological impairment in humans (MeCP2-duplication syndrome) and in transgenic mice12,13. We find that over-expression of MeCP2 in mice leads to the down-regulation of long genes in the brain5–7 (Fig. 1b and Extended Data Fig. 1). This further suggests that MeCP2 directly represses transcription in a length-dependent manner.
We next investigated if the length-dependent changes in gene expression correlate with onset and severity of RTT pathology. We found that misregulation of long gene expression in the brain of MeCP2 KO mice is more dramatic at nine weeks of age than at four weeks of age8, thus correlating with disease progression (Extended Data Fig. 2). In addition, when comparing two disease-causing MeCP2 mutations (MeCP2-R270X and MeCP2-G273X) that differ in the rate and severity with which they cause disease, we find that the magnitude of length-dependent gene misregulation correlates with the severity of RTT phenotypes8 (Extended Data Fig. 2, Supplementary Discussion). Furthermore, we find by microarray, nCounter, and qRT-PCR analysis, that a subtle missense mutation of MeCP2 (arginine 306 to cysteine, R306C) that causes RTT in humans and disrupts the interaction of MeCP2 with the NCoR co-repressor complex14 leads to length-dependent gene up-regulation in the mouse brain (Extended Data Fig. 1). Finally, we detect length-dependent gene up-regulation in cultured human neurons derived from embryonic stem cells lacking MECP215 and the cortex of humans with RTT16 (Fig. 1d, Extended Data Fig. 2, Supplementary Discussion). The close correlation between the occurrence of length-dependent gene misregulation and RTT-associated phenotypes across mice and humans suggests that this misregulation contributes to RTT pathology.
To characterize the mechanism by which MeCP2 tempers the expression of long genes, we asked if the binding of MeCP2 to methylated DNA is important for this process. MeCP2 was identified based on its high affinity for methylated cytosine in the context of a CpG dinucleotide (mCG)17. In addition to binding mCG, MeCP2 has been suggested to bind two additional forms of methylated DNA that are enriched in the brain, hydroxymethylcytosine (hmC)18 and methylated cytosine followed by a nucleotide other than guanine (mCH, where H = A or T or C)19. Notably, the frequency of hmCG and mCH in the neuronal genome increases significantly during the same postnatal period in which the level of MeCP2 protein increases dramatically20–24. This suggests that as neurons mature MeCP2 could function by binding to hmCG and/or mCH marks. Using a DNA electrophoretic mobility shift assay (EMSA) we assessed the binding of MeCP2 to various forms of methylated DNA. Consistent with previous studies, we find that MeCP2 shows high affinity for DNA containing mCG but not hmCG, suggesting that MeCP2 may not bind preferentially to hmCG in neurons (Fig. 2a, Extended Data Fig. 3, Supplementary Discussion). By contrast, MeCP2 binds to mCA, hmCA, and mCG with relatively high affinity, but binds to mCC and mCT with low affinity similar to that of unmethylated DNA. This selective, tight binding of MeCP2 to mCG, mCA, and hmCA suggests that MeCP2 may regulate long gene expression in the brain by binding to these sites. We note that thin-layer chromatography and Tet-assisted bisulfite sequencing (TAB-seq) analysis suggest that hmCA is very rare in the brain21,24. Therefore, in our subsequent investigation of MeCP2 binding to CA sequences in vivo we focused our analysis on mCA. However, at genomic sites where CA sequences are hydroxymethylated, MeCP2 might also be predicted to bind and regulate gene expression (see Supplementary Discussion).
Figure 2. MeCP2 represses long genes containing high levels of mCA.

a, EMSA analysis of the MeCP2 methyl-binding domain (amino acids 78–162) binding to 32P-end-labeled mCA-containing DNA probe incubated with 100-fold excess of unlabeled competitor oligonucleotides containing unmodified, methylated, or hydroxymethylated cytosines at the dinucleotides indicated in bold; no competitor indicated by “−” (see Methods, Extended Data Fig. 3). b, Boxplots of MeCP2 ChIP-seq read density within genes >100 kb plotted by quartile of mCA/CA in the cortex and cerebellum. c, Mean fold-change in gene expression binned according to gene length in MeCP2 KO cortical tissue for genes with high (mCA/CA > 0.034, top 25%) and low (mCA/CA < 0.031, bottom 66%) mCA levels (left), or binned according to gene-body mCA/CA levels for long (>62 kb, top 25%) and short (<16.8 kb, shortest 25%) genes (right). Lines represent mean fold-change in expression for each bin (200 gene bins, 40 gene step), and the ribbon is S.E.M. of each bin. d, Bar plots of the mean fold-change in expression for all genes >100 kb compared to subsets of genes >100 kb containing low mCA (bottom 50% mCA/CA) or high mCA (top 25% mCA/CA) within their gene body. Values shown for mice with the indicated Mecp2 genotypes (left) and human RTT brain (right). CTX, Cortex; HC, Hippocampus; CB, cerebellum; KO, MeCP2 Knockout; OE, MeCP2 overexpression; R306C, MeCP2 arginine 306 to cysteine missense mutation; ***, p < 1×10−10; **, p < 1×10−3; *, p < 0.01; two-tailed t-test, Bonferroni correction. Error bars represent S.E.M.
To examine if MeCP2 binds mCA in the brain, we performed chromatin immunoprecipitation sequencing analysis (ChIP-seq) of MeCP2, comparing the MeCP2 binding profile across the genome to base-pair resolution DNA methylation data (see Methods)24. As previously reported20,25, we find that MeCP2 binds broadly across the genome. Nevertheless, within the context of this broad binding, we detect a relative enrichment of MeCP2 at gene bodies that have a high level of mCA (level = (h)mCN/CN within the gene, see Methods), and a depletion of MeCP2 binding at gene bodies where the level of hmCG is high (Extended Data Fig. 4). Notably, long genes (>100 kb) display a strong relationship between mCA levels and MeCP2 ChIP-seq read density (Fig. 2b, Extended Data Fig. 4). Higher resolution analysis of MeCP2 ChIP and mCA levels in the frontal cortex revealed increased mCA under sites of local MeCP2 enrichment in the genome, supporting the conclusion that MeCP2 binds to mCA in vivo (Extended Data Fig. 4). We note that genes containing the highest level of hmCA are also enriched for the MeCP2 ChIP signal (Extended Data Fig. 4). Therefore, if due to limitations of the methods of analysis the amount of hmCA within gene bodies is being underestimated, some of the effects of MeCP2 deletion that are being attributed to MeCP2 binding to mCA might be due to MeCP2 binding to hmCA (see Supplementary Discussion).
To investigate if length-dependent gene repression by MeCP2 requires binding to mCA, we assessed whether there is a correlation between the degree of misregulation of gene expression upon the disruption of MeCP2 function and the levels of DNA methylation within the transcribed regions of genes (See Supplementary Discussion). We noted a trend whereby genes containing high levels of mCA, but not mCG or hmCG, are up-regulated in the MeCP2 KO (Extended Data Fig. 5, 6). We reasoned that if mCA within genes is required for length-dependent repression by MeCP2, long genes containing low levels of mCA should be largely unaffected in the MeCP2 KO mice. Consistent with this prediction, little to no length-dependent up-regulation of gene expression is observed in MeCP2 KO brain for genes containing low levels of mCA, while long genes with a high density of mCA are significantly up-regulated in MeCP2 KO brains. In addition, we found that the shortest genes in the genome are not up-regulated when MeCP2 function is disrupted, even when the average level of mCA within their gene body is relatively high (Fig. 2c, Extended Data Fig. 6). The requirement for the presence of mCA within long genes for the gene to be repressed by MeCP2 is reproducible, as it is detected across three MeCP2 KO brain regions, in gene expression data from MeCP2 R306C and MeCP2 OE mice, and in human RTT brain (Fig. 2d, Extended Data Fig. 6). Strikingly, when we plotted the level of mCA versus gene length, we found that the density of mCA is higher on average in longer genes compared to shorter genes (Extended Data Figs. 5, 6). The enrichment of mCA within long genes may explain why most of these genes are repressed by MeCP2 and up-regulated in the MeCP2 KO.
To further test if MeCP2 tempers long gene transcription by binding to mCA within genes we asked if elimination of mCA in the brain has an effect on gene expression that is similar to that observed in the MeCP2 KO. Recent evidence suggests that Dnmt3a is the enzyme that catalyzes the deposition of mCA in maturing neurons19,24. We therefore conditionally disrupted the Dnmt3a gene26 in the brain to block the accumulation of mCA (Nestin-Cre; Dnmt3aflx/flx mice, designated Dnmt3a cKO, Extended Data Fig. 7, Supplementary Discussion). Bisulfite sequencing of cerebellum DNA indicated that methylation of DNA at CA, but not CG, is eliminated from the genome in the Dnmt3a cKO (Fig. 3a). Microarray analysis of cerebella from Dnmt3a cKO mice revealed a length- and mCA-dependent up-regulation of gene expression that is similar to the gene misregulation detected in MeCP2 KO mice (Fig. 3b, Extended Data Fig. 8). While the deletion of Dnmt3a also leads to a decrease in methylation at CT and CC, given that MeCP2 selectively binds to mCA in vitro, we conclude that reduction of mCA within gene bodies in the Dnmt3a cKO likely disrupts length-dependent gene repression by MeCP2. Taken together, these findings support a model in which Dnmt3a catalyzes the methylation of CA in the neuronal genome. MeCP2 then binds to these sites within the transcribed regions of genes to restrain transcription in a length-dependent manner.
