

Abstract
Hepatocellular carcinoma (HCC) is the one of the leading causes of cancer mortality in the world, mainly due to the difficulty of early detection and limited therapeutic options. The implementation of HCC surveillance programs in well-defined, high-risk populations were only able to detect about 40–50% of HCC at curative stages (Barcelona Clinic Liver Cancer stages 0 & 1) due to the low sensitivities of the current screening methods. The advance of sequencing technologies has identified numerous modifications as potential candidate DNA markers for diagnosis/surveillance. Here we aim to provide an overview of the DNA alterations that result in activation of cancer pathways known to potentially drive HCC carcinogenesis and to summarize performance characteristics of each DNA marker in the periphery (blood or urine) for HCC screening.
Keywords: circulating DNA biomarkers, HBV-associated HCC DNA markers, HCC DNA markers, HCC surveillance and early detection, hepatocellular carcinoma, non-invasive
Hepatocellular carcinoma (HCC) represents 90% of all adult primary liver cancers and is one of the leading causes of cancer mortality in the world, responsible for over 746,000 deaths in 2012 and accounting for 9.1% of the global cancer mortality rate [1]. While overall cancer mortality has decreased, liver cancer related mortality has been rising steadily by 2.4% yearly, with 782,000 new HCC cases each year [1]. This high mortality rate is mainly due to limited therapeutic options and difficulty in the early detection, thus undermining the prognosis [2].
The 5-year survival rate for HCC is 29% when detected at an early localized stage, 10% at a regional stage and as low as 3% at a distant metastasized stage [3,4]. Up to 90% of HCC cases arise in individuals with an established risk factor. Given that there is a long period, sometimes over a decade in length, between the establishment of risk factors and the development of HCC, surveillance of patients at risk for HCC is a critical component of any measure adopted for early detection of cancer and improvement of patient prognosis, as recommended by both the American Association fur Study of Liver Diseases (AASLD) and the European Association for Study of Liver Diseases (EASL) [2,5,6]. Despite the implementation of HCC surveillance programs in the well-defined, high-risk populations, early detection of HCC remains difficult to achieve with current screening methods, due to low sensitivities. The most widely used biomarker for HCC screening is the serum level of α-fetoprotein (AFP), which has a sensitivity of 40 60% for all stages and 40–50% for early stages HCC [2,7,8]. Thus, the need is urgent fur biomarkers with increased sensitivities for the early detection of HCC.
Like other cancers, HCC is a disease of the genome, caused by genetic and epigenetic DNA modifications [9–11]. Identification of the DNA modifications (mutation or methylation) underlying the development of HCC should permit unambiguous diagnosis of HCC cases. The introduction of noct generation sequencing (NGS) has lead to the identification of many of such modifications, associating with HCC tissues. A significant amount of work has fucused on distinguishing the drivers from the passengers of these DNA modifications, in combination with studying the pathways involved in carcinogenesis to determine how these DNA modifications can be used as potential markers for HCC detection [11–19]. However, these candidate DNA markers will not be useful for either diagnosis or surveillance unless they can be detected in the peripherals, without liver biopsy. The objectives of this article are to provide an overview of DNA alterations that result in activation of cancer pathways known to potentially drive HCC carcinogenesis and to summarize performance characteristics of each DNA marker as a potential marker for HCC screening. Special attention is drawn to the markers that have been previously detected in the peripherals (blood or urine) in association with HCC. Finally, we discuss how these HCC markers can be used in HCC surveillance and personalized management of patients.
HCC surveillance
Almost 90% of HCC cases arise in individuals with an estab lished risk factor. These risk factors include hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol intake, aflatoxin B 1 exposure, hemochromatosis, α-1-antitrypsin deficiency, primary biliary cirrhosis and non-alcoholic fatty liver disease [2.5.20]. An HCC-surveillance program has been recommended by both AASLD and EASL to patients with high-risk factors, in order to detect HCC earlier for better prognosis [2,5]. The current recommended test for surveillance of HCC is an abdominal ultrasound performed by an experienced operator every 6 months, in subjects at risk for HCC. Ultrasound, as a screening test, has a sensitivity in the range of 40–89% and a specificity of 90%. This wide range of sensitivity is mainly due to the skill of the operator and the challenge of detecting HCC in a back ground of cirrhotic liver with micro- and macro-regenerative nodules [5,21-23]. Serum AFP levels is one of the most commonly performed and best-studied serological test. At a cutoff of 20 ng/ml, serum AFP has a sensitivity of 40–60% and a specificity of 90%, but an unacceptable positive predictive value of 25% at 5% disease prevalence [7]. Thus, AFP as a screening test at a 20 ng/ml cutoff has both a high false positive rate and a poor efficacy for surveillance, and is therefore not recommended as a surveillance tool, either alone or in combination with ultrasound, by both AASLD and EASL. Other serological tests such as des-γ-carboxy prothrombin and AFP-L3 fraction have low sensitivities and are also not recommended for screening [5,24,25]. Additional tests include CT scan and MRI, which are both sensitive imaging methods, but they too are not used for HCC-surveillance because of the high cost and the inherent risk of radiation. With the current surveillance program for HCC detection, an average of 50% of cases are identified at early stages of HCC [26]. Consequently, better screening tests are needed to improve the efficacy of HCC surveillance.
Potentials of circulating DNA markers for HCC surveillance & the early detection
Malignant transformation of hepatocytes occurs when DNA alterations either activate or inactivate certain cancer pathways that induce uncontrolled growth of the cells, or clonal expansion, which is initiated from primary alterations that occur in genes as ‘drivers’. The development of HCC, as with other solid tumors, is believed to require the dysregulation of at least three core cellular pathways (cell cycle, apoptosis/cell survival, genome maintenance) within the cell [27-30]. Thus, identification of the DNA modifications underlyin g the development of HCC should permit unambiguous diagnosis of HCC. A great deal of work has been devoted to identifying such DNA modi fications as potential biomarkers for the early detection of cancer. In general, the number of candidate cancer biomarkers has exploded since the introduction of genome wide sequencing of diseased tissue DNA, using NGS [11–15,18,19,31–33] and genome wide methylation study using methylation arrays [34–37]. However, such identification will only be useful for cancer screening and the early detection of cancer, if it can be done in the periphery in a non-invasive or minimally invasive manner.
DNA containing cancer ‘signatures’ (mutations or hypermethylation) has been found in the plasma, serum and urine of patients with many cancers, including HCC. In almost all instances where tumor tissue was available, the tumor associated DNA modification s in the plasma [38–40] or urine [41–43] were generally consistent to those modifications detected in the primary tumor, demonstrating that the tumor-derived DNA can be detected in the circulation via blood or urine, if a tumor is present. Several reviews have addressed the possible mechanisms of the presence of cell-free tumor DNA in the circulation [44–47]. Briefly, this cell-free circulating tumor DNA could be either from primary tumor or from circulating tumor cells due to cell death by either necrosis or apoptosis, or even microvesicle-released DNA in the circulation originating from tumor cells. Regardless of the various sources of cell-free circulating tumor DNA, the presence of tumor DNA in circulation provides a great promise to use blood or urine as a liquid biopsy to profile cancer genetics for cancer screening or early detection [44,48–52].
One advantage of using DNA biomarkers is that an HCC DNA test can be very sensitive if the test can detect all the possible DNA modifications that drive hepatocarcinogenesis in a non invasive or minimally invasive manner. Furthermore, DNA tests can also provide information on cancer genetics, helping to determine a possible prognosis and even tailor personalized treatments for patients, if a tumor is detected. Another clear advantage of using circulating DNA biomarkers for HCC screening as the detection of the DNA markers are not affected by the background liver disease, location of the lesions, number of nodules or obesity, which are all technical challenges for ultrasound. Thus, if using DNA markers alone does not provide sufficient sensitivity for HCC screening, then these markers can at least increase the performance of ultrasound .
The major challenge, however, is the specificity of circulating DNA markers for HCC. As mentioned, the sensitivity of the test can be extremely high, thus the specificity of the test will always be a challenge because most driver modifications are not liver-cancer specific. For instance, mutations of TP53 or methylations of tumor suppressors (such as the adenomatous polyposis coli gene [APC], RASSF1A and CDKN2A) are found not only in HCC, but also in many other cancers [53-58]. Fortunately, the specificity of the test can be improved if organ-specific markers, such as TP53 249T mutations or HBV-associated HCC DNA markers for patients with HBV-infections (discussed in a latter section), are included [42,59–61]. Taking advantage of a high sensitivity for early detection in a minimally or non invasive manner, the circulating-DNA markers, remain extremely valuable as a screening tool, even with compromised specificity. These markers can be used to guide patients toward more sophisticated imaging diagnosis, or be used in combination with patient history, to provide a molecular characterization of the tumor for better management, if the tumor is diagnosed.
Strategy for constructing a panel of multiple DNA makers for HCC screening
As discussed, DNA markers should provide high sensitivity for HCC screening, if a test can detect all the drivers of hepatocarcinogenesis. HCC has a multifactorial etiology resulting in a disease with high heterogeneity, as different risk factors disrupt different signaling pathways. Thus, a single DNA marker is unlikely to have sufficient sensitivity for HCC screening, whereas a panel of biomarkers encompassing the frequently altered genes in the key pathways associated with HCC patho genesis would be needed to provide sufficient sensitivity to detect most of the HCC Previous studies have shown that mutations in cancer pathways exhibit an ‘exclusivity principle’, where if a certain gene is mutated in a pathway, other genes in that pathway are excluded from mutations [62–64]. The best known example is the exclusive existence of the K-ras and BRAF mutations in a tumor since these two genes are within the Ras pathway [65]. Similarly, in HCC, studies have suggested that the CTNNB1, AXIN1 and APC gene mutations are mutu ally exclusive [18,66,67]. Although correlated biomarkers (biomarkers that belong to only one pathway) can underscore the importance of a biological pathway, they often do not provide a substantial increase in predictive value when they are combined in a panel, unless the pathway is an essential pathway [68–70]. Thus, a large number of correlated biomarkers are substantially less informative than a small number of uncorrelated biomarkers. For instance, when we combined four uncorrelated biomarkers, the methylated APC gene (mAPC), the methylated RASSF1A gene ( mRASSF1A), the methylated glutathione S-transferase P1 gene (mGSTP1) and TP53 249T mutation in one panel, an additive effect of sensitivity was obtained with 93% sensitivity and approximately 80% specific ity to distinguish early HCC from non-HCC tissue (FIGURE 1).
