

Abstract
Phosphonates are a large class of organophosphorus compounds with a characteristic carbon-phosphorus bond. The genes responsible for phosphonate utilization in gram-negative bacteria are arranged in an operon of 14 genes. The carbon-phosphorus lyase complex, encoded by the genes phnGHIJKLM, catalyzes the cleavage of the stable carbon-phosphorus bond of organophosphonates to the corresponding hydrocarbon and inorganic phosphate. Recently, complexes of this enzyme containing five subunits (PhnG-H-I-J-K), four subunits (PhnG-H-I-J), and two subunits (PhnG-I) were purified after expression in Escherichia coli. Here we demonstrated using mass spectrometry, ultracentrifugation, and chemical crosslinking experiments that these complexes are formed from a PhnG2I2 core that is further elaborated by the addition of two copies each of PhnH and PhnJ to generate PhnG2H2I2J2. This complex adds an additional subunit of PhnK to form PhnG2H2I2J2K. Chemical crosslinking of the 5-component complex demonstrated that PhnJ physically interacts with both PhnG and PhnI. We were unable to demonstrate the interaction of PhnH or PhnK with any other subunits by chemical crosslinking. Hydrogen-deuterium exchange was utilized to probe for alterations in the dynamic properties of individual subunits within the various complexes. Significant regions of PhnG become less accessible to hydrogen/deuterium exchange from solvent within the PhnG2I2 complex compared with PhnG alone. Specific regions of PhnI exhibited significant differences in the H/D exchange rates in PhnG2I2 and PhnG2H2I2J2K.
Phosphorus is essential for life but the availability of this element can be limited in various ecosystems. Today, the most abundant sources of phosphorus are orthophosphate and various phosphate esters (1). However, organophosphonates may have been the most predominant phosphorus source from the prebiotic earth (2). In contrast to phosphate esters, which contain a hydrolysable C-O-P bond, the C-P bond in organophosphonates is highly resistant to chemical hydrolysis, thermal decomposition and photolysis. The C-P bond in organophosphonate-containing lipids and exopolysaccharides helps to stabilize the cell membrane in some marine organisms (3). Organophosphonate moieties have also been employed within antibiotic, such as fosfomycin, and the herbicides phosphinothricin and phosphonothrixin (4). Every year more than 20,000 tons of phosphonates are discharged in the United States, primarily as herbicides and detergents, (5).
The bacterial C-P lyase complex catalyzes the complex biochemical pathway for the metabolism of phosphonates and these transformations are illustrated in Scheme 1. In bacteria such as Escherichia coli, the catalytic machinery for the C-P lyase complex is governed by an operon of 14 genes (phnCDEFGHIJKLMNOP) that is up-regulated by the PhoR/PhoB two-component signaling system (3). Four of the expressed proteins (PhnC, PhnD, PhnE, and PhnF) are necessary as regulatory or transport proteins (6, 7). Seven of the proteins (PhnG through PhnM) are critical for the enzymatic steps during the conversion of organophosphonates to phosphate (8) and three enzymes (PhnN, PhnO, and PhnP) catalyze auxiliary reactions (9-11).
Scheme 1.

The individual chemical reactions involved in the cleavage of the carbon phosphorus bond in methylphosphonate by the individual components of the C-P lyase complex from E. coli have been reconstituted in vitro (12). PhnI, in the presence of PhnG, PhnH, and PhnL catalyzes the displacement of adenine from ATP by methylphosphonate (MePn) to form ribose-1-methylphosphonate-5-triphosphate. In the second step this product is hydrolyzed by PhnM to generate ribose-1-methylphosphonate-5-phosphate and in the last step PhnJ catalyzes the cleavage of this product to form methane and ribose-1,2-cyclic phosphate-5-phosphate. PhnG, PhnH, and PhnM have been expressed and purified via standard methods. However, PhnI, PhnJ, PhnK, and PhnL have only been expressed and purified as glutathione S-transferase conjugates (12). The instability of these proteins in the absence of chaperones has significantly hampered the structural elucidation of those enzymes responsible for phosphonate metabolism.
Zechel, Hove-Jensen and colleagues have recently demonstrated that multi-subunit fragments of the C-P lyase complex can be expressed from plasmids in E. coli and subsequently purified to apparent homogeneity (13). Partial complexes containing PhnG and PhnI (PhnG-I); PhnG, PhnH, PhnI, and PhnJ (PhnG-H-I-J); and PhnG, PhnH, PhnI, PhnJ, and PhnK (PhnG-H-I-J-K) were expressed and purified in high yield with high solubility and stability without the need for solubility tags (13). However, the catalytic properties, stoichiometry, and three-dimensional structural information are not currently available for any of these fragments from the C-P lyase complex. Here we use high-resolution mass spectrometry, chemical crosslinking, hydrogen/deuterium exchange, analytical ultracentrifugation and N-terminal protein sequencing to structurally characterize the protein-protein interactions within the C-P lyase complex from E. coli.
Materials and Methods
Materials
LB broth was purchased from Tpi Research Products International Corporation. The HisTrap and gel filtration columns were obtained from GE Healthcare. Mini-PROTEAN precast gels were purchased from Bio-Rad. Bis-sulfosuccinimidyl suberate (BS3), immobilized pepsin resin and trypsin were purchased from Thermo Scientific. The C18 reverse phase column was purchased from Waters Corporation. D2O was obtained from Cambridge Isotope Laboratories. All other buffers, purification reagents, and chemicals used in this project were purchased from Sigma/Aldrich, unless otherwise noted.