Figure 3. Disruption of Dnmt3a in the brain leads to length-dependent up-regulation of genes containing high levels of mCA.
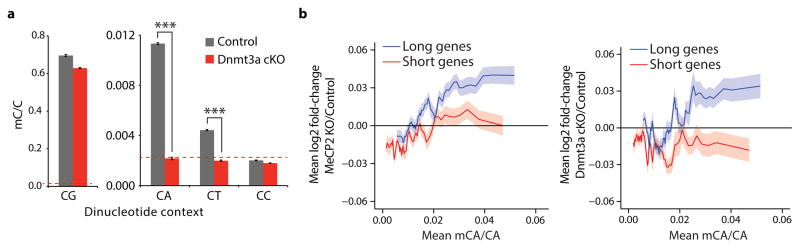
a, Summary of genome-wide bisulfite-sequencing analysis of mCN (where N = G, A, T, or C) in control and Dnmt3a cKO cerebella (n=2 per genotype). Dashed line represents mean background non-conversion rate of the bisulfite-seq assay (see Methods). b, Mean fold-change in gene expression versus gene-body mCA for MeCP2 KO (left) or Dnmt3a cKO (right) cerebella. Long (top 25%, >60kb) and short (bottom 25%, <14.9kb) genes were binned according to gene-body mCA/CA levels. Lines represent mean fold-change in expression for each bin (200 gene bins, 40 gene step), and the ribbon is S.E.M. of genes within each bin. ***, p < 0.005; two-tailed t-test, Bonferroni correction. Error bars represent S.E.M.
To characterize how the misregulation of long gene expression contributes to RTT pathology, we identified a representative set of genes that is consistently misregulated in multiple gene expression datasets when MeCP2 function is perturbed. Combined analysis of microarray studies across multiple brain regions identified 466 MeCP2-repressed genes whose expression is consistently up-regulated in MeCP2 KO mice and down-regulated in MeCP2 OE mice (Supplementary Discussion, Supplementary Table 3). Consistent with the conclusion that MeCP2-repressed genes are targets of gene-length- and mCA-dependent repression, these genes are exceptionally long and are enriched for mCA (Fig. 4a, Extended Data Fig. 8). Disruption of the expression of this geneset is specific to RTT, as these genes were not misregulated in datasets obtained from six other mouse models of neurological dysfunction (Extended Data Fig. 8).
Figure 4. Analysis of long gene expression and regulation in the brain.

a, Cumulative distribution function of gene lengths for all genes in the genome (black), MeCP2-repressed genes (red), and genes encoding putative FMRP target mRNAs29 (blue); p < 1×10−15 for each geneset vs all genes, 2-sample Kolmogorov-Smirnov (KS) test. b, Mean expression of genes binned according to length in mouse for neural and non-neural tissues. Line indicates mean expression for genes within each bin (200 gene bins, 40 gene step); the ribbon represents the S.E.M. of each bin.
We examined the functional annotations of the 466 MeCP2-repressed genes to gain insight into how their disruption might contribute to RTT pathology. Many of these MeCP2-repressed genes encode proteins that modulate neuronal physiology (e.g. calcium/calmodulin-dependent kinase Camk2d and the voltage-gated potassium channel Kcnh7). In addition, multiple genes involved in axon guidance and synapse formation were identified, including Epha7, Sdk1 and Cntn4 (Extended Data Fig. 8). Consistent with these observations, gene ontology analysis of MeCP2-repressed genes indicates that they are enriched for annotated neuronal functions (e.g. post-synaptic density, axonogenesis, voltage-gated cation channel activity; Extended Data Table 1). These findings suggest that RTT results from a subtle, yet widespread over-expression of long genes that have specific functions in the nervous system.
We next considered why the misregulation of long genes as a population in RTT leads specifically to neuronal dysfunction. Many genes with neuronal function are very long27,28, raising the possibility that long genes as a population might be enriched for functions in the nervous system relative to other tissues. If so, the high level of mCA and MeCP2 in neurons may have evolved to temper the expression of long genes specifically in the brain. Indeed, gene ontology analysis of all genes in the genome above 100 kb indicates that the longest genes in the genome are enriched for neuronal annotations (Extended Data Table 1). Moreover, by examining tissue-specific gene expression datasets, we find that long genes as a population are preferentially expressed in mouse and human brain relative to other tissues (Fig. 4b, Extended Data Fig. 9). We note that, while long genes typically have brain-specific function and expression, brain-specific expression is not a prerequisite for regulation of long genes by MeCP2 in neurons: some long genes are ubiquitously expressed but selectively repressed by MeCP2 in the brain. (Extended Data Fig. 8, Supplementary Discussion).
To explore if disruption of proteins that regulate long gene expression may broadly contribute to autism spectrum disorders (ASDs), we asked if a similar misregulation of gene expression occurs in a prominent ASD, Fragile X syndrome (FXS). FXS is caused by inactivation of FMRP, a protein that represses mRNA translation in neurons29. Strikingly, we find that FMRP target mRNAs and the genes that encode them are significantly longer than the genome average29 (Fig. 4a, Extended Data Fig. 8, Supplementary Discussion). Moreover, we detect significant overlap between MeCP2-repressed genes and genes encoding FMRP target mRNAs (Extended Data Fig. 8). These results suggest that up-regulation of long gene function, either through increased transcription (RTT) or mRNA translation (FXS), may represent a common cause of pathology in neurodevelopmental disorders.
A recent study demonstrated that pharmacological inhibition of topoisomerases leads to the broad down-regulation of long genes in neurons10, suggesting that topoisomerase inhibitors might reverse the up-regulation of long gene expression observed in the absence of MeCP2. To test this, we knocked-down MeCP2 expression in cultured cortical neurons with RNAi and treated these cells with the topoisomerase inhibitor topotecan. We found that MeCP2 knockdown leads to the up-regulation of long genes and that exposure of MeCP2-deficient neurons to topotecan results in a dose-dependent reversal of long gene misregulation (Extended Data Fig. 9).
The disruption of MeCP2 function in both mouse and human neurons leads to an overall reduction in cell health that can be measured as a decrease in the level of ribosomal RNA and cell size15,30. Strikingly, we found that the concentration of topotecan that most effectively reverses overexpression of long genes (50nM) partially reverses the decreased ribosomal RNA content observed in neurons lacking MeCP2 (Extended Data Fig. 9). This result suggests that the rebalancing of long gene expression improves cell health in MeCP2 knockdown neurons, leading to increased cellular rRNA content. Taken together, these data suggest that rebalancing long gene expression in neurons lacking MeCP2 may attenuate the cellular dysfunction observed in these cells.
Our finding that long genes are misregulated in RTT, and that this misregulation can be reversed by topotecan treatment complements recent studies from Zylka, Philpot and colleagues10 implicating topoisomerases in the regulation of long genes in the brain. Thus, our study provides additional evidence that disruption of long gene expression may be a general mechanism underlying ASDs, and suggests that developing methods to rebalance long gene expression may be a strategy to correct neural dysfunction in these disorders.
Methods
Analysis of published MeCP2-regulated gene lists
To search for unique characteristics of genes found to be misregulated in Mecp2 mutant mice we interrogated the list of genes found to be significantly activated or repressed by MeCP2 in the cerebellum of MeCP2 KO and MeCP2 OE mice6. Using published datasets for the mouse cerebellum from ENCODE and other sources, these genes were assessed for epigenetic marks at promoters and gene bodies, including histone acetylation and methylation as measured by ChIP-seq analysis, as well as DNA methylation and hydroxymethylation as measured by affinity purification methods18. In addition, we interrogated sequence attributes of genes, including dinucleotide frequencies, exon number, repeat density within genes and gene length. To determine if the misregulated genes were exceptional with respect to any epigenetic marks or sequence attributes, they were compared to several sets of control genes selected to be matched for gene expression levels (data not shown). While no obvious epigenetic differences were apparent from this analysis, we detected the extreme length of genes (measured as total basepairs from Refseq transcription start site to transcription termination site) repressed by MeCP2 (up-regulated in the MeCP2 KO and down-regulated in the MeCP2 OE). We note that affinity-based measures of DNA methylation that were used in this initial unbiased search are now known to be insensitive to low level methylation at individual cytosines and thus do not report mCA levels with high fidelity. This likely explains why we did not detect a methylation signature for MeCP2-repressed genes using the affinity-based data in our initial analysis. Subsequent analysis of multiple published gene lists from several brain regions revealed the consistent, extreme length of the genes identified as repressed by MeCP2 in each brain region. These findings are presented in Figure 1a as boxplots where each plot depicts the median (line), the 2nd through 3rd quartiles (box), 1.5x the interquartile range (whiskers), and 1.58x the interquartile range/(√# genes) (notches). The notches on each box approximate a 95% confidence interval for the median value31. Note that opposing changes in MeCP2 KO and MeCP2 OE published gene lists were used to define genes significantly activated or repressed by MeCP2 for hypothalamus5, cerebellum6, and amygdala7 tissues. For hippocampus8, striatum9 and liver9 MeCP2 KO data alone had been used to identify gene lists.