Figure 1. Multi-marker receiver operating curves for mAPC, mGSTP1, mRASSF1A and TP53 249T mutation individually and in combination for distinguishing hepatocellular carcinoma (n = 120) from cirrhosis (n = 35) and hepatitis (n = 35) tissues.
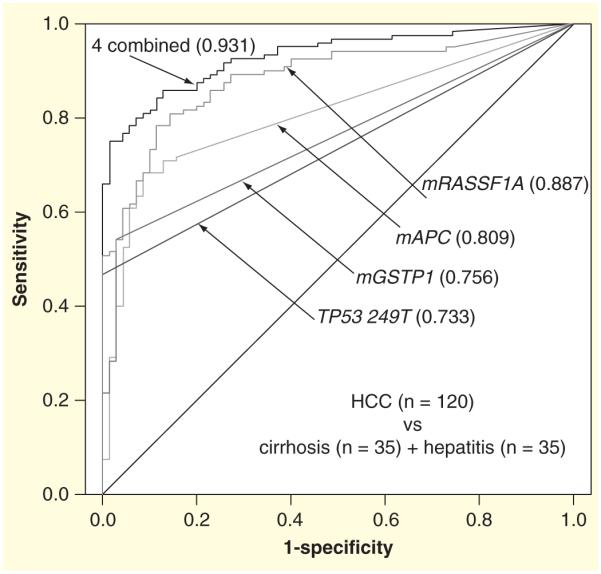
The area under the ROC is indicated in parentheses following each marker.
HCC: Hepatocellular carcinoma; mAPC: Methylated APC; mGSTP1: Methylated glutathione S-transferase P1; mRASSF1A: Methylated RASSF1A.
In addition, while most cases of HCC are associated with known etiologies, it would be ideal to have a single panel that can provide sufficient sensitivity and specificity for HCC screening, regardless of etiology. However, it may be necessary to construct etiologically-specific panels to improve the performance of markers. Recent developments in NGS have enabled the genome-wide sequencing studies of liver cancers, which cover mutational signatures with distinctive etiological backgrounds [13]. For instance, a significant co-relation has been reported between somatic substitution patterns and habitual alcohol drinking [19]. Additionally, two previously suggested copy number variations at 13q12.11 and 11q11 of non-alcoholic fatty liver disease were also identified by the genome wide study approach [71–74). Whether these risk markers are risk markers for non-alcoholic fatty liver disease-associated HCC needs to be studied.
In the next section, we briefly summarize HCC-associated cancer pathways, including whether or not there is a significant impact due to known etiologies, potential DNA modifications detected in tissue and most importantly, if these modifications have been detected in peripheral body fluids, which would serve as the foundation for the assembly of a single sensitive panel or etiologically specific panels for HCC screening.
HCC-associated cancer pathways & candidate DNA biomarkers for HCC detection
The multistep sequences of genetic and epigenetic alterations in liver cancer pathogenesis disrupt core cellular processes, such as cell cycle control, apoptosis and genome maintenance. The advent of NGS has allowed detailed mapping of the liver cancer genome, revealing extensive molecular alterations. Unlike some solid tumors, such as breast, lung or colon, development of HCC lacks a clear correlation to an oncogene. However, highly recurrent somatic mutations or methylations are found in the genes involved in some major cancer pathways, such as TP53 and Wnt/β-catenin pathways. This echoes the need for multiple markers to achieve a sufficient sensitivity for HCC screening. Here, we briefly review the key pathways and the DNA alterations, both genetic mutations and epigenetic DNA methylation, which have been reported in tissue as candidate DNA markers for building a panel of markers for molecular diagnosis of HCC. For this purpose, TABLE 1 lists the DNA alterations, both genetic and epigenetic, which are found in at least 5% of HCC tissue and have been reported by at least two independent studies, and these alterations are listed in order of incidence within each pathway. Also noted is whether or not each modification was previously detected in circulation.
Table 1.
Frequently reported genetic mutations (G) and epigenetic aberrant DNA methylations (M) in hepatocellular carcinoma tissues.
| Pathway | Gene | DNA alteration |
Incidence (%) | Reported in circulation (Yes/No) |
Ref. |
|---|---|---|---|---|---|
| Wnt signaling | APC | M/G | 80–93/1–2 | Yes | [11,18,38,75–80,82,176] |
| SFRP1 | M | 42–63 | Yes | [27,80,83–86,119] | |
| CTNNB1 | G | 19–33 | No | [11,18,87–94,177] | |
| AXIN1 | G | 13–15 | No | [11,12,18] | |
|
| |||||
| Cell cycle/ apoptosis |
RASSF1A | M | 55–85 | Yes | [80,120,178–180] |
| CDKN2A | M/G | 50–70/5–10 | Yes | [17,18,81,101,102,120,176,181] | |
| TP53 | G | 20–50 | Yes | [11,18,60,87,182–185] | |
|
| |||||
| Telomere maintenance |
TERT | G | 29–59 | No | [33,111] |
|
| |||||
| mTOR signaling | PTEN | M/G | 16–43/4–16 | No | [18,117,118] |
|
| |||||
| Chromatin remodeling |
ARID1A | G | 10–20 | No | [11,18,19] |
| ARID1B | G | 5–10 | No | [11,18,19] | |
| ARID2 | G | 5–18 | No | [11,18,113] | |
|
| |||||
| JAK/STAT | SOCS1 | M | 50–93 | No | [14,119,120] |
| JAK1 | G | 9 | No | [14,186] | |
|
| |||||
| Detoxification | GSTP1 | M | 38–80 | Yes | [80,121,122] |
|
| |||||
| Protease inhibitor |
TFPI2 | M | 47 | Yes | [124,125] |
Wnt/β-catenin signaling pathway
The Wnt pathway is the most frequently altered pathway found in HCC, and activation of which leads to the nuclear translocation of the β-catenin and initiates cell proliferation. Genes that are commonly altered in this pathway that are also detected in HCC are APC, CTNNB1, AXIN1 and Secreted Frizzled related protein 1 (SFRP1) genes. At least 75% of HCC was found to contain DNA modifications in one of these genes [11]. Among these alterations, aberrant DNA methylation in the promoter region of the APC gene is the most frequent alteration found in HCC for approximately 50–80% of cases [75–78]. Genetic muta tions of the APC genes have also been reported, but at a lower frequency (1.6%) [18]. The mAPC gene has been detected in the circulation of patients with HCC [38,79–82].
The next most frequently detected DNA marker is the epige netic alteration of the SFRP1 gene. SFRP1 protein forms an inhibitory complex with the frizzled receptor and acts as a Wnt pathway inhibitor. It is also known to be a negative regulator of cell invasion and could possibly predict the metastatic potential of a tumor [83]. Hypermethylation of the SFRP1 promoter is observed in more than 50% of HCC [27,84–86] and loss of heterozygosity is reported to be seen in 17.4% of cases [84].
As for genetic mutations in the Wnt pathway, the only fre quent mutation also found in HCC is in the CTNNB1 gene. Both point mutations and deletions were identified at the phosphorylation site in the region from codon 32 to codon 451 of exon 3 of the gene, accounting for 30–35% of HCC [11,18,67,87–94]. Interestingly, it was found that downregulation of the SRFP1 gene acted synergistically with the mutation of the CTNNB1 gene, effectively causing an upregulation of Wnt signaling in liver cancer [86]. Additionally, it was recently discovered via NGS analysis that mutations in the AXIN1 gene, a downstream protein in the pathway, were found in approximately 15% of HCC [12,18,19].
The cell cycle control pathways
The second most altered pathway found in HCC tissue is the cell cycle regulation pathway. A frequently altered gene of this pathway is the RASSF1A gene, which is a member of the Ras-association domain family of genes that can associate with the Ras family of GTPases to regulate the cell cycle and trigger apo ptosis [95]. It can also regulate microtubule dynamics and serve as a scaffold for multiple rumor suppressor complexes [96–100]. Abnormal signaling in Ras oncogenes was found in approximately 70% of all cancers [56,80,100]. Another frequently identified DNA modification in this pathway is methylation of CDKN2A (pl6), which occurs in 50 –70% of HCC. In addition to DNA methylation, mutations of the gene were also found, albeit less frequently (5–10%) [18,101,102]. Also within this pathway, mutations of the TP53 gene, which are associated with chromosomal instability, occur in 20–50% of HCC. TP53 mutations in codon 249 occur frequently (up to 50%) in aflatoxin-exposed populations [59,103,104]. Furthermore, IRF2, a gene upstream of TP53, was recently identified to be mutated in approximately 5% of HCC [18]. Among these, mRASSF1A and TP53 mutations were detected in the serum and urine of HCC patients at rates up to 90 and 50% respectively, suggesting these genes could be promising bio markers for non invasive HCC screening [38,42,60,80,86,105–107].
Telomere maintenance
Loss of telomere is one of the hallmarks of cancer. Nault et al. recently described frequent mutations (59% in HCC and 25% in cirrhosis) in the promoter region of the telomerase reverse transcriptase (TERT) gene [33]. Preferential integration of the HBV genome in the regions near or in the TERT gene has also been reported in five of the six HBV-HCC NGS studies [16,19,108–110]. Interestingly, higher rates of mutations in TERT (46% in HCV-HCC vs 16% in HBV-HCC) [111] and CTNNB1 (62.5% in HCV-HCC vs 37.5% in non-HCV-HCC) [15] were reported in HCV-associated HCC. Recently, it was suggested that TERT is the gate keeper of HCC [112].
The chromatin remodeling pathway
Approximately 50% of HCC cases were found to have mutations in the chromatin remodeling pathway [19]. Among the mutations detected, ARID1A, ARID1B and ARID2 are the most frequently detected, all of which are part of the SWI/SNF related chromatin-remodeling complex. It has been shown that siRNA targeting ARID1A, ARID1B and ARID2 genes in HCC cell lines resulted in increased cell proliferation, suggesting their role as drivers of hepatocarcinogenesis [113]. ARID1A mutations are linked to HCC with alcoholic liver disease and overlap with tumors harboring CTNNB1 mutations. ARID2 mutations were associated with HCV-HCC [18]. Additional mutations, such as in the SMARCA and MLL families [18], and copy number losses, such as in BRG and BRM [114], have also been identified in this pathway associated with HCC, but have not been recurrent across multiple studies. Since these mutations were identified recently, no report as of yet shows the detection of these mutations in the periphery.
The mTOR pathway
The PI3K/AKT/mTOR signaling pathway, which plays an important regulatory role in cell proliferation, migration, survival and angiogenesis, is frequently dysregulated in HCC [115,116]. Elevated Akt phosphorylation is seen in 15–41% of HCC. However, the pathway activation is predominantly the result of ligand-based activation due to growth factor and growth factor receptor overexpression in HCC cells, and this is usually mediated by copy number alterations of growth factors or receptors. The most frequent recurrent modifications identified from this pathway are PTEN mutations and methylation [18,117,118]. In addition, RPS6KA3 mutation was recently reported in about 10% of cases [11,18].