To test if long gene misregulation is specific to Mecp2 mutants we surveyed gene expression studies profiling models of neurological dysfunction, asking if long gene length is a common attribute in genesets from these studies. We analyzed the lengths of the lists of up and down regulated genes identified in these studies, or if “called” misregulated gene lists were not available, we generated lists using the Genespring 12.6 software package (Agilent Technologies) or the Geo2R analysis tool (http://www.ncbi.nlm.nih.gov/geo/geo2r/). This analysis did not uncover any additional genesets with similar long length to that of MeCP2 mutant studies (Extended Data Fig. 1a), suggesting that misregulation of extremely long genes is not a common consequence of cell dysfunction in models of neurodegeneration or several other neurological diseases.
To analyze gene expression genome-wide with respect to gene length, CEL files containing the raw hybridization data in from multiple MeCP2 KO and MeCP2 OE gene expression studies were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/; study details, sample numbers and genotypes are provided in Supplementary Table 1) and analyzed for expression at the gene level using using the GeneSpring software suite (Agilent Technologies) with RMA summarization of “Core” probesets. To facilitate unambiguous analysis of individual genes, expression values for transcript cluster IDs were filtered to include only transcript clusters that map to single Refseq genes, and expression values for genes with multiple transcript clusters were derived by taking the average log2 expression value across all transcript clusters corresponding to each gene. To facilitate comparison between microarray platforms, throughout this study we present analysis only for genes represented on all microarray platforms; this corresponds to 14,168 genes for mouse, and 17,989 genes for human. While this represents a subset of genes in each genome, we have obtained similar results for length-dependent changes in gene expression for expanded gene sets covered by individual platforms (data not shown). In addition, similar results were obtained using the Affymetrix Power Tools pipeline with PLIER as an alternative summarization method. For consistency, microarray data for gene expression in human cells was presented using a comparable array summarization scheme as the mouse microarray data (RMA). Similar qualitative results showing length-dependent gene misregulation were obtained from gene expression values generated by Li and colleagues using a normalization scheme that included spike-controls15 (summarized transcript expression values were downloaded directly from GEO). However, with this normalization procedure, the absolute values of fold-change of all genes across the entire genome were downshifted in MECP2 null neurons relative to wild-type. For analysis of RTT patient samples, raw CEL files from Deng et al.16 were downloaded from GEO, and summarized using the RMA function in the R “affy” package.
To quantify the relationship between fold-change and gene length, we sorted genes by the lengths of their immature transcripts (RefSeq annotation) and employed a sliding window containing 200 consecutive genes in steps of 40 genes. The log2 fold-change values for the 200 genes within each length bin were averaged and plotted; displayed standard errors (SEs) for a bin were calculated by propagating the SE deduced from the bin’s log2 fold-change values and the mean SE of the individual genes reflecting their sample variability. Null distributions displayed on fold-change plots were constructed for each bin from 10,000 random samples of 200 genes selected without regard to transcript length.
RNA sequencing and analysis
Total RNA was prepared from cortex of male wild-type and MeCP2 KO mice at 8–9 weeks of age. Sample size (3 per genotype) was determined based on previous detection of length-dependent gene expression effects from datasets that used similar sample sizes (see Fig. 1b, c; Extended Data Fig. 1; Supplementary Table 1). Animals were preselected based on genotype before collection to insure that paired samples were taken within litters, but collection was randomized and the experimenter was uninformed of genotype during collection, sample processing, and analysis. Brain samples were dissected on ice in HBSS and immediately frozen in liquid nitrogen. To extract RNA, the tissue was thawed in trizol (Ambion), homogenized, extracted with chloroform, and further purified on RNeasy Columns (Qiagen) using on-column DNAse treatment to remove residual DNA as specified in the manufacturer’s instructions. High-throughput sequencing of total RNA was performed as a service by BGI America. Briefly, ERCC control RNAs (Ambion) were added to samples, and total RNA was depleted of ribosomal RNA using the Ribozero rRNA removal kit (Epicentre), heat-fragmented to 200–700 bp in length and cloned using Uricil-N-Glycosylase-based strand-specific cloning. cDNA fragments were sequenced using an Illumina HiSeq 2000, typically yielding 20M–40M usable 49 bp single-end reads per sample (Supplementary Table 1 for details). Gene expression levels were assessed using an in-house analysis pipeline previously developed for RNA-seq quantification32. After filtering out adapter and low quality reads, reads were mapped using BWA33 to the mm9 genome augmented by an additional set of splicing targets (~3M sequences of length ≤ 98 bp representing all possible mm9 sequences that could cross at least one exon-exon junction based on the RefSeq annotation). Samples were normalized based on uniquely mapped reads that fell outside of rRNA and noncoding genes in order to avoid skewing by spikes in incompletely depleted ribosomal and transfer RNA. Normalization of each sample was referred to an in-house standard of 10M 35-bp reads. Gene expression within exons and other features was quantified as “Density,” defined as read coverage of that feature, equal to the total number of read bases per total number of feature bases multiplied by the overall normalization coefficient. Units of Density are always proportional to RPKM (Density = 0.35 × RPKM).
Average read Density within a gene’s exons was taken as a proxy for gene expression (for genes with multiple annotated transcripts, exonic loci were unioned together). For a given set of samples, a quantile distribution (QD) was constructed from all samples’ sorted expression levels, and values from the QD were reassigned to each gene according to its rank in each sample. Within each subset of samples corresponding to wild type (WT), knockout (KO), etc., each gene was assigned its mean log QD value and a standard error (SE) over its values for this subset in order to quantify its sample-to-sample variability within the subset. Precisely zero expression levels were ignored in constructing the QD. The log of the fold-change (FC) between subsets for each gene, e.g., log (KO/WT), was set to the difference of the means of the KO and WT log values for the gene, along with a propagated SE of the log values (variance equal to the sum of KO and WT variances). For consistency, the RNA-seq analysis in this study is presented for the common set of genes covered by microarray analyses in previous studies (see above). Similar results were obtained for larger sets of genes defined by all RefSeq genes.
To confirm that our findings with RNA-seq were robust to the method of quantification used, we also performed analysis using the Spliced Transcripts Alignment to a Reference (STAR)34 software to align reads to the mm9 genome and Cufflinks35 to estimate gene-level expression values as Fragments Per Kilobase of exon model per Million mapped fragments (FPKM). This analysis yielded results that were nearly identical to those generated using our in-house RNA-seq analysis pipeline. In addition, we derived similar results using transcripts per million (TPM)36 as the measure mRNA levels in place of FPKM (data not shown).
MeCP2 has previously been implicated in the repression of repeat elements across the mammalian genome, raising the possibility that the up-regulation of long genes we observe in our analysis is a reflection of increased transcription from repeat elements or possibly cryptic promoters. To look for changes in the expression of repeat RNAs in the MeCP2 KO brain, RNA-seq reads were mapped to the genome using Bowtie, keeping reads mapping to multiple sites in the genome. Each read was assigned a score of 1/n (n=number of sites a read mapped to). Expression values for each repeat family was calculated by adding the scores within each repeat (annotated using Repeatmasker) and normalizing to sequencing depth. This analysis did not reveal evidence of up-regulation of specific repeat classes in the MeCP2 KO brain. In addition, to look for evidence of increased expression of repeats in connection with longer genes we assessed whether there was increased antisense transcription in these genes using our in-house RNA-seq analysis pipeline. This analysis failed to provide evidence of increased antisense transcription. Another alternative explanation for our results was that the increase in expression of long genes we observe is due to spurious transcription, which might initiate from cryptic promoters within genes to generate sense coding, incomplete RNAs. In this case the up-regulated RNAs would not reflect mature protein coding mRNA levels. To assess the expression of mature mRNA directly we measured mRNA expression by quantifying only RNA-seq reads that map across exon splice junctions. Consistent with there being an up-regulation of mature mRNAs from long genes in the MeCP2 KO, this analysis yielded genome-wide length-dependent up-regulation of gene expression that is highly similar to our whole-exon-based approach described above (data not shown). We conclude from this analysis that functional, protein-coding mRNAs derived from long genes are up-regulated in the MeCP2 KO, and that this increase is likely due to an alteration in canonical genic transcription mechanisms, not an increase in spurious transcripts coming from long gene loci.
Gene expression analysis of MeCP2 R306C mice
Consistent with nomenclature from past descriptions of RTT missense mutations, the R306C nomenclature refers to the mouse MeCP2 isoform 2 (MeCP2_e2; NCBI Reference Sequence NP_034918). For gene expression analysis brain regions were dissected from male Mecp2R306C/y mice14 and wild type littermates at 8–10 weeks of age and RNA was isolated as described above. Animals were preselected based on genotype before collection to insure that paired samples were taken within litters, but collection was randomized and the experimenter was uninformed of genotype during collection, sample processing, and analysis. Microarray analysis of cerebellar RNA was performed using the Affymetrix Mouse Exon 1.0 ST array platform. Analysis was performed in the Dana Farber microarray core facility following manufacturer’s recommendations. Analysis of hybridization data was performed as described above. Sample size (4 per genotype) was determined based on previous detection of length-dependent gene expression effects from datasets that used similar sample sizes (see Extended Data Fig. 1; Supplementary Table 1).