The JAK/STAT pathway
Genomic alterations in the JAK/STAT pathway have been reported in 45.5% of the HBV-HCC tumors, which are mainly caused by the aberrant methylation of the SOCS1 gene (50–90%) [14,119,120]. This modification is consistent with the notion that activation of the JAK-STAT pathway could be a driving factor in HBV-associated HCC [14]. In addition to DNA methylation, the activating mutations of JAK1 were also found, however, these modifications occurred less frequently, and were identified in only approximately 9% of HCC [14].
Other pathways
In addition to disrupting core cellular pathways, the aberrant methylation of one of the detoxification genes, GSTP1, and a protease inhibitor, TFPI12, were identified in a significant portion of HCC (~50% for both genes) [80,121–125]. Since these genes are distinct from the major pathways discussed, they should also be included for added value when a sensitive HCC DNA marker panel is assembled. Other pathways such as the oxidative stress pathway/NRF2-KEAP1 and the MLL pathway have been reported with several infrequent modifications to be involved in HCC pathogenesis [15,29], thus they are not included in this discussion.
Identification of AFP-negative HCC by DNA markers
As discussed earlier, the current ‘gold standard’ serum marker, AFP, and its fucosylated glycoform, L3, are of limited value because they have sensitivities of only 25–50% for early HCC [126]. Moreover, there is currently no biochemical marker that can detect AFP-negative HCC, in which the serum AFP level is <20 ng/ml, as suggested by the American Association of Liver Diseases [126]. Because of this, it was of interest to see whether DNA biomarkers could detect AFP negative HCC. DNA marker distribution was plotted for the HCC tissue from patients with serum AFP values available for analysis, as shown in FIGURE 2. In this study population (n = 112), 61 patients (54%, 61/112) with HCC had AFP serum levels <20 ng/ml, and their HCC samples were therefore considered to be AFP-negative. Encouragingly, DNA markers were found in 92% (56/61) of the AFP-negative HCC tissues, suggesting the potential of these markers to be used to detect AFP-negative HCC, should they be detectable in the periphery.
Figure 2. Detection of four DNA markers, mAPC, mGSTP1, mRASSF1A and TP53 249T mutation in hepatocellular carcinoma tissues (n = 112).

Marker distribution in individual HCC with available AFP value in the study population.
Filled boxes represent a marker detected or positive.
Empty boxes represent a marker not detected or negative by cutoff.
AFP: α-Fetoprotein; HCC: Hepatocellular carcinoma; mAPC: Methylated APC; mGSTP1: Methylated glutathione S-transferase P1; mRASSF1A: Methylated RASSF1A.
Specificity of HCC DNA markers for cancer screening & early detection
More than 80% of HCC cases arise from cirrhotic liver [127]. The process of carcinogene sis is a continuous process and is believed to occur over a course of years, initiating from a cirrhotic liver and evolving into the size of a tumor that can be detected via ultrasound imaging, which is approximately 2 cm in diameter [128]. The DNA mutations and aberrant epigenetic DNA methylation patterns that drive carcinogenesis, the drivers, are expected to exist at the onset of carcinogenesis, thus, occur mostly in cirrhotic liver. Many, if not all, of these early markers were also detected in adjacent non-HCC tissue, which, as part of the cancer microenvironment, is the field where HCC nodules are expected to arise. DNA modifications of these drivers would be good candidates as DNA markers for the early detection. However, these markers would not be very specific to HCC, as the observed modifications could already exist in cirrhotic liver or pre neoplastic nodules. As for HCC screening or early detection, these early markers are important to identify subjects for more sophisticated imaging diagnosis.
As discussed above, one approach to potentially improve the specificity of the test for screening and early detection is to combine early, organ-specific, and highly-specific late markers into a panel evaluated with a trained algorithm. Another approach to improve the specificity of the marker is to detect the region of the gene that is more specific for HCC. This is of particular importance for methylated DNA markers as there is emerging evidence suggesting that the specificity of methylation of different regions of the CpG island of the tumor suppressor genes could vary the performance of biomarkers for cancer detection, or in its association with clinicopathology [75,121,129,130]. For instance, we noticed a variation between studies of the specificity for distinguishing tumor from non-tumor tissues of three exten sively studied HCC-associated aberrantly methylated DNA markers, mAPC [75], mGSTP1 [121] and mRASSF1A (manuscript submitted). After performing a comprehensive analysis of the DNA methylation profile of these three genes, using bisulfite-specific (BS)-PCR sequencing and methylation-specific PCR assays, comparing tissue DNA from normal-liver, hepatitis, cirrhosis, HCC and a dozen non liver, normal tissue samples, we demonstrated that little specificity for HCC was found when mGSTP1 was analyzed at the 3′ region of the −48-nt position and when mAPC was analyzed on the antisense strand of the DNA Conversely, a high specificity was obtained when mGSTP1 at the 5′ region of the 48 nt position and the sense strand of DNA of the mAPC gene were examined [75,121]. Furthermore, among three previously defined promoter regions of RASSF1A, P1, P2 and E1, we found that methylation of the P1 region was more specific to HCC as compared with P2 and E1 (data not shown). Collectively, previous studies suggest that the choice of the pro moter region to examine DNA methylation is a critical step that should be carefully considered in the development of DNA methylation markers for cancer screening [129].
Detection of HCC DNA markers in peripheral body fluids
As previously discussed, DNA markers can only be useful for cancer screening or early detection if the marker can be detected in the periphery. TABLE 1 lists major genetic and epige netic DNA modifications that have been identified to have an association with HCC as compared with hepatitis and cirrhosis. There have been many attempts to evaluate the performance of each marker separately, or in combination with other markers, in the periphery. We have summarized these data in TABLE 2, which lists the markers that have been reported to be found in the body fluid and the performance of each marker for HCC screening. Please note, the ranges of the sensitivities and specificities listed are the collection of the results from references. Note, these values of sensitivity and specificity reported by each study were dependent on the cutoff value of the marker used in the study, and when the sensitivity was reported to be low, it was often associated with a high specificity. Moreover, the difference of the study populations and sizes of the studies could also impact the marker performance which is one of many reasons why 99% of biomarkers failed to be translated into clinic.
Table 2.
Hepatocellular carcinoma DNA markers detected in circulation.
| Pathway | Marker | Sensitivity (%) |
Specificity (%) |
Periphery | Cases/controls† | Ref. |
|---|---|---|---|---|---|---|
| Wnt signaling | mAPC | 24–68 | 88–97 | Plasma | 26/16, 72/41, 108/60, 28/0 | [38,80–82] |
| 100 | 100 | Serum | 23/8 | [79] | ||
| mSFRP1 | 28.7–55.6 | 87.8 | Plasma | 72/41, 108/60 | [80,81] | |
|
| ||||||
| Cell cycle/ apoptosis |
TP53 249T
mutation |
15–47 | 46–86 | Plasma | 39/0, 186/98, 14/5, 20/10, 53/53, 89/131, 84/56, 25/20, 55/52, 198/325, 176/133, 41/74 |
[39,60,61,103,132,133,187–194] |
| 4–18 | 83.3 | Serum | 76/110, 158/0, 108/0 | [106,195,196] | ||
| 53 | 75 | Urine | 17/15 | [42] | ||
| mCDKN2A | 19–59 | 68–100 | Plasma | 26/16, 39/0, 28/0 | [38,39,82] | |
| 44–92 | 68–96 | Serum | 50/50, 25/17,66/43 | [197–199] | ||
|
| ||||||
| mTOR signaling | mRASSF1A | 27–94 | 64–100 | Plasma | 26/16, 72/41, 40/10 | [38,80,200] |
| 70–100 | 52–100 | Serum | 35/10, 50/50 | [179,197] | ||
|
| ||||||
| Detoxification | mGSTP1 | 19–55 | 90–93 | Plasma | 26/16, 72/41 | [38,80] |
| 50 | 62.5 | Serum | 32/8 | [136] | ||
|
| ||||||
| Protease inhibitor |
mTFPI2 | 47 | 80 | Serum | 28/0, 43/26 | [82,125] |
|
| ||||||
| Cell adhesion | mCDH1 | 17–27 | 75–97 | Plasma | 43/26 | [125] |
Number of cases and controls used in each study are listed corresponding to the order of the references listed in this table.
mAPC: Methylated APC; mGSTP1: Methylated glutathione S-transferase P1; mRASSF1A: Methylated RASSF1A; mSFRP1: Secreted Frizzled-related protein 1.
Among the genetic mutations that have been associated with HCC, mutation of TP53 is the only mutation reported in the circulation of HCC subjects. However, TP53 mutations were detected in the circulation of patients with HCC with an excel lent concordance [103,106,131–133]. We have further shown that the TP53 249T mutation can be successfully detected in the urine from approximately 50% of HCC patients [42].
Contrary to genetic mutations, almost all frequently methyl ated genes in HCC have been reported to be detectable in the circulation of patients with liver cancer with a good concor dance between tissue and serum/plasma specimens, underscor ing the potential of these methylated DNA markers for HCC screening [82,105,134–136].
As mentioned above, multiple markers are needed to obtain a sufficient sensitivity for screening. Several recent studies have investigated the performance of various combinations of methylated markers m cirrulation for HCC screening [80,81,119,137] with promising results. For instance, Huang et al. performed quantitative analysis of a circulating methylated DNA marker panel (APC, RASSF1A, GSTP1 and SFRP1) in a study of 150 patients with and without HCC, which demonstrated a significantly better performance of the panel (92.7% sensitivity, 81.9% specificity with area under the curve of 0.933), as opposed to individual markers [80]. Recent advancements of NGS have allowed the comprehensive comparison of DNA alterations between tumor and non tumor in the circulation [138-141]. Thus, the number of potential markers used to distinguish HCC from non-HCC has greatly pro gressed to hundreds or even thousands. However, to be the most effective, the development of an algorithm to be used with these markers will be necessary in order to process the large amount of data that would be produced. Overall, this approach provides an extremely powerful tool in detecting tumor derived mutations and copy number aberrations for not only cancer detection and monitoring, but also for providing cancer genetics for better disease management.
HBV-based viral DNA markers
Chronic HBV infection is a major risk factor for developing HCC, and it is associated with over 50% of HCC cases world-wide and up to 70–80% of HCC cases in HBV-endemic areas. The contribution of HBV infection to the pathogenesis of HCC is believed to be multifactorial, further compounding this risk factor. During carcinogenesis of HBV-related HCC, it is believed that modifications of the HBV genome, both genetic and epigenetic, ocrur not only in the host genome, but also in the viral genome. Although detection of HBV-DNA markers will only be useful for patients with HBV-related infections, the inclusion of HCC associated HBV-DNA markers provides potential screening markers that are liver specific. The most commonly studied HCC associated HBV mutation is the basal core promoter double mutation A1762T/G1764A [142–149]. This mutation causes an increase in the host immune response and diminishes HBeAg production by suppressing transcription of precore mRNA [143]. It has also been suggested to be an early marker event during carcinogenesis, as detection can occur up to 10 years before HCC diagnosis [150,151]. Furthermore, it is a particularly useful marker in a specific subset of male patients who are HBsAg+ [152]. This double mutation marker has been detected in both the circulation and the trans renal DNA of patients with HBV-HCC [153], lending itself to potentially be a good marker for screening or risk stratification of HBV-HCC patients. This mutation has also been implicated in HCC-risk prediction in HBV genotype B or C patients [143,154,155].
The preS region of the HBV genome plays a role in the viral interaction with the host immune response due to the B-cell and T-cell epitopes in this region [142,156]. PreS deletions will decrease the expression of surface proteins and may contribute to immune escape. Studies have linked progressive liver disease with a higher frequency of preS deletions [157–159]. Particularly, the 5′ terminal (nt. 3206–60) of the preS region was found to be the most frequently deleted portion in HCC, compared to non-HCC patients (43 vs 28%) [142]. Also, preSl and preS2 promoter mutations have been reported to be significandy higher in HCC patients (19.7 vs 3% and 15.3 vs 8.9%, respectively) [142]. Additionally, a precore mutation, G1899A, has been found to be significantly associated with HCC in Thailand [160,161].
The performance of these mutations in predicting HCC varies in such a way that no single genetic mutation is able to sufficiently identify all HBV-related HCC. In recent studies, when at least six of eight particular HBV mutations (G1613A, C1653T, T1753V, A1762T, G1764A, A1846T, G1896A and G1899A), are used in combination, the panel has a predictive power of 94.3% with a specificity of 97.3%, although the sen sitivity was only 44% (n = 75) [162,163]. While these studies looked at only HBV infected patients with genotype C2, the area under the rurve of mutation number versus AFP was com parable (0.825 vs 0.869).
There have been increasing studies in determining both mutation frequency and mutation correlation with HCC in HBV patients with genotype D, as prior studies are often only applicable for those HBV patients with genotypes B or C [164,165]. These emerging studies may shed light on new mutations associated with HCC that are specific for HBV genotype D patients. Mutations in HBV genotype D patients who have been found to be associated with HCC are G1727 (5 vs 35%), C1741 (7.5 vs 30%), C1761 (2.5 vs 15%) and T1773 (52.5 vs 95%) (non HCC, n = 107 vs HCC, n = 45) [164].
Lastly, there have been emerging studies looking at epigenetic modifications in the HBV genome. The HBV genome has 2–3 conventional CpG islands depending on the geno type [166,167]. Interestingly, these CpG islands are found at strate gic locations in the regulatory elements of the HBV genome [168]. To better understand the methylation status of HBV DNA in hepatitis, cirrhosis and HCC patients, a comprehensive methyla tion profile of three CpG islands in the HBV genome was assessed via BS sequencing. From which, the methylation pattern of each of the three CpG islands was compared by using BS specific PCR and methylation-specific PCR assays. In a sample size of 94 patients (hepatitis, n = 11, cirrhosis, n = 9 and HCC, n = 74), we observed that the methylation of CpG Island 1 and 3 of HBV DNA from liver tissue was significantly higher in HCC, as compared with hepatitis and cirrhosis (manuscript in preparation). Furthermore, the extent of the HBV genome meth ylation was significantly co related to the disease progression of the infected liver (p < 0.05 by student's t-test). While these results are promising, co-relation of HBV genome methylation and HCC carcinogenesis in the periphery warrants further research to explore its potential as a cancer biomarker.
Challenges for the development of biomarkers for clinical applications
The development of a biomarker is a process that begins with biomarker discovery and is concluded with the evaluation of the impact of the biomarker on a clinical outcome. Unfortunately, only 1%, possibly less, of published cancer biomarkers actually enter clinical practice; most published biomarkers have failed. To facilitate biomarker development, supportive plat forms have been established by NIH that offers to improve the development process, including the creation of the Early Detection Research Network. As a result, a five phase guideline for cancer screening biomarker development has been gener ated [169], and standards have been suggested for study designs of biomarker evaluation for either classification or prediction of the outcome of a specific cancer [170]. Instructive anecdotes relating specifically to cancer biomarkers have been collected, and the commentary has been published [171–173]. With the application of NGS in the cancer genome, the number of DNA modifications for HCC increase exponentially. Therefore, more attention is necessary so as to avoid developing bio markers for modifications that lead to failed biomarker development for either molecular diagnostics or as targets for liver cancer therapy.
Expert commentary
All cancers, including HCC, result from accumulation of genomic and epigenomic modifications. Identification of DNA modifications (markers) underlying the development of HCC should permit an unambiguous diagnosis and provide genetic characterization of the tumor to tailor a more precise treatment plan. Thus, DNA-based markers can serve not only as a first-line test for screening and diagnosis of high-risk individuals, but can also play important roles in the clinical management of HCC, such as prognosis prediction and selection of targeted therapies [174]. Given the high heterogeneity of HCC, different combinations of cancer pathways could be altered in a manner dependent on patient etiologies, and therefore a multi-gene panel approach will be needed to obtain a high sensitivity in this DNA marker based screening/surveillance test. Genetic and epigenetic alterations are two unique facets of DNA damage that go hand in hand toward contributing to tumor formation. Thus, an ideal test should utilize a technology capable of detecting both genetic and epigenetic DNA modifications. While genome wide sequencing and methylation profiling are best suitable for discovery of the alterations of the drivers responsible for tumor initiation, a targeted sequencing approach is more applicable for screening and diagnostics due to a greater cost–effectiveness and easier data manage ment. An example of this targeted NGS approach has been applied in a breast cancer study using plasma DNA [175]. In order to optimize this approach for molecular diagnostics, it will be critical to develop well-defined algorithms that can analyze DNA marker profiles with etiologies and other risk factors, including age, gender and liver function, which will be needed in order to translate the wealth of knowledge for the DNA markers obtained from the advent of NGS technology.
Five-year view
In the near future, we anticipate that a large amount of NGS data, of both genetic and epigenetic DNA modifications , comparing HCC to non-HCC in tissue of distinct etiologies, as well as in the periphery (plasma and urine), will become available from an adequately powered sample size to provide the genetic/epigenetic landscape of HCC, while comparing various etiological factors. This will allow the identification of driver mutations across the spectrum of liver emerging from these very diverse etiological factors. The abundant DNA biomarker information obtained from tissue studies will allow molecular characterization of HCC, once the tumor is confirmed via biopsy or removed through surgery, to tailor the treatment plan. Given the recent breakthroughs in HCV treatment, we envision the incidence of HCC will be reduced and the reduction of this risk factor might also result in a decrease in the overall incidence of HCV related DNA modifications in HCC. NGS data from DNA bio markers detected in the periphery will allow for strategic designs of a marker panel for screening and diagnosis. Such a liquid biopsy will provide the molecular characterization of a tumor, in order to enable improved disease treatment plans and to monitor for recurrence. We envision a panel of genetic and epigenetic DNA markers with other clinical vari ables such as patient risk factors (age, gender, hepatitis B/C, alcohol, NASH, etc.), clinical laboratory information (AFP, liver function) and radiographic studies (ultrasound), to be integrated into an algorithm for both screening and early detection. The continuous advances in sequencing technology will lower costs, making it possible to use DNA sequencing as a routine screening or surveillance tool. Simultaneously, efforts will be made for the standardization and quality con trol of clinical NGS. We also anticipate the validation of sur veillance values of these HCC DNA biomarkers with prospective cohort studies for their translation into actual clinical use.
Key issues.
The current methods of surveillance are inadequate and there is an urgent need for better methods.
A non-invasive sensitive approach is critical for surveillance, given the chronic nature of the disease and well-defined high-risk populations.
Due to hepatocellular carcinoma (HCC) heterogeneity, a panel of multiple DNA biomarkers is required for a highly efficacious screening test.
DNA biomarkers are promising screening tools for AFP-negative HCC.
A comprehensive algorithm integrating data from targeted NGS, various risk factors and laboratory tests could be a very powerful tool, not only for HCC screening and molecular diagnosis, but also for personalized management of patients.