Validation of microarray and RNA-seq findings
For reverse transcription-quantitative PCR expression analysis candidate genes were selected for analysis in the visual cortex based on consistent up-regulation in the MeCP2 KO (log2 fold-change greater than zero) and down-regulation in the MeCP2 OE (log2 fold-change less than zero) across eight published microarray datasets in five brain regions (hypothalamus, cerebellum, amygdala, striatum, hippocampus). For Nanostring nCounter validation genes were selected based on the above criteria and evidence of up-regulation in the visual cortex RNA-seq analysis. Genes with this profile were selected for qPCR assessment in the visual cortex. cDNA was generated from 500 ng of visual cortex total RNA (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems), and quantitative PCR was performed using transcript-specific primers (designed with the universal probe library design center, Roche, Supplementary Table 2) and SYBR green detection on the Lightcycler 480 platform (Roche). Relative transcript levels and fold-changes were calculated by normalizing qPCR signal within each sample to six genes that do not show evidence of altered expression across published microarray data sets (Supplementary Table 2). Similar results were obtained by analyzing raw Cp values for test transcripts without normalization to control genes (data not shown).
For non-amplification-based gene expression analysis, Nanostring nCounter reporter CodeSets were designed to detect candidate MeCP2-repressed genes in 250 ng of total RNA extracted from MeCP2 KO and R306C mice. Samples were processed at Nanostring Technologies, Inc. following the nCounter Gene Expression protocol. Briefly, total RNA was incubated at 65°C with reporter and capture probes in hybridization buffer overnight, and captured probes were purified and analyzed on the nCounter Digital Analyzer. The number of molecules of a given transcript was determined by normalizing detected transcript counts to the geometric mean of ERCC control RNA sequences and a set of control genes that do not show evidence of altered expression across published microarray data sets. Hotelling T2 test for small sample size37 was used to calculate significance in order to incorporate variance across both samples and genes. Significant differences between wild-type and MeCP2 KO or MeCP2 R306C samples (p < 0.01) were also detected by paired two-tailed t-test comparing the paired mean values for each gene (averaged across samples within each genotype) between genotypes.
Electromobility shift assays
Oligonucleotide probes (Integrated DNA Technologies) were 5′-32P-end-labeled by T4 polynucleotide kinase (New England Biolabs) with [γ-32P]ATP (Perkin Elmer) under conditions recommended by the enzyme supplier. 5′-32P-end-labeled upper strands were purified over NucAway Spin Columns (Ambion) and annealed to equal molar concentration of the appropriate unlabeled complement strand in 10 mM Tris, pH 8.0, 50 mM NaCl, 1 mM EDTA at 95°C for 5 minutes, followed by slow cooling to room temperature. Similarly, unlabeled competitors were annealed. Proper annealing of probes and competitors was verified by native gel electrophoresis.
For binding reactions using the MBD fragment of MeCP2, each reaction contained 180 ng of protein (AA 81 – 170, Abnova or AA 78–162, Diagenode), 50 fmol of 5′-32P-end-labeled probe with an excess of an unlabeled competitor in the presence of 1 μg of poly-dIdC (Sigma), 1X Tris-borate-EDTA (TBE) buffer, 1 mM DTT, 20 mM HEPES, pH 7.5, 0.5 mM EDTA, 0.2% Tween-20, 30 mM KCl, and 1X Orange DNA loading dye (Thermo Scientific). Binding was carried out in a 10 μl volume for 10 minutes at room temperature. Each reaction was loaded on a 10% non-denaturing polyacrylamide (37.5:1, acrylamide/bis-acrylamide) gel in 1X TBE buffer and electrophoresed for 30 minutes at 240 V on ice. For binding reactions using the full-length MeCP2 protein, each reaction contained 60 ng of protein (AA 1–486, Millipore), 100 fmol of 5′-32P-end-labeled probe with an excess of unlabeled competitor in the presence of 250ng of pdIdC (Sigma), 0.5X Tris-borate-EDTA (TBE) buffer, 1mM DTT, 20mM HEPES, pH 7.5, 0.5mM EDTA, 0.2% Tween-20, 30mM KCl, and 1X Orange DNA loading dye (Thermo Scientific) in a 10 μl reaction volume for 10 minutes at room temperature. Each reaction was loaded on a 6% non-denaturing polyacrylamide gel (Life Technologies) in 0.5X TBE buffer and electrophoresed for 25 minutes at 300 V on ice. Gels were then dried on Whatman filter paper on a gel drier at 80°C for 1 hour. For imaging, dried gels were exposed to film overnight (Kodak X-Omat XB film) at −80°C.
Whole-genome bisulfite sequencing and analysis
For bisulfite sequencing analysis cerebella and cortices from four, eight-week-old mice were dissected and genomic DNA extracted. Starting with 25 ng of genomic DNA, 0.25 ng of unmethylated lambda DNA was added and libraries were generated using the Ovation Ultralow Methyl-Seq Library System (Nugen). Bisulfite treatment was performed using the EpiTect bisulfite conversion kit (Qiagen) following manufacturer’s instructions. Libraries were constructed using TruSeq reagents (Illumina) and sequenced on the Hiseq 2000 or Miseq instruments (Illumina). Reads were mapped to the mm9 genome using BS seeker38, allowing up to four mismatches. Duplicate reads were removed and only uniquely mapping reads were kept (Supplementary Table 1 for details). For analysis of published bisulfite sequencing datasets19,24, short read files were downloaded from GEO, mapped, and analyzed as described above, or processed data files showing number of reads and number of non-converted reads per cytosine base were used (Supplementary Table 1 for details). Methylation levels in all datasets were calculated as # of cytosine base calls/(# of cytosine + # of thymine base calls) within mapped reads at genomic sites where the reference genome encodes cytosine. For hydroxymethylation analysis, the same approach was applied to Tet-assisted Bisulfite sequencing (TAB-seq) data from cortical tissue24. To examine the effects of gene body methylation independently of promoters, only genes greater than 4.5 kb and with a minimal coverage of CGs and CHs were used in our analysis, and methylation levels within regions of the transcription start site +3 kb to transcription end site were calculated by taking the average methylation levels for all reads mapping within this region. Comparison to gene expression data was performed using corresponding microarray expression values for the hippocampus and the cerebellum or RNA-seq from the cortex. To facilitate fold-change analysis of RNA-seq data, the genes analyzed were filtered for minimal (non-zero) expression values.
MeCP2 Chromatin immunoprecipitation analysis
MeCP2 ChIP analysis was performed on cortex and cerebella dissected from 8-week-old wild-type male mice as previously described25,39. To facilitate direct comparison of MeCP2 ChIP to published frontal cortex DNA methylation and hydroxymethylation data24, we also performed MeCP2 ChIP analysis using the same brain region at the same developmental stage (frontal cortex isolated from 6-week-old mice). ChIP DNA was cloned into libraries and sequenced on the Illumina HiSeq 2000 or Hiseq 2500 platform to generate 49 or 50 bp single-end reads. Reads were mapped to mouse genome mm9 using BWA33 and custom perl scripts were employed to quantify read density (reads/kb) for each gene. Normalized read density values were calculated as reads/kb in each genomic feature (e.g. gene), normalized to the total number of reads sequenced for each sample, and divided by the reads/kb in that feature for the input DNA that was isolated prior to the ChIP and sequenced in parallel. As with the methylation analysis, gene bodies were defined as +3000 bp to the predicted transcription termination site in the Refseq gene model. To ensure sufficient coverage and accurate assessment of density in gene bodies, only genes greater than 4500 bp in total length with at least one read in the input sample were included in the analysis.
To explore the relationship between MeCP2 binding and mCA at high resolution, we also quantified the MeCP2 ChIP signal from the frontal cortex in 500 bp bins tiled for all genes in the genome and compared it to mCA levels derived from high-coverage DNA methylation analysis of this brain region (Extended Data Fig. 4)24. In addition, we employed the MACS40 algorithm to identify sites of MeCP2 ChIP enrichment, or “summits”, across the genome and looked for evidence of mCN at these sites. Due to the broad binding of MeCP2 across the genome, MeCP2 ChIP yields numerous sites of modest local enrichment (~2-fold), not isolated, highly-enriched peaks (>10-fold) that are characteristic of transcription factors. Thus, to define MeCP2 summits, we utilized a low threshold of MeCP2 ChIP over input enrichment (> 1-fold) and a low stringency p-value threshold (p < 0.2), which yielded 31,479 summits of MeCP2 ChIP signal. Aggregate plots across all 31,479 MeCP2 summits were generated using the annotatePeaks.pl program in the Hypergeometric Optimization of Motif EnRichment (HOMER)41 software. Input-normalized MeCP2 ChIP signal was calculated as the ratio of MeCP2 ChIP/Input read coverage. Log2 enrichment of mCN under MeCP2 summits was determined by calculating the level of methyl-cytosine (# non-converted cytosines sequenced)/(# converted and non-converted cytosines sequenced) occurring at CA, CC, CT, or CG positions in the genome, normalized to the flanking region (mean of −4kb to −3kb and 3kb to 4kb region relative to the MeCP2 summit). The average value for the ChIP signal or relative mCN was then calculated for windows (100 bp for ChIP, 10 bp for mCN) tiled across each summit location and averaged across all of the 31,479 summits of MeCP2 ChIP enrichment identified using the MACS peak-calling algorithm40 (red) and 31,479 randomly selected control sites (gray).