Acknowledgments
The work was supported by National Institutes of Health R43 CA165312 (WS and YHS), R44 CA165312 (SJ, WS and YHS). S Jain is an employee of JBS Science, Inc. W Song is President and share-holder of JBS-Science, Inc.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing assistance was provided by A Clemens, JBS Science, Inc.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Ferlay J, Soerjomataram I, Ervik MDR, et al. GLOBOCAN 2012 v1.0, Cancer incidence and mortality worldwide: IARC Cancerbase No. 11. Internet International Agency for Research on Cancer; Lyon, France: 2013. Available from: http://globocan.iarc.fr [Last accessed on 23 April 2014] [Google Scholar]
- 2.Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–2. doi: 10.1002/hep.24199. • Provides a detailed report on the recommendations of the American Association for Study of Liver Diseases for the management of hepatocellular carcinoma (HCC) including screening and surveillance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer facts and figures. American Cancer Society; 2012. [Google Scholar]
- 4.Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2010. National Cancer Institute; Bethesda MD: 2013. Available from: http://seer.cancer.gov/csr/1975_2010/ based on November 2012 SEER data submission, posted to the SEER web site. [Google Scholar]
- 5.European Association for the Study of the Liver, European Organization for Research, Treatment of Cancer EASL–EORTC Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908–43. doi: 10.1016/j.jhep.2011.12.001. • Provides a detailed report on the recommendations of the European Association for Study of Liver Diseases for the management of HCC including screening and surveillance. [DOI] [PubMed] [Google Scholar]
- 6.Trevisani F, Cantarini MC, Wands JR, Bernardi M. Recent advances in the natural history of hepatocellular carcinoma. Carcinogenesis. 2008;29(7):1299–305. doi: 10.1093/carcin/bgn113. [DOI] [PubMed] [Google Scholar]
- 7.Trevisani F, D’Intino PE, Morselli-Labate AM, et al. Serum α-fetoprotein for diagnosis of hepatocellular carcinoma in patients with chronic liver disease: influence of HBsAg and anti-HCV status. J Hepatol. 2001;34(4):570–5. doi: 10.1016/s0168-8278(00)00053-2. [DOI] [PubMed] [Google Scholar]
- 8.Moriya S, Morimoto M, Numata K, et al. Fucosylated fraction of alpha-fetoprotein as a serological marker of early hepatocellular carcinoma. Anticancer Res. 2013;33(3):997–1001. [PubMed] [Google Scholar]
- 9.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 11.Ozen C, Yildiz G, Dagcan AT, et al. Genetics and epigenetics of liver cancer. New Biotechnol. 2013;30(4):381–4. doi: 10.1016/j.nbt.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Nault JC, Zucman-Rossi J. Genetics of hepatocellular carcinoma: the next generation. J Hepatol. 2014;60(1):224–6. doi: 10.1016/j.jhep.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa H, Shibata T. Comprehensive genome sequencing of the liver cancer genome. Cancer Lett. 2013;340(2):234–40. doi: 10.1016/j.canlet.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 14.Kan Z, Zheng H, Liu X, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23(9):1422–33. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cleary SP, Jeck WR, Zhao X, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58(5):1693–702. doi: 10.1002/hep.26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sung WK, Zheng H, Li S, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44(7):765–9. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 17.Shen J, Wang S, Zhang YJ, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55(6):1799–808. doi: 10.1002/hep.25569. •• Profiles aberrant DNA methylation of HCC in a genome-wide methylation array of a large sample size (n = 62) with validation of identified hypermethylation markers in plasma DNA by pyrosequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–8. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760–4. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 20.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–55. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 21.Bolondi L. Screening for hepatocellular carcinoma in cirrhosis. J Hepatol. 2003;39(6):1076–84. doi: 10.1016/s0168-8278(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 22.Bolondi L, Gaiani S, Celli N, et al. Characterization of small nodules in cirrhosis by assessment of vascularity: the problem of hypovascular hepatocellular carcinoma. Hepatology. 2005;42(1):27–34. doi: 10.1002/hep.20728. [DOI] [PubMed] [Google Scholar]
- 23.Kim CK, Lim JH, Lee WJ. Detection of hepatocellular carcinomas and dysplastic nodules in cirrhotic liver: accuracy of ultrasonography in transplant patients. J Ultrasound Med. 2001;20(2):99–104. doi: 10.7863/jum.2001.20.2.99. [DOI] [PubMed] [Google Scholar]
- 24.Sterling RK, Jeffers L, Gordon F, et al. Clinical utility of AFP-L3% measurement in North American patients with HCV-related cirrhosis. Am J Gastroenterol. 2007;102(10):2196–205. doi: 10.1111/j.1572-0241.2007.01405.x. [DOI] [PubMed] [Google Scholar]
- 25.Koike Y, Shiratori Y, Sato S, et al. Des-γ-carboxy prothrombin as a useful predisposing factor for the development of portal venous invasion in patients with hepatocellular carcinoma. Cancer. 2001;91(3):561–9. doi: 10.1002/1097-0142(20010201)91:3<561::aid-cncr1035>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 26.Llovet JM, Bruix J. Novel advancements in the management of hepatocellular carcinoma in 2008. J Hepatol. 2008;48(Suppl 1):S20–37. doi: 10.1016/j.jhep.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 27.Nomoto S, Kinoshita T, Kato K. Hypermethylation of multiple genes as clonal markers in multicentric hepatocellular carcinoma. Br J Cancer. 2007;97(9):1260–5. doi: 10.1038/sj.bjc.6604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 29.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29(36):4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 30.Marquardt JU, Thorgeirsson SS. SnapShot: hepatocellular carcinoma. Cancer Cell. 2014;25(4):550–0.e551. doi: 10.1016/j.ccr.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Su Z, Ning B, Fang H, et al. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev Mol Diagn. 2011;11(3):333–43. doi: 10.1586/erm.11.3. [DOI] [PubMed] [Google Scholar]
- 32.Klee EW, Hoppman-Chaney NL, Ferber MJ. Expanding DNA diagnostic panel testing: is more better? Expert Rev Mol Diagn. 2011;11(7):703–9. doi: 10.1586/erm.11.58. [DOI] [PubMed] [Google Scholar]
- 33.Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4 doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One. 2010;5(3):e9749. doi: 10.1371/journal.pone.0009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ammerpohl O, Pratschke J, Schafmayer C, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer. 2011;130(6):1319–28. doi: 10.1002/ijc.26136. [DOI] [PubMed] [Google Scholar]
- 36.Revill K, Wang T, Lachenmayer A, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145(6):1424–1435.e1425. doi: 10.1053/j.gastro.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shitani M, Sasaki S, Akutsu N, et al. Genome-wide analysis of DNA methylation identifies novel cancer-related genes in hepatocellular carcinoma. Tumor Biol. 2012;33(5):1307–17. doi: 10.1007/s13277-012-0378-3. [DOI] [PubMed] [Google Scholar]
- 38.Chang H, Yi B, Li L, et al. Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Exp Mol Pathol. 2008;85(2):96–100. doi: 10.1016/j.yexmp.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Zhang YJ, Rossner P, Jr, Chen Y, et al. Aflatoxin B 1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int J Cancer. 2006;119(5):985–91. doi: 10.1002/ijc.21699. [DOI] [PubMed] [Google Scholar]
- 40.Mora J, Urgell E, Farre A, et al. Agreement between K-ras sequence variations detected in plasma and tissue DNA in pancreatic and colorectal cancer. Clin Chem. 2006;52(7):1448–9. doi: 10.1373/clinchem.2006.067140. [DOI] [PubMed] [Google Scholar]
- 41.Su YH, Wang M, Block TM, et al. Transrenal DNA as a diagnostic tool: important technical notes. Ann N Y Acad Sci. 2004;1022(1):81–9. doi: 10.1196/annals.1318.014. [DOI] [PubMed] [Google Scholar]
- 42.Lin SY, Dhillon V, Jain S, et al. A locked nucleic acid clamp-mediated PCR assay for detection of a p53 codon 249 hotspot mutation in urine. J Mol Diagn. 2011;13(5):474–84. doi: 10.1016/j.jmoldx.2011.05.005. •• First report of detection of a HCC-associated mutation, p53 codon 249 T→C, in the urine of HCC patients, suggesting the potential of a urine-based DNA test for HCC screening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aaltonen LA, Salovaara R, Kristo P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338(21):1481–7. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 44.Chan AKC, Chiu RWK, Lo YMD. Cell-free nucleic acids in plasma, serum and urine: a new tool in molecular diagnosis. Ann Clin Biochem. 2003;40:122–30. doi: 10.1258/000456303763046030. [DOI] [PubMed] [Google Scholar]
- 45.Giacona MB, Ruben GC, Iczkowski KA, et al. Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas. 1998;17:89–97. doi: 10.1097/00006676-199807000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Lui YYN, Woo KS, Wang AYM, et al. Origin of plasma cell-free DNA after solid organ transplantation. Clin Chem. 2003;49(3):495. doi: 10.1373/49.3.495. [DOI] [PubMed] [Google Scholar]
- 47.Larson CJ, Moreno JG, Pienta KJ, et al. Apoptosis of circulating tumor cells in prostate cancer patients. Cytometry A. 2004;62A(1):46–53. doi: 10.1002/cyto.a.20073. [DOI] [PubMed] [Google Scholar]
- 48.Chan KCA, Leung SF, Yeung SW, et al. Quantitative analysis of the transrenal excretion of circulating EBV DNA in nasopharyngeal carcinoma patients. Clin Cancer Res. 2008;14(15):4809–13. doi: 10.1158/1078-0432.CCR-08-1112. [DOI] [PubMed] [Google Scholar]
- 49.Fleischhacker M, Schmidt B. Cell-free DNA resuscitated for tumor testing. Nat Med. 2008;14(9):914–15. doi: 10.1038/nm0908-914. [DOI] [PubMed] [Google Scholar]
- 50.Narayan A, Carriero NJ, Gettinger SN, et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012;72(14):3492–8. doi: 10.1158/0008-5472.CAN-11-4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papadopoulou E, Davilas E, Sotiriou V, et al. Cell-free DNA and RNA in plasma as a new molecular marker for prostate cancer. Oncol Res. 2004;14:439–45. doi: 10.3727/0965040041791473. [DOI] [PubMed] [Google Scholar]
- 52.Pathak AK, Bhutani M, Kumar S, et al. Circulating cell-free DNA in plasma/serum of lung cancer patients as a potential screening and prognostic tool. Clin Chem. 2006;52(10):1833–42. doi: 10.1373/clinchem.2005.062893. [DOI] [PubMed] [Google Scholar]
- 53.Iacopetta B. TP53 mutations in colorectal cancer. Hum Mutat. 2003;21:271–6. doi: 10.1002/humu.10175. [DOI] [PubMed] [Google Scholar]
- 54.Su P, Zhang L, Wan W, et al. Detection of p53 gene mutation in the plasma of gastric cancer patients. Beijing Da Xue Xue Bao. 2005;37(5):523–6. [PubMed] [Google Scholar]
- 55.Cohen Y, Singer G, Lavie O, et al. The RASSF1A tumor suppressor gene is commonly inactivated in adenocarcinoma of the uterine cervix. Clin Cancer Res. 2003;9(8):2981–4. [PubMed] [Google Scholar]
- 56.Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers. 2007;23(1):73–87. doi: 10.1155/2007/291538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan PS, Shi H, Rahmatpanah F, et al. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res. 2003;63(19):6178–86. [PubMed] [Google Scholar]
- 58.Brucher BLDM, Gedder H, Langner C, et al. Hypermethylation of hMLH1, HPP1, p14 ARF, p16 INK4A, and APC in primary adenocarcinomas of the small bowel. Int J Cancer. 2008:1298–302. doi: 10.1002/ijc.21990. [DOI] [PubMed] [Google Scholar]
- 59.Hsu IC, Metcaf RA, Sun T, et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–8. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 60.Kirk GD, Lesi OA, Mendy M, et al. 249ser TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene. 2005;24(38):5858–67. doi: 10.1038/sj.onc.1208732. [DOI] [PubMed] [Google Scholar]
- 61.Kuang SY, Lekawanvijit S, Maneekarn N, et al. Hepatitis B 1762T/1764A mutations, hepatitis C infection, and codon 249 p53 mutations in hepatocellular carcinomas from Thailand. Cancer Epidemiol Biomarkers Prev. 2005;14(2):380–4. doi: 10.1158/1055-9965.EPI-04-0380. [DOI] [PubMed] [Google Scholar]
- 62.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochem Biophys Acta. 2002;1602(1):73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 63.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2(12):910–17. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 64.Sherr CJ. The pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60(14):3689–95. [PubMed] [Google Scholar]
- 65.Ulivi P, Capelli L, Valgiusti M, et al. Predictive role of multiple gene alterations in response to cetuximab in metastatic colorectal cancer: a single center study. J Transl Med. 2012;10(1):87. doi: 10.1186/1479-5876-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishida N, Nishimura T, Nagasaka T, et al. Extensive methylation is associated with Î2-catenin mutations in hepatocellular carcinoma: evidence for two distinct pathways of human hepatocarcinogenesis. Cancer Res. 2007;67(10):4586–94. doi: 10.1158/0008-5472.CAN-06-3464. [DOI] [PubMed] [Google Scholar]
- 67.Edamoto Y, Hara A, Biernat W, et al. Alternations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int J Cancer. 2003;106:334–41. doi: 10.1002/ijc.11254. [DOI] [PubMed] [Google Scholar]
- 68.Gerszten RE, Wang TJ. The search for new cardiovascular biomarkers. Nature. 2008:949–52. doi: 10.1038/nature06802. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Z, Yu Y, Xu F, et al. Combining multiple serum tumor markers improves detection of stage I epithelial ovarian cancer. Gynecol Oncol. 2007;107(3):526–31. doi: 10.1016/j.ygyno.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pepe MS, Thompson ML. Combining diagnostic test results to increase accuracy. Biostatistics. 2000;1:123–40. doi: 10.1093/biostatistics/1.2.123. [DOI] [PubMed] [Google Scholar]
- 71.Zain SM, Mohamed R, Cooper DN, et al. Genome-wide analysis of copy number variation identifies candidate gene loci associated with the progression of non-alcoholic fatty liver disease. PLoS One. 2014;9(4):e95604. doi: 10.1371/journal.pone.0095604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hernaez R. Genetic factors associated with the presence and progression of nonalcoholic fatty liver disease: a narrative review. Gastroenterol Hepatol. 2012;35(1):32–41. doi: 10.1016/j.gastrohep.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 73.Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jain S, Chang TT, Hamilton JP, et al. Methylation of the CpG Sites only on the sense strand of the APC gene is specific for hepatocellular Carcinoma. PLoS One. 2011;6(11):e26799. doi: 10.1371/journal.pone.0026799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163(3):1101–7. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Csepregi A, Rocken C, Hoffmann J, et al. APC promoter methylation and protein expression in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134(5):579–89. doi: 10.1007/s00432-007-0321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee S, Lee HJ, Kim JH, et al. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163(4):1371–8. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishida N, Arizumi T, Takita M, et al. Quantification of tumor DNA in serum and vascular invasion of human hepatocellular carcinoma. Oncology. 2013;84:82–7. doi: 10.1159/000345895. [DOI] [PubMed] [Google Scholar]
- 80.Huang ZH, Hu Y, Hua D, et al. Quantitative analysis of multiple methylated genes in plasma for the diagnosis and prognosis of hepatocellular carcinoma. Exp Mol Pathol. 2011;91(3):702–7. doi: 10.1016/j.yexmp.2011.08.004. • Demonstrates the quantitative analysis of a panel of methylated genes in plasma as a valuable diagnostic and prognostic tool for HCC. [DOI] [PubMed] [Google Scholar]
- 81.Liu JB, Zhang YX, Zhou SH, et al. CpG island methylator phenotype in plasma is associated with hepatocellular carcinoma prognosis. World J Gastroenterol. 2011;17(42):4718–24. doi: 10.3748/wjg.v17.i42.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iyer P, Zekri AR, Hung CW, et al. Concordance of DNA methylation pattern in plasma and tumor DNA of Egyptian hepatocellular carcinoma patients. Exp Mol Pathol. 2009;88(1):107–11. doi: 10.1016/j.yexmp.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu Y, Li J, Sun CY, et al. Epigenetic inactivation of the canonical Wnt antagonist secreted frizzled-related protein 1 in hepatocellular carcinoma cells. Neoplasma. 2012;59(3):326–32. doi: 10.4149/neo_2012_042. [DOI] [PubMed] [Google Scholar]
- 84.Huang J, Zhang YL, Teng XM, et al. Down-regulation of SFRP1 as a putative tumor suppressor gene can contribute to human hepatocellular carcinoma. BMC Cancer. 2007;7(1):126. doi: 10.1186/1471-2407-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shih YL, Shyu RY, Hsieh CB, et al. Promoter methylation of the secreted frizzled-related protein 1 gene SFRP1 is frequent in hepatocellular carcinoma. Cancer. 2006;107(3):579–90. doi: 10.1002/cncr.22023. [DOI] [PubMed] [Google Scholar]
- 86.Takagi H, Sasaki S, Suzuki H, et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J Gastroenterol. 2008;43(5):378–89. doi: 10.1007/s00535-008-2170-0. [DOI] [PubMed] [Google Scholar]
- 87.Boyault S, Rickman DS, de Reynies A, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–5. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 88.Cieply B, Zeng G, Proverbs-Singh T, et al. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology. 2009;49(3):821–31. doi: 10.1002/hep.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de La Coste A, Romagnolo B, Billuart P. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci USA. 1998;95(15):8847–51. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang H, Fujii H, Sankila A. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155(6):1795–801. doi: 10.1016/s0002-9440(10)65496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Legoix P, Bluteau O, Bayer J. Beta-catenin mutations in hepatocellular carcinoma correlate with a low rate of loss of heterozygosity. Oncogene. 1999;18(27):4044–6. doi: 10.1038/sj.onc.1202800. [DOI] [PubMed] [Google Scholar]
- 92.Miyoshi Y, Iwao K, Nagasawa Y. Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res. 1998;58(12):2524–7. [PubMed] [Google Scholar]
- 93.Taniguchi K, Roberts LR, Aderca IN. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene. 2002;21(31):4863–71. doi: 10.1038/sj.onc.1205591. [DOI] [PubMed] [Google Scholar]
- 94.Terris B, Pineau P, Bregeaud L. Close correlation between beta-catenin gene alterations and nuclear accumulation of the protein in human hepatocellular carcinomas. Oncogene. 1999;18(47):6583–8. doi: 10.1038/sj.onc.1203051. [DOI] [PubMed] [Google Scholar]
- 95.Gordon M, Baksh S. RASSF1A: not a prototypical Ras effector. Small GTPases. 2011;2(3):148–57. doi: 10.4161/sgtp.2.3.16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Song MS, Song SJ, Ayad NG, et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol. 2004;6(2):129–37. doi: 10.1038/ncb1091. [DOI] [PubMed] [Google Scholar]
- 97.Vos MD, Martinez A, Elam C, et al. A Role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer Res. 2004;64(12):4244–50. doi: 10.1158/0008-5472.CAN-04-0339. [DOI] [PubMed] [Google Scholar]
- 98.Liu L, Tommasi S, Lee DH, et al. Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene. 2003;22(50):8125–36. doi: 10.1038/sj.onc.1206984. [DOI] [PubMed] [Google Scholar]
- 99.Shivakumar L, Minna J, Sakamaki T, et al. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. MolCell Biol. 2002;22(12):4309–18. doi: 10.1128/MCB.22.12.4309-4318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci. 2007;120(18):3163–72. doi: 10.1242/jcs.010389. [DOI] [PubMed] [Google Scholar]
- 101.Matsuda Y, Ichida T, Matsusawa J, et al. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology. 1999;116:394–400. doi: 10.1016/s0016-5085(99)70137-x. [DOI] [PubMed] [Google Scholar]
- 102.Kaneto H, Sasaki S, Yamamoto H, et al. Detection of hypermethylation of the p16 (INK4A) gene promoter in chronic hepatitis and cirrhosis associated with hepatitis B or C virus. Gut. 2001;48:372–7. doi: 10.1136/gut.48.3.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jackson PE. Specific p53 mutations detected in plasma and tumors of hepatocellular carcinoma patients by electrospray ionization mass spectrometry. Cancer Res. 2001;61:33–5. [PubMed] [Google Scholar]
- 104.Stern MC, Umbach DM, Yu MC, et al. Hepatitis B, aflatoxin B1, and p53 codon 249 mutation in hepatocellular carcinomas from Guangxi, People’s Republic of China, and a meta-analysis of existing studies. Cancer Epidemiol Biomarkers Prev. 2001;10(6):617–25. [PubMed] [Google Scholar]
- 105.Yeo W, Wong N, Wong WL, et al. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int. 2005;25(2):266–72. doi: 10.1111/j.1478-3231.2005.01084.x. [DOI] [PubMed] [Google Scholar]
- 106.Hosny G, Farahat N, Tayel H, Hainaut P. Ser-249 TP53 and CTNNB1 mutations in circulating free DNA of Egyptian patients with hepatocellular carcinoma versus chronic liver diseases. Cancer Lett. 2008;264(2):201–8. doi: 10.1016/j.canlet.2008.01.031. [DOI] [PubMed] [Google Scholar]
- 107.Igetei R, Otegbayo J, Ndububa D, et al. Detection of p53 codon 249 mutation in Nigerian patients with hepatocellular carcinoma using a novel evaluation of cell-free DNA. Ann Hepatol. 2008;7(4):339–44. [PubMed] [Google Scholar]
- 108.Toh ST, Jin Y, Liu L, et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34(4):787–98. doi: 10.1093/carcin/bgs406. [DOI] [PubMed] [Google Scholar]