Analysis of Dnmt3aflx/flx; Nestin-Cre+/− mice
Female Dnmt3aflx/flx mice26 (kindly provided by M. Goodell) were bred to male Nestin-Cre+/− mice42 to generate Dnmt3aflx/+; Nestin-Cre+/− animals. To ensure expression of the imprinted Nestin-Cre transgene, male Dnmt3aflx/+ Tg(Nes-cre)1Kln/J animals were bred to Dnmt3aflx/flx females to generate Dnmt3aflx/flx Tg(Nes-cre)1Kln/J conditional knockout mice (“Dnmt3a cKO”) and Dnmt3aflx/flx control animals (“Control”). For western blot, DNA methylation and gene expression analyses, cerebella were dissected from 10–11-week-old animals. Proteins were resolved by SDS-PAGE and immunoblotted using the following antibodies: Dnmt3a (abcam, ab13888), MeCP2 (custom antisera43) and Gapdh (Sigma Aldrich, #G9545-25UL). Genotyping for the Dnmt3a locus was performed by PCR with primers flanking both loxP sites (F: 5′-GCAGCAGTCCCAGGTAGAAG-3′, R: 5′-ATTTTTCATCTTACTTCTGTGGCATC-3′) on DNA derived from tails. The presence of the cre allele was detected using primers to this transgene (F: 5′-GCAAGTTGAATAACCGGAAATGGTT-3′, R: 5′-AGGGTGTTATAAGCAATCCCCAGAA-3′). This genotyping scheme allows for simultaneous assessment of the presence of the floxed allele and the relative level of loxP recombination that has occurred in the sample. Brain-specific recombination was confirmed by PCR of tail DNA compared to cerebellar DNA (see Extended Data Fig. 7). For gene expression analysis RNA was extracted and analyzed as described above for MeCP2 R306C cerebellum samples.
Identification and analysis of MeCP2-repressed genes
To facilitate identification of genes repressed by MeCP2 in the context of extremely small changes in gene expression, we analyzed the 14168 common genes quantified across eight published microarray “training datasets” in five brain regions (hypothalamus, cerebellum, amygdala, striatum, hippocampus), applying the lowest possible threshold for fold-change (fold-change > 0 in the MeCP2 KO, fold-change < 0 in the MeCP2 OE) but demanding consistent misregulation in the predicted direction (at least 7 out of 8 datasets). Genes meeting this minimal threshold for direction of change were then filtered for minimum average change in gene expression (>7.5%), yielding 466 MeCP2-repressed genes (Supplementary Table 3). To determine if there 466 genes represent a significant population of reproducibly affected genes in MeCP2 mutants above what would be expected by chance we performed 7×105 resampling iterations, calculating the number of genes meeting the MeCP2-repressed criteria when the gene identity was randomized with respect to the calculated fold-change. This analysis yielded an average of 31 genes per iteration (observed/expected = 466/31 = 15) and did not detect an instance of 466 or more genes meeting the MeCP2-repressed criteria (maxium of 60 genes per iteration), thus yielding a significance of p < 1.5×10−6. The robustness of this gene list for predicting misregulation in Mecp2 mutants is demonstrated by the reproducible up-regulation of these genes in the “test datasets” in Extended Data Figure 8. Negative control datasets used in this analysis to test for specificity were identified through a survey of available GEO datasets. To qualify for analysis they were required to have a minimum number of biological replicates similar to the MeCP2 datasets (>4) and to have been analyzed on either of the microarray platforms used for the training datasets (Affymetrix MoGene 1.0 ST, or MoExon 1.0 ST). For individual gene analysis we calculated the significance of misregulation for individual example genes across the 10 Mecp2 mutant datasets displayed in Extended Data Figure 8 as follows: after confirming a normal distribution of fold-change values in each dataset, we calculated a z-score for the fold-change of each gene in each dataset. Assuming the null hypothesis that each gene would be randomly sampled from a standard normal distribution, a t-statistic was derived from the mean and standard error of the gene’s z-scores across the datasets, and this sample’s p-value was calculated from the t-distribution for 9 degrees of freedom. While the analysis presented here utilizes these 466 genes identified on the criteria described above, similar results for gene length, enriched overlap with FMRP target genes, and enrichment for neuronal annotations were obtained with gene lists generated using alternative criteria (e.g. up in MeCP2 KO, down in MeCP2 OE in 8 out 8 datasets without minimum expression threshold).
Gene ontology analysis was performed using the DAVID v6.7 bioinformatics resource44 (http://david.abcc.ncifcrf.gov/), using the 14168 genes covered in our analysis as background. Overlap of MeCP2-repressed genes with FMRP target genes was performed by mapping putative FMRP target lists29,45 to the 14168 genes used for identification of MeCP2-repressed genes. Data processing, plotting, and statistical analysis were performed using available packages and custom scripts in R.
Brain-specific expression of long genes
To assess expression of long genes across neural and non-neural tissues, RNA-Seq datasets for seven mouse tissues dissected from eight-week-old mice46 and ten human tissues47 were mapped and quantified as described above. Similar results of brain-specific long gene expression were obtained for microarray data from the wild type samples of the five brain regions analyzed in Mecp2 mutant studies compared to the wild type liver (data not shown).
Neuronal Cell Culture and Topotecan treatment
Primary cortical neurons were prepared from E16.5 mouse embryos and cultured as described by Kim et al.33. For lentiviral-mediated shRNA knockdown, virus was prepared as described in Tiscornia et al.48 using the MeCP2 shRNA and control shRNA plasmids previously validated in Zhou et al.49. Virus was concentrated and titrated using the GFP signal expressed from IRES GFP in the virus. After one day in vitro (DIV), cells were infected with lentivirus (knockdown or control) at an MOI of ~5, such that >90% of cells were infected. On DIV 4 cells were fed (neurobasal media with AraC, 2 μM final concentration) and subsequently treated with various dilutions of topotecan in DMSO (0.05% DMSO final concentration). At DIV 10, cells were collected in trizol for RNA analysis, or protein gel loading buffer for protein. RNA samples were processed and analyzed using the Nanostring nCounter assay as described above, with the exception that 6 control genes were used for normalization. Western blot analysis to confirm knockdown of MeCP2 was performed as described in Chen et al.43. Mean values shown in Extended Data Figure 9 (n = 3–5) are derived from separate cultures obtained from independent litters of mice (independent biological replicates), dissected on separate days, cultured and collected independently.
Regulatory Approval
All animal experiments were performed in accordance with regulations and procedures approved by the Harvard Medical Area Standing Committee on Animals (HMA IACUC).
Extended Data
Extended Data Figure 1. Analysis of gene expression changes in Mecp2 mutant mice.

a, Heatmap of median gene lengths for genes identified as misregulated in Mecp2 mutant studies or sixteen different studies of neurological dysfunction and disease in mice. Mouse model and GEO accession number, or reference, are listed (for Strand et al. (1), 3NP treatment; (2), Human HD brain; (3), R2/6 Htt transgenic). b, Scatter plots of fold-change in gene expression in the MeCP2 KO for the amygdala (left), which shows robust length-dependent misregulation, and the liver (right), which does not. Fold-change values for genes (black points) and mean fold-change for 200 genes per bin with a 40 gene step are shown (mean, red line; ribbon, S.E.M.). c, The fraction of genes showing fold-change > 0 for datasets in b; genes binned by length (100 gene bins, 50 gene step). d–f, Analysis of published microarray5–9 (d, e) or RNA sequencing (RNA-seq)18 (f) datasets from MeCP2 KO (d, f) or OE (e) mice. Mean fold-change in expression (200 gene bins, 40 gene step), red line; ribbon, S.E.M. For d–f, mean (black line) and two standard deviations (gray ribbon) are shown for 10,000 resamplings in which gene lengths were randomized with respect to fold-change. The spike in mean fold-change at ~1 kb in several plots corresponds to the olfactory receptor genes (Supplementary Discussion). g, Mean changes in expression of genes binned by length from RNA-seq analysis of MeCP2 KO cortex (n = 3 per genotype). h, Mean changes in expression from microarray analysis of genes binned by length in MeCP2 R306C cerebellum (n = 4 per genotype) i, Heatmap summary of fold-changes in gene expression from RNA-seq analysis of Mecp2 mutant mean in g compared to Nanostring nCounter (18 genes, top) or RT-qPCR (17 genes, bottom) analysis from cortex (n=4 per genotype). Selected long genes (>100 kb) consistently up-regulated in the MeCP2 KO or down-regulated in MeCP2 OE mutant mice across brain tissues were tested (Supplementary Table 2). A statistically significant up-regulation of these genes is observed in the cortex for both MeCP2 KO (nCounter, p = 0.00073; qPCR, p < 1×10−15) and MeCP2 R306C (nCounter, p = 0.0482; qPCR, p = 1.69×10−6; Hotelling T2 test for small sample size37). Note that for completeness, data from other figures have been re-presented here.
Extended Data Figure 2. Timing and severity of gene expression changes in models of RTT.

a, Mean fold-change in gene expression versus gene length in the hippocampus of MeCP2 KO mice compared to wild type at four and nine weeks of age reveals increasing magnitude of length-dependent gene misregulation that parallels the onset of RTT-like symptoms in these animals8. b, Mean fold-change in gene expression versus gene length in hippocampus of mice expressing truncated forms of MeCP2 mimicking human disease-causing alleles at four weeks of age. Re-expression of a longer truncated form of MeCP2 (G273X) in the MeCP2 KO normalizes expression of long genes more effectively than expression of a shorter truncation of MeCP2 (R270X), and parallels the higher degree of phenotypic rescue observed in MeCP2 G273X-expressing mice compared to MeCP2 R270X-expressing mice8. c, Mean fold-change in gene expression versus gene length in hippocampus of mice expressing truncated MeCP2 at nine weeks of age. Consistent with the eventual onset of symptoms of these mouse strains, length-dependent gene misregulation is evident in both strains. d, Changes in gene expression for genes binned by length in human MECP2 null ES cells differentiated into neural progenitor cells, neurons cultured for 2 weeks, or neurons cultured for 4 weeks15. In all plots, lines represent mean fold-change in expression for each bin (200 gene bins, 40 gene step), and the ribbon is S.E.M. of genes within each bin.