- 109.Li W, Zeng X, Lee NP, et al. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics. 2013;102(4):338–44. doi: 10.1016/j.ygeno.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 110.Ding D, Lou X, Hua D, et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing–based approach. PLoS Genet. 2012;8(12):e1003065. doi: 10.1371/journal.pgen.1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen YL, Jeng YM, Chang CN, et al. TERT promoter mutation in resectable hepatocellular carcinomas: a strong association with hepatitis C infection and absence of hepatitis B infection. Int J Surg. 2014;12(7):659–65. doi: 10.1016/j.ijsu.2014.05.066. [DOI] [PubMed] [Google Scholar]
- 112.Pinyol R, Tovar V, Llovet JM. TERT promoter mutations: gatekeeper and driver of hepatocellular carcinoma. J Hepatol. 2014 doi: 10.1016/j.jhep.2014.05.028. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 113.Li M, Zhao H, Zhang X, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43(9):828–9. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Endo M, Yasui K, Zen Y, et al. Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and BRM in hepatocellular carcinoma. Liver Int. 2013;33(1):105–17. doi: 10.1111/liv.12005. [DOI] [PubMed] [Google Scholar]
- 115.Grabinski N, Ewald F, Hofmann B, et al. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer. 2012;11(1):85. doi: 10.1186/1476-4598-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Villanueva A, Chiang DY, Newell P, et al. Pivotal Role of mTOR Signaling in Hepatocellular Carcinoma. Gastroenterology. 2008;135(6):1972–83.e1911. doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang L, Wang WL, Zhang Y, et al. Epigenetic and genetic alterations of PTEN in hepatocellular carcinoma. Hepatol Res. 2007;37(5):389–96. doi: 10.1111/j.1872-034X.2007.00042.x. [DOI] [PubMed] [Google Scholar]
- 118.Yu J, Ni M, Xu J, et al. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer. 2002;2(1):29. doi: 10.1186/1471-2407-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hua D, Hu Y, Wu YY, et al. Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma. Exp Mol Pathol. 2011;91(1):455–60. doi: 10.1016/j.yexmp.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 120.Nishida N, Nagasaka T, Nishimura T, et al. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47(3):908–18. doi: 10.1002/hep.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jain S, Chen S, Chang KC, et al. Impact of the location of CpG methylation within the GSTP1 gene on its specificity as a DNA marker for hepatocellular carcinoma. PLoS One. 2012;7(4):e35789. doi: 10.1371/journal.pone.0035789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harder J, Opitz OG, Brabender J, et al. Quantitative promoter methylation analysis of hepatocellular carcinoma, cirrhotic and normal liver. Int J Cancer. 2008;122(12):2800–4. doi: 10.1002/ijc.23433. [DOI] [PubMed] [Google Scholar]
- 123.Wong CM, Ng IOL. Molecular pathogenesis of hepatocellular carcinoma. Liver Int. 2007;28(2):160–74. doi: 10.1111/j.1478-3231.2007.01637.x. [DOI] [PubMed] [Google Scholar]
- 124.Wong CM, Ng YL, Lee JM, et al. Tissue factor pathway inhibitor-2 as a frequently silenced tumor suppressor gene in hepatocellular carcinoma. Hepatology. 2007;45:1129–38. doi: 10.1002/hep.21578. [DOI] [PubMed] [Google Scholar]
- 125.Sun FK, Fan YC, Zhao J, et al. Detection of TFPI2 methylation in the serum of hepatocellular carcinoma patients. Dig Dis Sci. 2013;58(4):1010–15. doi: 10.1007/s10620-012-2462-3. [DOI] [PubMed] [Google Scholar]
- 126.Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42(5):1208–36. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- 127.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–27. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 128.Lencioni R, Piscaglia F, Bolondi L. Contrast-enhanced ultrasound in the diagnosis of hepatocellular carcinoma. J Hepatol. 2008;48(5):848–57. doi: 10.1016/j.jhep.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 129.Jain S, Wojdacz TK, Su YH. Challenges for the application of DNA methylation biomarkers in molecular diagnostic testing for cancer. Expert Rev Mol Diagn. 2013;13(3):283–94. doi: 10.1586/erm.13.9. [DOI] [PubMed] [Google Scholar]
- 130.van Vlodrop IJH, Niessen HEC, Derks S, et al. Analysis of promoter CpG island hypermethylation in cancer: location, location, location! Clin Cancer Res. 2011;17(13):4225–31. doi: 10.1158/1078-0432.CCR-10-3394. [DOI] [PubMed] [Google Scholar]
- 131.Jackson PE, Kuang SY, Wang JB, et al. Prospective detection of codon 249 mutations in plasma of hepatocellular carcinoma patients. Carcinogenesis. 2003;24(10):1657–63. doi: 10.1093/carcin/bgg101. [DOI] [PubMed] [Google Scholar]
- 132.Kirk GD. Ser-249 p53 mutations in plasma DNA of patients with hepatocellular carcinoma from The Gambia. J Natl Cancer Inst. 2000;92:148–53. doi: 10.1093/jnci/92.2.148. [DOI] [PubMed] [Google Scholar]
- 133.Lleonart ME, Kirk GD, Villar S, et al. Quantitative analysis of plasma TP53 249Ser-mutated DNA by electrospray ionization mass spectrometry. Cancer Epidemiol Biomarkers Prev. 2005;14(12):2956–62. doi: 10.1158/1055-9965.EPI-05-0612. [DOI] [PubMed] [Google Scholar]
- 134.Wong IHN, Dennis Lo YM, Zhang J, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999;59(1):71–3. [PubMed] [Google Scholar]
- 135.Usadel H, Brabender J, Danenberg KD, et al. Quantitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer Res. 2002;62(2):371–5. [PubMed] [Google Scholar]
- 136.Wang J, Qin Y, Li B, et al. Detection of aberrant promoter methylation of GSTP1 in the tumor and serum of Chinese human primary hepatocellular carcinoma patients. Clin Biochem. 2006;39(4):344–8. doi: 10.1016/j.clinbiochem.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 137.Iizuka N, Oka M, Sakaida I, et al. Efficient detection of hepatocellular carcinoma by a hybrid blood test of epigenetic and classical protein markers. Clin Chim Acta. 2011;412(1-2):152–8. doi: 10.1016/j.cca.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 138.Chan KCA, Jiang P, Chan CWM, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci USA. 2013;110(47):18761–8. doi: 10.1073/pnas.1313995110. •• Demonstrates detection of genome-wide hypomethylation in plasma using massively parallel bisulfite sequencing as a promising approach for cancer detection and tumor-associated copy number aberrations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Swanton C. Plasma-derived tumor DNA analysis at whole-genome resolution. Clin Chem. 2013;59(1):6–8. doi: 10.1373/clinchem.2012.197053. [DOI] [PubMed] [Google Scholar]
- 140.Chan KCA, Jiang P, Zheng YW, et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem. 2013;59(1):211–24. doi: 10.1373/clinchem.2012.196014. •• Provides evidence that shotgun sequencing of plasma DNA has the ability to non-invasively detect not only tumor signatures, but can also reflect on tumor hetergeneity in a genome-wide fashion. [DOI] [PubMed] [Google Scholar]
- 141.Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra168. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 142.Liu S, Zhang H, Gu C, et al. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101(15):1066–82. doi: 10.1093/jnci/djp180. • Demonstrates that PreS mutations, C1653T, T1753V and A1762T/ G1764A were each associated with an increased risk of HCC and in combination, were more than 80% specific for the prediction of HCC based on an extensive meta-analysis of over 11,582 hepatitis B virus-infected patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kao JH, Chen PJ, Lai MY, Chen DS. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology. 2003;124(2):327–34. doi: 10.1053/gast.2003.50053. [DOI] [PubMed] [Google Scholar]
- 144.Guo X, Jin Y, Qian G, Tu H. Sequential accumulation of the mutations in core promoter of hepatitis B virus is associated with the development of hepatocellular carcinoma in Qidong, China. J Hepatol. 2008;49(5):718–25. doi: 10.1016/j.jhep.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 145.Wang Z, Tanaka Y, Huang Y, et al. Clinical and virological characteristics of hepatitis B virus subgenotypes Ba, C1, and C2 in China. J Clin Microbiol. 2007;45(5):1491–6. doi: 10.1128/JCM.02157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhu R, Zhang HP, Yu H, et al. Hepatitis B virus mutations associated with in situ expression of hepatitis B core antigen, viral load and prognosis in chronic hepatitis B patients. Pathol Res Pract. 2008;204(10):731–42. doi: 10.1016/j.prp.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 147.Chou YC, Yu MW, Wu CF, et al. Temporal relationship between hepatitis B virus enhancer II/basal core promoter sequence variation and risk of hepatocellular carcinoma. Gut. 2008;57(1):91–7. doi: 10.1136/gut.2006.114066. [DOI] [PubMed] [Google Scholar]
- 148.Tong MJ, Blatt LM, Kao J-H, et al. Precore/basal core promoter mutants and hepatitis B viral DNA levels as predictors for liver deaths and hepatocellular carcinoma. World J Gastroenterol. 2006;2(41):6620. doi: 10.3748/wjg.v12.i41.6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jang J, Lee Y, Kim M, et al. A 13-year longitudinal study of the impact of double mutations in the core promoter region of hepatitis B virus on HBeAg seroconversion and disease progression in patients with genotype C chronic active hepatitis. J Viral Hepat. 2007;14(3):169–75. doi: 10.1111/j.1365-2893.2006.00788.x. [DOI] [PubMed] [Google Scholar]
- 150.Yuan JM, Ambinder A, Fan Y, et al. Prospective evaluation of hepatitis B 1762T/ 1764A mutations on hepatocellular carcinoma development in Shanghai, China. Cancer Epidemiol Biomarkers Prev. 2009;18(2):590–4. doi: 10.1158/1055-9965.EPI-08-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang HI, Yeh SH, Chen PJ, et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J Natl Cancer Inst. 2008;100(16):1134–43. doi: 10.1093/jnci/djn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fang ZL, Sabin CA, Dong BQ, et al. HBV A1762T, G1764A mutations are a valuable biomarker for identifying a subset of male HBsAg carriers at extremely high risk of hepatocellular carcinoma: a prospective study. Am J Gastroenterol. 2008;103(9):2254–62. doi: 10.1111/j.1572-0241.2008.01974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lin SY, Jain S, Song W, et al. Strategic assay developments for detection of HBV 1762T/1764A double mutation in urine of patients with HBV-associated hepatocellular carcinomas. In: Lau WY, editor. Hepatocellular carcinoma - clinical research. Intech; Croatia: 2012. [Google Scholar]
- 154.Yuan J, Zhou B, Tanaka Y, et al. Hepatitis B virus (HBV) genotypes/subgenotypes in China: mutations in core promoter and precore/core and their clinical implications. J Clin Virol. 2007;39(2):87–93. doi: 10.1016/j.jcv.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 155.Lindh M, Hannoun C, Dhillon AP, et al. Core promoter mutations and genotypes in relation to viral replication and liver damage in East Asian hepatitis B virus carriers. J Infect Dis. 1999;179(4):775–82. doi: 10.1086/314688. [DOI] [PubMed] [Google Scholar]