Extended Data Figure 3. High affinity of MeCP2 for mCG, mCA and hmCA in electrophoretic mobility shift assays.

a, Binding of the recombinant methyl-binding domain (MBD) of MeCP2 (amino acids 81–170) to 32P-end-labeled oligonucleotides containing a methylated cytosine in a CA (left) or a CG (right) context competed with unlabeled competitor substituted with unmethylated, methylated, or hydroxymethylated cytosine in a CG or CA context (indicated in bold). Representative full gels showing shifted and unshifted probe in the presence of 50-fold excess of unlabeled competitor (top); close-up of shifted bands over a range of unlabeled competitor (bottom). A mCA-containing oligonucleotide competes for MeCP2 binding with equal or higher efficacy to that of a symmetrically methylated CG oligonucleotide. While hmCG-containing probes compete with similar efficacy to an unmethylated probe, a hmCA-containing probe competes with high efficacy. This difference in affinity of MeCP2 for hmCA- and hmCG-containing probes may explain conflicting results reported for the affinity of MeCP2 for hydroxymethylated DNA18,50–53 (Supplementary Discussion). b, Binding and competition of recombinant MeCP2 MBD (amino acids 78–162, left) or full-length MeCP2 (amino acids 1–486, right) incubated with 32P-end-labeled oligonucleotides containing a methylated cytosine in a CA context and competed with oligonucleotides containing unmethylated, methylated, or hydroxymethylated cytosine in a CG, CA, CT, or CC context. Representative full gels showing 100-fold excess of unlabeled competitor (top); close-up of shifted bands over a range of unlabeled competitor (bottom). The results obtained from competitors containing mCG, mCA, hmCG and hmCA are similar to those shown in a. In addition, both (h)mCT- and (h)mCC-containing oligonucleotides compete for MeCP2 binding with similar efficacy to that of an unmethylated probe.
Extended Data Figure 4. ChIP-seq analysis of MeCP2 binding in vivo.

a, Boxplots of input-normalized read density within gene bodies (TSS +3 kb to TTS) for MeCP2 ChIP from the mouse frontal cortex plotted for genes according to quartile of mCA/CA, mCG/CG, hmCA/CA and hmCG/CG in the frontal cortex24 for all genes and genes >100 kb. b, Similar analysis of MeCP2 ChIP from the mouse cortex (left) or cerebellum (right) plotted for genes according to quartile of mCA/CA or mCG/CG for all genes and genes >100 kb. MeCP2 ChIP-signal is correlated with mCA/CA levels from the frontal cortex, cortex, and cerebellum for all genes and this correlation is more prominent among genes >100 kb. mCG does not show as prominent a correlation with MeCP2 ChIP signal, and hmCG trends toward anti-correlation with MeCP2 ChIP. These results suggest that MeCP2 has a lower affinity for hmCG than mCG, suggesting that, in vivo, hmCG is associated with reduced MeCP2 occupancy (Supplementary Discussion). c, High resolution analysis of high-coverage bisulfite sequencing data from the frontal cortex showing a correlation between MeCP2 ChIP signal and mCA. Input-normalized ChIP signal plotted for mCA levels for 500 bp bins tiled across all genes. d, Aggregate plots of MeCP2 input-normalized ChIP signal (top) and relative methylation (log2 enrichment in mC as compared to the flanking regions) for mCA, mCC, mCT, and mCG (bottom) are plotted around the 31,479 summits of MeCP2 ChIP enrichment identified using the MACS peak-calling algorithm40 (red) or 31,479 randomly selected control sites (gray, see Methods).
Extended Data Figure 5. Genomic analysis of mCG, hmCG, and hmCA in length-dependent gene regulation by MeCP2.

a–c, Mean methylation of CG dinucleotides (mCG/CG) within gene bodies (transcription start site +3 kb, up to transcription termination site) in the cortex (a), hippocampus (b) and cerebellum (c) for genes binned according to length. d–f, Mean fold-change in gene expression in MeCP2 KO compared to wild type in the cortex (d), hippocampus (e), and cerebellum (f) for genes binned according to mCG levels (mCG/CG) within gene bodies. g, Mean hmCG levels (hmCG/CG) within gene bodies in the frontal cortex24 for genes binned according to length. h, Mean fold-change in gene expression in MeCP2 KO compared to wild type for genes binned according to hmCG levels (hmCG/CG) within gene bodies in the frontal cortex24 i, Mean hmCA levels (hmCA/CA) within gene bodies in the frontal cortex24 for genes binned according to length. j, Mean fold-change in gene expression in MeCP2 KO compared to wild type genes binned according to hmCA levels (hmCA/CA) within gene bodies in the frontal cortex24. In all panels, mean values for each bin are indicated as a line (200 gene bins, 40 gene step); ribbon depicts S.E.M. for genes within each bin.
Extended Data Figure 6. Genomic analysis supports a role for mCA in length-dependent gene regulation by MeCP2.

a–c, Mean methylation at CA dinucleotides (mCA/CA) within gene bodies (TSS +3 kb to TTS) in cortex (a), hippocampus (b), and cerebellum (c) for genes binned by length. d–f, Mean changes in gene expression in cortex (d), hippocampus (e), and cerebellum (f) of MeCP2 KO for high mCA genes (top 25% mean gene body mCA/CA) and low mCA genes (bottom 66% mean gene body mCA/CA) binned by length. g–i, Mean changes in gene expression in cortex (g), hippocampus (h), and cerebellum (i) of MeCP2 KO for genes binned according to average gene body mCA/CA levels. j–l, Mean changes in gene expression in cortex (j), hippocampus (k), and cerebellum (l) of MeCP2 KO mice for long genes (top 25%) and short genes (bottom 25%) in each brain region binned by gene body mCA/CA level. A correlation between fold-change in the MeCP2 KO and mCA/CA for all genes is less prominent, or not observed, in the hippocampus and cerebellum for all genes together (h, i), but it is clear for the longest genes in the genome analyzed alone (k, l). Note that average levels of mCA appear lower in hippocampus and cerebellum compared to cortex (compare y-axis in a, b and c), and may explain why a correlation across all genes in not detected in these brain regions. In long genes analyzed alone the cumulative effect of higher mCA levels and integration across the gene may be larger, resulting in a detectable effect. In all panels, the line indicates the mean for 200 gene bins, with a 40 gene step; ribbon depicts S.E.M. for genes within each bin. Note that, for completeness, data from analysis of the cortex presented in Figure 2 are re-presented here.
Extended Data Figure 7. Conditional knockout of Dnmt3a in vivo.

a, Diagram of the Dnmt3a locus and Cre-dependent conditional knockout strategy for Dnmt3a26. LoxP sites (green triangles) flank exon 17, which is removed following Cre-mediated recombination. Primers (purple arrows) were designed to flank exons 17 and 18. The wild-type (WT), floxed (FLX), and knockout (KO) allele are depicted. b, Representative PCR genotyping for tail DNA samples indicates presence or absence of the floxed (flx, ~800 bp), wild-type (WT, ~750 bp), and knockout (KO, ~500 bp) alleles. Separate genotyping reaction for the Nestin-cre transgene (~250 bp) is shown. c, Efficient excision of the floxed exon is detected in cerebellar DNA from conditional knockout (Dnmt3aflx/flx; Nestin-Cre+/−, Dnmt3a cKO) mice but not from and control animals (Dnmt3aflx/flx, Control). d, Western blot analysis of Dnmt3a, MeCP2, and Gapdh (loading control) protein from the cerebellum of control and Dnmt3a cKO adult mice.
Extended Data Figure 8. Analysis of MeCP2-repressed genes and FMRP target genes.

a, Mean fold-change in mRNA expression for examples of MeCP2-repressed genes across three different Mecp2 mutant genotypes (KO, OE, and R306C) and six brain regions. p-values for each gene are derived from the mean z-scores for fold-change across all datasets (see Methods). b, Gene expression and CA methylation data from the cerebellum for selected MeCP2-repressed genes from a (right), as well as examples of extremely long genes (>100kb) that are not enriched for mCA and are not misregulated (left). Fold-changes in mRNA expression in Mecp2 mutants and the Dnmt3a cKO are shown (left axis), as well as mean mCA levels (gray; right axis). Red line indicates genomic median for gene body mCA/CA c, Boxplots of mCA levels in MeCP2-repressed genes compared to all genes. d, Mean fold-change for MeCP2-repressed genes in eight “training datasets” used to define these genes (see Methods), and nine “test datasets”: three Mecp2 mutant datasets not used to define MeCP2-repressed genes (CTX MeCP2 KO and CB MeCP2 R306C, generated in this study; HC MeCP2 KO 4wk, analyzed from Baker et al.8), and six datasets from brains of mouse models of neurological dysfunction generated using the same microarray platforms as the MeCP2 datasets (Geo accession # in order: GSE22115, GSE27088, GSE43051, GSE47706, GSE44855, GSE52584). Error bars are SEM of MeCP2–repressed gene expression across samples (n=4–8 microarrays per genotype per dataset); ** p <0.01, one-tailed t-test, Benjamini-Hochberg correction. Note that significance testing was not performed on training datasets. Brain regions indicated as in Figure 1, (WB, whole brain). e, Cumulative distribution function (CDF) of gene lengths plotted exclusively for genes that are among the top 60% of expression levels in the brain (Supplementary Discussion). The extreme length of MeCP2-repressed genes and genes encoding FMRP target mRNAs29 when controlling for expression level indicates that the long length of these genesets is not a secondary effect of the preferential expression of long genes in the brain (p < 1×10−15 for each geneset versus all expressed genes; 2-sample Kolmogorov-Smirnov (KS) test). f, The CDF of gene lengths for all genes compared to an independent set of FMRP targets identified by Brown and colleagues45 (p < 1 ×10−15, KS-test). g, CDF of gene lengths for genes expressed at similar levels in the brain and other somatic tissues (Supplementary Discussion). The extreme length of each geneset (p < 1×10−15, KS-test) when filtering for genes that are expressed in all tissues indicates that regulation of long genes by MeCP2 and FMRP is not dependent on brain-specific expression. h, CDF of mature mRNA lengths for MeCP2-repressed genes, and FMRP target genes (p < 1×10−11 for each geneset versus all genes, KS-test). i, Overlap of MeCP2-repressed genes and putative FMRP target mRNAs29 (p < 5×10−5, hypergeometric test). Expected overlap was calculated by dividing the expected overlap genome-wide (hypergeometric distribution) according to the distribution of all gene lengths in the genome.