- 156.Kay A, Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007;127(2):164–76. doi: 10.1016/j.virusres.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 157.Sugauchi F, Ohno T, Orito E, et al. Influence of hepatitis B virus genotypes on the development of preS deletions and advanced liver disease. J Med Virol. 2003;70(4):537–44. doi: 10.1002/jmv.10428. [DOI] [PubMed] [Google Scholar]
- 158.Fang ZL, Sabin CA, Dong BQ, et al. Hepatitis B virus pre-S deletion mutations are a risk factor for hepatocellular carcinoma: a matched nested case–control study. J Gen Virol. 2008;89(11):2882–90. doi: 10.1099/vir.0.2008/002824-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen CH, Hung CH, Lee CM, et al. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology. 2007;133(5):1466–74. doi: 10.1053/j.gastro.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 160.Tangkijvanich P, Sa-nguanmoo P, Mahachai V, et al. A case–control study on sequence variations in the enhancer II/core promoter/precore and X genes of hepatitis B virus in patients with hepatocellular carcinoma. Hepatol Int. 2010;4(3):577–84. doi: 10.1007/s12072-010-9197-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cho EY, Choi CS, Cho JH, Kim HC. Association between hepatitis B virus X gene mutations and clinical status in patients with chronic hepatitis B infection. Gut Liver. 2011;5(1):70–6. doi: 10.5009/gnl.2011.5.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jang JW, Chun JY, Park YM, et al. Mutational complex genotype of the hepatitis B virus X/precore regions as a novel predictive marker for hepatocellular carcinoma. Cancer Sci. 2012;103(2):296–304. doi: 10.1111/j.1349-7006.2011.02170.x. [DOI] [PubMed] [Google Scholar]
- 163.Park Y, Jang J, Yoo S, et al. Combinations of eight key mutations in the X/preC region and genomic activity of hepatitis B virus are associated with hepatocellular carcinoma. J Viral Hepat. 2014;21(3):171–7. doi: 10.1111/jvh.12134. [DOI] [PubMed] [Google Scholar]
- 164.Khan A, Al Balwi MA, Tanaka Y, et al. Novel point mutations and mutational complexes in the enhancer II, core promoter and precore regions of hepatitis B virus genotype D1 associated with hepatocellular carcinoma in Saudi Arabia. Int J Cancer. 2013;133(12):2864–71. doi: 10.1002/ijc.28307. [DOI] [PubMed] [Google Scholar]
- 165.Ouneissa R, Bahri O, Alaya-Bouafif NB, et al. Frequency and clinical significance of core promoter and precore region mutations in Tunisian patients infected chronically with hepatitis B. J Med Virol. 2012;84(11):1719–26. doi: 10.1002/jmv.23394. [DOI] [PubMed] [Google Scholar]
- 166.Zhang Y, Li C, Zhang Y, et al. Comparative analysis of CpG Islands among HBV genotypes. PLoS One. 2013;8(2):e56711. doi: 10.1371/journal.pone.0056711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fernandez AF, Rosales C, Lopez-Nieva P, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009;19(3):438–51. doi: 10.1101/gr.083550.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Vivekanandan P, Thomas D, Torbenson M. Hepatitis B viral DNA is methylated in liver tissues. J Viral Hepatitis. 2008;15(2):103–7. doi: 10.1111/j.1365-2893.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- 169.Pepe MS, Etzioni R, Feng Z, et al. Phases of Biomarker Development for Early Detection of Cancer. JNCI Cancer Spectrum. 2001;93(14):1054–61. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- 170.Pepe MS, Feng Z, Janes H, et al. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. J Natl Cancer Inst. 2008;100(20):1432–8. doi: 10.1093/jnci/djn326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ransohoff DF. Developing molecular biomarkers for cancer. Science. 2003;299(5613):1679–80. doi: 10.1126/science.1083158. [DOI] [PubMed] [Google Scholar]
- 172.Diamandis EP. Cancer biomarkers: can we turn recent failures into success? J Natl Cancer Inst. 2010;102(19):1462–7. doi: 10.1093/jnci/djq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kern SE. Why your new cancer biomarker may never work: recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012;72(23):6097–101. doi: 10.1158/0008-5472.CAN-12-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hoshida Y, Moeini A, Alsinet C, et al. Gene signatures in the management of hepatocellular carcinoma. Semin Oncol. 2012;39(4):473–85. doi: 10.1053/j.seminoncol.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 175.Dawson SJ, Tsui DWY, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368(13):1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 176.Um TH, Kim H, Oh BK, et al. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J Hepatol. 2011;54(5):939–47. doi: 10.1016/j.jhep.2010.08.021. [DOI] [PubMed] [Google Scholar]
- 177.Wong CM, Fan ST, Ng IO. Beta-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92(1):136–45. doi: 10.1002/1097-0142(20010701)92:1<136::aid-cncr1301>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 178.Xu B, Di J, Wang Z, et al. Quantitative analysis of RASSF1A promoter methylation in hepatocellular carcinoma and its prognostic implications. Biochem Biophys Res Commun. 2013;438(2):324–8. doi: 10.1016/j.bbrc.2013.07.070. [DOI] [PubMed] [Google Scholar]
- 179.Hu L, Chen G, Yu H, Qiu X. Clinicopathological significance of RASSF1A reduced expression and hypermethylation in hepatocellular carcinoma. Hepatol Int. 2010;4(1):423–32. doi: 10.1007/s12072-010-9164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Di Gioia S, Bianchi P, Destro A, et al. Quantitative evaluation of RASSF1A methylation in the non-lesional, regenerative and neoplastic liver. BMC Cancer. 2006;6(1):89. doi: 10.1186/1471-2407-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nishida N, Nagasaka T, Nishimura T, et al. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908–18. doi: 10.1002/hep.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Honda K, Sbisà E, Tullo A. p53 mutation is a poor prognostic indicator for survival in patients with hepatocellular carcinoma undergoing surgical tumour ablation. Br J Cancer. 1998;77(5):776–82. doi: 10.1038/bjc.1998.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hayashi H, Sugio K, Matsumata T, et al. The clinical significance of p53 gene mutation in hepatocellular carcinomas from Japan. Hepatology. 1995;22(6):1702–7. [PubMed] [Google Scholar]
- 184.Hussain SP, Schwank J, Staib F, et al. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26(15):2166–76. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- 185.Laurent-Puig P, Legoix P, Bluteau O, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763–73. doi: 10.1053/gast.2001.24798. [DOI] [PubMed] [Google Scholar]
- 186.Xie HJ, Bae HJ, Noh JH, et al. Mutational analysis of JAK1 gene in human hepatocellular carcinoma. Neoplasma. 2009;56(2):136–42. doi: 10.4149/neo_2009_02_136. [DOI] [PubMed] [Google Scholar]
- 187.Kuang SY, Jackson PE, Wang JB, et al. Specific mutations of hepatitis B virus in plasma predict liver cancer development. Proc Natl Acad Sci USA. 2004;101(10):3575–80. doi: 10.1073/pnas.0308232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Villar S, Ortiz-Cuaran S, Abedi-Ardekani B, et al. Aflatoxin-induced TP53 R249S mutation in hepatocellular carcinoma in Thailand: association with tumors developing in the absence of liver cirrhosis. PLoS One. 2012;7(6):e37707. doi: 10.1371/journal.pone.0037707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang XH, Sun LH, Lu DD, et al. Codon 249 mutation in exon 7 of p53 gene in plasma DNA: maybe a new early diagnostic marker of hepatocellular carcinoma in Qidong risk area, China. World J Gastroenterol. 2003;9(4):692–5. doi: 10.3748/wjg.v9.i4.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Igetei R, Otegbayo JA, Lesi OA, et al. P53 codon 249 mutation and other risk factors among Nigerians with hepatocellular carcinoma. J Afr Cancer. 2010;2(3):133–9. [Google Scholar]
- 191.Gouas DA, Villar S, Ortiz-Cuaran S, et al. TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis. 2012;33(6):1–6. doi: 10.1093/carcin/bgs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ortiz-Cuaran S, Villar S, Gouas D, et al. Association between HBX status, aflatoxin-induced R249S TP53 mutation and risk of hepatocellular carcinoma in a case-control study from Thailand. Cancer Lett. 2013;331(1):46–51. doi: 10.1016/j.canlet.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 193.Carvalho F, Almeida Pereira T, Gonçalves P, et al. Hepatocellular carcinoma and liver cirrhosis TP53 mutation analysis reflects a moderate dietary exposure to aflatoxins in Espírito Santo State, Brazil. Mol Biol Rep. 2013;40(8):4883–7. doi: 10.1007/s11033-013-2587-2. [DOI] [PubMed] [Google Scholar]
- 194.Özdemir F, Tiftikci A, Sancak S, et al. The prevalence of the mutation in codon 249 of the P53 gene in patients with hepatocellular carcinoma (HCC) in Turkey. J Gastrointest Canc. 2010;41(3):185–9. doi: 10.1007/s12029-010-9140-5. [DOI] [PubMed] [Google Scholar]
- 195.Kimbi GC, Kew MC, Yu MC, et al. 249serp53 mutation in the serum of black southern African patients with hepatocellular carcinoma. J Gastroenterol Hepatol. 2005;20(8):1185–90. doi: 10.1111/j.1440-1746.2005.03951.x. [DOI] [PubMed] [Google Scholar]
- 196.Mah YH, Hsu CS, Liu CH, et al. Serum p53 gene polymorphisms and severity of hepatitis B or C-related chronic liver diseases in Taiwan. Hepatol Int. 2011;5(3):814–21. doi: 10.1007/s12072-010-9248-5. [DOI] [PubMed] [Google Scholar]
- 197.Zhang YJ, Wu HC, Shen J, et al. Predicting hepatocellular carcinoma by detection of aberrant promoter methylation in serum DNA. Clin Cancer Res. 2007;13(8):2378–84. doi: 10.1158/1078-0432.CCR-06-1900. •This is the first prospective study that examined hypermethylation of RASSF1A, p16 and p15 promoters in serum DNA samples, which were collected up to 9 years before clinical diagnosis. [DOI] [PubMed] [Google Scholar]
- 198.Ahmed R, Salama H, Fouad A, et al. Detection of aberrant p16INK4A methylation in sera of patients with HCV-related liver diseases: an Egyptian study. Med Sci Monit. 2010;19(9):CR410–15. [PubMed] [Google Scholar]
- 199.Huang G, Krocker JD, Kirk JL, et al. Evaluation of INK4A promoter methylation using pyrosequencing and circulating cell-free DNA from patients with hepatocellular carcinoma. Clin Chem Lab Med. 2014:1–11. doi: 10.1515/cclm-2013-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mohamed NA, Swify EM, Amin NF, et al. Is serum level of methylated RASSF1A valuable in diagnosing hepatocellular carcinoma in patients with chronic viral hepatitis C? Arab J Gastroenterol. 2012;13(3):111–15. doi: 10.1016/j.ajg.2012.06.009. [DOI] [PubMed] [Google Scholar]