Extended Data Figure 9. Consequences of long gene misregulation in neurons.

a, Mean expression of genes binned according to length in human neural and non-neural tissues. Mean expression for genes within each bin (200 gene bins, 40 gene step) is indicated by the line; ribbon represents the S.E.M. of genes within each bin. b, Western blot analysis of MeCP2 from primary cortical neurons after control or MeCP2 shRNA knockdown (KD) and treatment with DMSO vehicle (−) or topotecan (+). c, Heatmap summary of nCounter analysis for the expression of selected MeCP2-repressed (MR) genes from primary neurons treated with control or MeCP2 shRNA and topotecan (n = 3–4). Normalized log2 fold-change relative to the DMSO-treated, control KD is shown. MeCP2 KD conditions are significantly different from control, (p = 1e-4, repeated measures ANOVA across 8 genes). Newman-Keuls corrected, post-hoc comparisons: p < 0.05 control KD, 0 nM drug versus MeCP2 KD, 0 nM drug; p > 0.05, control KD, 0 nM drug versus MeCP2 KD, 50 nM drug; p < 0.05 MeCP2 KD, 0 nM drug versus MeCP2 KD, 50 nM drug. d, Bioanalyzer profiles of 18S and 28S ribosomal RNA (top) and total RNA quantification (bottom) for treated neurons (n=3–5). Total RNA values normalized to DMSO-treated control KD, red dashed line. Two-way repeated measures ANOVA indicates a significant effect of KD (p < 0.01) and drug treatment (p < 0.05). Rescue assessed by one-tailed t-test, Bonferroni multiple testing correction, * p < 0.05.
Extended Data Table 1.
Gene ontology analysis of MeCP2-repressed genes and genes >100 kbFunctional annotation clustering analysis of genes identified as MeCP2-repressed and the longest genes in the genome (> 100 kb) was performed using the David bioinformatics resource (David v6.7) 44. The top fifteen enriched gene ontology terms with p < 0.01 (Benjamini multiple testing correction) are listed for “Biological Process”, “Cellular Component”, and “Molecular Function”, respectively.
| MeCP2 Repressed Genes (466 genes) | |||||
|---|---|---|---|---|---|
| GO Term | Gene Count | EASE pval | Fold Enriched | Benjamini pval | GO Accession |
| Biological Process | |||||
| axon guidance | 17 | 3.7E-08 | 5.6 | 6.3E-05 | GO:0007411 |
| axonogenesis | 21 | 6.5E-08 | 4.3 | 5.5E-05 | GO:0007409 |
| cell morphogenesis involved in differentiation | 23 | 2.4E-07 | 3.7 | 1.4E-04 | GO:0000904 |
| neuron projection morphogenesis | 21 | 2.6E-07 | 4 | 1.1E-04 | GO:0048812 |
| cell morphogenesis involved in neuron differentiation | 21 | 3.6E-07 | 3.9 | 1.2E-04 | GO:0048667 |
| neuron projection development | 23 | 3.9E-07 | 3.6 | 1.1E-04 | GO:0031175 |
| neuron development | 26 | 9.3E-07 | 3.1 | 2.3E-04 | GO:0048666 |
| cell projection morphogenesis | 21 | 1.3E-06 | 3.6 | 2.8E-04 | GO:0048858 |
| cell morphogenesis | 26 | 2.1E-06 | 3 | 4.0E-04 | GO:0000902 |
| cell part morphogenesis | 21 | 3.3E-06 | 3.4 | 5.5E-04 | GO:0032990 |
| phosphate metabolic process | 49 | 3.3E-06 | 2 | 5.1E-04 | GO:0006796 |
| phosphorus metabolic process | 49 | 3.3E-06 | 2 | 5.1E-04 | GO:0006793 |
| cellular component morphogenesis | 27 | 4.2E-06 | 2.8 | 5.9E-04 | GO:0032989 |
| cell projection organization | 26 | 5.2E-06 | 2.8 | 6.8E-04 | GO:0030030 |
| enzyme linked receptor protein signaling pathway | 24 | 8.5E-06 | 2.9 | 1.0E-03 | GO:0007167 |
| Cellular Component | |||||
| plasma membrane | 110 | 2.00E-05 | 1.4 | 5.3E-03 | GO:0005886 |
| cell junction | 29 | 4.30E-05 | 2.3 | 5.7E-03 | GO:0030054 |
| cytoskeleton | 50 | 6.00E-05 | 1.8 | 5.4E-03 | GO:0005856 |
| postsynaptic density | 8 | 2.40E-04 | 6.2 | 1.6E-02 | GO:0014069 |
| synapse | 21 | 3.80E-04 | 2.4 | 2.0E-02 | GO:0045202 |
| plasma membrane part | 64 | 8.20E-04 | 1.5 | 3.6E-02 | GO:0044459 |
| cell fraction | 29 | 2.10E-03 | 1.8 | 7.9E-02 | GO:0000267 |
| basement membrane | 8 | 2.60E-03 | 4.2 | 8.3E-02 | GO:0005604 |
| neuron projection | 16 | 3.10E-03 | 2.4 | 8.7E-02 | GO:0043005 |
| synapse part | 14 | 3.20E-03 | 2.6 | 8.3E-02 | GO:0044456 |
| insoluble fraction | 26 | 3.50E-03 | 1.9 | 8.3E-02 | GO:0005626 |
| membrane fraction | 25 | 4.50E-03 | 1.8 | 9.7E-02 | GO:0005624 |
| postsynaptic membrane | 10 | 4.90E-03 | 3.1 | 9.7E-02 | GO:0045211 |
| Molecular Function | |||||
| cation binding | 148 | 5.00E-07 | 1.4 | 2.6E-04 | GO:0043169 |
| metal ion binding | 147 | 5.50E-07 | 1.4 | 1.4E-04 | GO:0046872 |
| ion binding | 149 | 7.50E-07 | 1.4 | 1.3E-04 | GO:0043167 |
| calcium ion binding | 47 | 8.50E-06 | 2 | 1.1E-03 | GO:0005509 |
| actin binding | 21 | 2.00E-04 | 2.6 | 2.1E-02 | GO:0003779 |
| cytoskeletal protein binding | 26 | 3.80E-04 | 2.2 | 3.3E-02 | GO:0008092 |
| protein kinase activity | 33 | 5.30E-04 | 1.9 | 3.9E-02 | GO:0004672 |
| cation channel activity | 18 | 8.40E-04 | 2.5 | 5.3E-02 | GO:0005261 |
| voltage-gated cation channel activity | 12 | 1.10E-03 | 3.2 | 6.3E-02 | GO:0022843 |
| alkall metal ion binding | 16 | 1.40E-03 | 2.6 | 6.8E-02 | GO:0031420 |
| metal ion transmembrane transporter activity | 19 | 1.90E-03 | 2.3 | 8.6E-02 | GO:0046873 |
| voltage-gated ion channel activity | 14 | 1.90E-03 | 2.7 | 7.9E-02 | GO:0005244 |
| voltage-gated channel activity | 14 | 1.90E-03 | 2.7 | 7.9E-02 | GO:0022832 |
| potassium ion binding | 11 | 2.20E-03 | 3.2 | 8.6E-02 | GO:0030955 |
| Genes Longer that 100KB (1431 genes) | |||||
|---|---|---|---|---|---|
| GO Term | Gene Count | EASE pval | Fold Enriched | Benjamini pval | GO Accession |
| Biological Process | |||||
| phosphate metabolic process | 150 | 1.2E-18 | 2 | 3.4E-15 | GO:0006796 |
| phosphorus metabolic process | 150 | 1.2E-18 | 2 | 3.4E-15 | GO:0006793 |
| protein modification process | 191 | 8.3E-18 | 1.8 | 1.2E-14 | GO:0006464 |
| protein amino acid phosphorylation | 120 | 4.1E-17 | 2.2 | 4.0E-14 | GO:0006468 |
| biopolymer modification | 191 | 1.4E-15 | 1.7 | 1.0E-12 | GO:0043412 |
| phosphorylation | 124 | 2.7E-15 | 2 | 1.6E-12 | GO:0016310 |
| cellular component organization | 247 | 1.1E-14 | 1.6 | 5.5E-12 | GO:0016043 |
| biological adhesion | 101 | 2.4E-14 | 2.2 | 1.0E-11 | GO:0022610 |
| cell adhesion | 101 | 2.4E-14 | 2.2 | 1.0E-11 | GO:0007155 |
| post-translational protein modification | 156 | 1.4E-13 | 1.8 | 5.0E-11 | GO:0043687 |
| cellular process | 849 | 1.6E-13 | 1.1 | 5.2E-11 | GO:0009987 |
| nervous system development | 137 | 4.9E-13 | 1.8 | 1.4E-10 | GO:0007399 |
| cell projection organization | 65 | 9.0E-11 | 2.3 | 2.4E-08 | GO:0030030 |
| cell morphogenesis | 62 | 2.3E-10 | 2.3 | 5.6E-08 | GO:0000902 |
| neuron development | 59 | 8.2E-10 | 2.3 | 1.8E-07 | GO:0048666 |
| Cellular Component | |||||
| synapse | 74 | 7.9E-17 | 2.8 | 4.4E-14 | GO:0045202 |
| cell junction | 86 | 2.5E-13 | 2.3 | 5.0E-11 | GO:0030054 |
| neuron projection | 58 | 3.4E-13 | 2.8 | 4.5E-11 | GO:0043005 |
| cell projection | 96 | 1.4E-12 | 2.1 | 1.4E-10 | GO:0042995 |
| cytoskeleton | 149 | 2.9E-12 | 1.7 | 2.3E-10 | GO:0005856 |
| plasma membrane | 325 | 5.3E-12 | 1.4 | 3.5E-10 | GO:0005886 |
| plasma membrane part | 205 | 5.5E-12 | 1.6 | 3.1E-10 | GO:0044459 |
| extracellular matrix part | 30 | 2.5E-11 | 4 | 1.2E-09 | GO:0044420 |
| basement membrane | 24 | 1.8E-09 | 4.1 | 8.0E-08 | GO:0005604 |
| synapse part | 43 | 1.1E-08 | 2.6 | 4.2E-07 | GO:0044456 |
| proteinaceous extracellular matrix | 56 | 1.4E-08 | 2.2 | 4.9E-07 | GO:0005578 |
| axon | 31 | 1.8E-08 | 3.1 | 6.1E-07 | GO:0030424 |
| extracellular matrix | 57 | 2.3E-08 | 2.2 | 7.1E-07 | GO:0031012 |
| dendrite | 29 | 4.8E-08 | 3.1 | 1.4E-06 | GO:0030425 |
| postsynaptic membrane | 29 | 2.2E-07 | 2.9 | 5.8E-06 | GO:0045211 |
| Molecular Funtion | |||||
| calcium ion binding | 138 | 1.4E-15 | 2 | 1.2E-12 | GO:0005509 |
| protein kinase activity | 111 | 1.6E-15 | 2.2 | 6.6E-13 | GO:0004672 |
| adenyl ribonucleotide binding | 209 | 9.3E-15 | 1.7 | 2.6E-12 | GO:0032559 |
| cytoskeletal protein binding | 85 | 1.5E-14 | 2.4 | 3.1E-12 | GO:0008092 |
| GTPase regulator activity | 72 | 1.2E-13 | 2.5 | 2.0E-11 | GO:0030695 |
| nucleoside binding | 215 | 1.5E-13 | 1.6 | 2.1E-11 | GO:0001882 |
| adenyl nucleotide binding | 212 | 2.6E-13 | 1.6 | 3.1E-11 | GO:0030554 |
| purine nucleoside binding | 213 | 2.8E-13 | 1.6 | 2.9E-11 | GO:0001883 |
| ATP binding | 202 | 3.0E-13 | 1.6 | 2.8E-11 | GO:0005524 |
| nucleoside-triphosphatase regulator activity | 72 | 3.9E-13 | 2.5 | 3.3E-11 | GO:0060589 |
| ion binding | 412 | 9.8E-12 | 1.3 | 7.5E-10 | GO:0043167 |
| metal ion binding | 403 | 2.0E-11 | 1.3 | 1.4E-09 | GO:0046872 |
| purine ribonucleotide binding | 229 | 2.5E-11 | 1.5 | 1.6E-09 | GO:0032555 |
| ribonucleotide binding | 229 | 2.5E-11 | 1.5 | 1.6E-09 | GO:0032553 |
| cation binding | 404 | 3.8E-11 | 1.3 | 2.3E-09 | GO:0043169 |
Supplementary Material
Acknowledgments
We thank E. Griffith and members of the Greenberg laboratory, A. Bird, G. Mandel and members of their laboratories for helpful discussions, M. Goodell for providing the Dnmt3a mice, N. Sharma and F. Dibiase for experimental support, and M. Mistry of the HSPH Bioinformatics Core, Harvard School of Public Health, Boston, MA for assistance with gene expression analysis. This work was supported by grants from the Rett Syndrome Research Trust and the NIH (1RO1NS048276) to M.E.G., fellowships from the Damon Runyon Cancer Research Foundation (DRG-2048-10) and the William Randolf Hearst fund to H.W.G., as well as NIH grant T32GM007753, and the HHMI Gilliam fellowship to B.Z.K., H.S. is a HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG-2194-14).
Footnotes
Author Contributions
H.W.G., and B.Z.K. performed or directed all experiments and analysis in the study. H.W.G., B.Z.K. and D.A.H. performed gene expression analysis. B.Z.K. performed EMSA assays. H.W.G. performed ChIP-seq analysis. H.W.G., H.S., N.R.K. performed bisulfite sequencing and DNA methylation analysis. H.W.G., D.A.H., H.S., N.R.K., and M.H. performed bioinformatics and statistical analysis. H.W.G., B.Z.K and C.S.G. performed Dnmt3a mouse experiments and neuronal culture experiments. D.H.E provided mouse reagents. H.W.G., B.Z.K. and M.E.G. wrote the manuscript. M.E.G. advised on all aspects of the study.
Raw data and processed values from RNA-seq, Microarray, ChIP-seq and bisulfite-seq experiments have been submitted to the NCBI Gene Expression Omnibus under accession number GSE60077.
The authors declare no competing financial interests.
References
- 1.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annual review of cell and developmental biology. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 3.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan C, Li HH, Kwan HC, Francke U. Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC medical genetics. 2007;8:36. doi: 10.1186/1471-2350-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Human molecular genetics. 2009;18:2431–2442. doi: 10.1093/hmg/ddp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samaco RC, et al. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nature genetics. 2012;44:206–211. doi: 10.1038/ng.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker SA, et al. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152:984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao YT, Goffin D, Johnson BS, Zhou Z. Loss of MeCP2 function is associated with distinct gene expression changes in the striatum. Neurobiology of disease. 2013;59:257–266. doi: 10.1016/j.nbd.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King IF, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugino K, et al. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:12877–12883. doi: 10.1523/JNEUROSCI.2674-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meins M, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. Journal of medical genetics. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins AL, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human molecular genetics. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 14.Lyst MJ, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nature neuroscience. 2013;16:898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell stem cell. 2013;13:446–458. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng V, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Human molecular genetics. 2007;16:640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JD, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 18.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo JU, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nature neuroscience. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Molecular cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nature neuroscience. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie W, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen S, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaneda M, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 27.Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raychaudhuri S, et al. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS genetics. 2010;6:e1001097. doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yazdani M, et al. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem cells. 2012;30:2128–2139. doi: 10.1002/stem.1180. [DOI] [PubMed] [Google Scholar]
- 31.McGill R, Tukey J, Larsen WA. Variations of Box Plots. The American Statistician. 1978;32:12–16. [Google Scholar]
- 32.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory in biosciences = Theorie in den Biowissenschaften. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 37.Wu Y, Genton MG, Stefanski LA. A multivariate two-sample mean test for small sample size and missing data. Biometrics. 2006;62:877–885. doi: 10.1111/j.1541-0420.2006.00533.x. [DOI] [PubMed] [Google Scholar]
- 38.Chen PY, Cokus SJ, Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC bioinformatics. 2010;11:203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebert DH, et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng J, Liu T, Zhang Y. Using MACS to identify peaks from ChIP-Seq data. Current protocols in bioinformatics/editoral board, Andreas D. Baxevanis … [et al.] 2011;Chapter 2(Unit 2):14. doi: 10.1002/0471250953.bi0214s34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagy G, Daniel B, Jonas D, Nagy L, Barta E. A novel method to predict regulatory regions based on histone mark landscapes in macrophages. Immunobiology. 2013;218:1416–1427. doi: 10.1016/j.imbio.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Tronche F, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature genetics. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 43.Chen WG, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 44.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 45.Brown V, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477–487. doi: 10.1016/s0092-8674(01)00568-2. [DOI] [PubMed] [Google Scholar]
- 46.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 47.Gray JM, et al. SnapShot-Seq: a method for extracting genome-wide, in vivo mRNA dynamics from a single total RNA sample. PloS one. 2014;9:e89673. doi: 10.1371/journal.pone.0089673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nature protocols. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valinluck V, et al. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic acids research. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashimoto H, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic acids research. 2012;40:4841–4849. doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spruijt CG, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 53.Khrapunov S, et al. Unusual characteristics of the DNA binding domain of epigenetic regulatory protein MeCP2 determine its binding specificity. Biochemistry. 2014;53:3379–3391. doi: 10.1021/bi500424z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.