Cloning and Expression of C-P Lyase Complexes
DNA containing the gene for PhnG through PhnK (his-PhnG-H-I-J-K and PhnG-H-I-J-K-his) from E. coli was amplified from the strain BW5328/pGY3 (CGSC, Yale University). For the his-PhnG-H-I-J-K construct, 5′-AGCTCGTCGAT GCGACATATGCACGCAGATACCGCGACCCGCC-3′ and 5′-TCAAGTGGATCCTCAATTCTGCAAAACCGATGACACCAGCAGCTG-3′ were utilized as the forward and reverse primers, respectively. For the PhnG-H-I-J-K-his construct, 5′-AGCTCGTCGAT GCGACATATGCACGCAGATACCGCGACCCGCC-3′ and 5′-TCAAGTCTCGAGATTCTGCAAAACCGATGACACCAGCAGCTG-3′ were utilized as the forward and reverse primers, respectively. The polymerase chain reaction (PCR) was performed using Platinum Pfx DNA Polymerase (Life Technology) and the following reaction conditions: 5 minutes at 95 °C, followed by 30 cycles of 30 seconds at 95 °C, 30 seconds at 66 °C, and 5 minutes at 72 °C. The his-PhnG-H-I-J-K PCR fragment was subsequently digested by the restriction enzymes Nde1 and BamH1 (New England Biolabs). The digested DNA fragment was ligated to a pET-28b vector for expression with an N-terminal 6x-His-tag for purification. The PhnG-H-I-J-K-his PCR fragment was subsequently digested by the restriction enzymes Nde1 and Xho1 (New England Biolabs). The digested DNA fragment was ligated to a pET-24b vector for expression with a C-terminal 6x-His-tag for purification. The DNA for the expression of his-PhnG, his-PhnH, his-PhnG-I and PhnG-I-his (containing only PhnG and PhnI) and PhnG-H-I-J-his (containing PhnG, PhnH, PhnI and PhnJ) was cloned as previously described (12, 13).
Protein Purification
The plasmids for the expression of the various complexes of C-P lyase were transformed into Rosetta2 (DE3) pLysS cells (Novagen) by electroporation and plated on LB agar. A single colony was used to inoculate 10 mL of LB medium containing 50 μg/mL kanamycin and allowed to grow overnight at 37 °C (250 rpm agitation). The 10 mL culture was subsequently used to inoculate 1 L of LB medium and incubated (250 rpm agitation) at 37 °C for 2-3 hours until the OD600 reached ~0.6. The temperature was then reduced to 18 °C and the culture induced with 0.5 mM isopropyl-β-thiogalactoside (IPTG). The cells were harvested by centrifugation after 16-18 hours of incubation and then stored at -80 °C. The frozen cells were thawed and then resuspended (4:1, v/w) in buffer A (50 mM HEPES, pH 8.5, 150 mM NaCl, and 20 mM imidazole) containing 0.10 mg/mL PMSF and a protease inhibitor cocktail. The cells were disrupted by sonication, the insoluble debris was removed by centrifugation at 4 °C, and the supernatant solution applied to a 5-mL HisTrap (GE Healthcare) column. The column was pre-equilibrated with 10 column volumes of Buffer A and the proteins were eluted with a gradient of Buffer B (500 mM imidazole in Buffer A). The fractions were pooled and applied to a High Load 26/60 Superdex 200 prep grade gel filtration column (GE Healthcare), which was previously equilibrated with Buffer C (Buffer A without imidazole). The fractions were pooled and analyzed by SDS-PAGE. Typical yields for the PhnG-H-I-J-K C-P lyase complexes were ~30 mg/L of culture. Typical yields for the his-PhnG, his-PhnH, his-PhnG-I, PhnG-I-his, and PhnG-H-I-J-his fragments were ~18 and ~25 mg per liter of culture, respectively. The N-terminal His-tag was cleaved by Thrombin CleanCleave Kit (Sigma-Aldrich) following the manufacturer's protocol.
N-terminal Protein Sequencing
The amino acid sequence and stoichiometry of the subunits of the two protein complexes were determined by amino terminal protein sequencing using a Procise 494A protein sequencer (Applied Biosystems). Eight cycles of automated Edman degradation of the PhnG-I-his complex was performed. The subunit stoichiometry was calculated by comparing the relative protein concentration of each subunit after each cycle of Edman degradation.
Analytical Ultracentrifugation
The molecular weight and oligomeric state of three C-P lyase complexes (his-PhnG-I, his-PhnG-H-I-J, and his-PhnG-H-I-J-K) were characterized by sedimentation velocity using an Optima XL-A analytical ultracentrifuge (Beckman Instruments). All experiments and analyses were conducted at the University of Texas Health Science Center, San Antonio, TX. Samples of the C-P lyase complexes contained 10 mM Tris (pH 8.5), 150 mM NaCl, and 1.0 mM DTT. The protein concentrations ranged from 0.25 to 0.75 mg/mL and the samples were centrifuged at 30,000 rpm at 20 °C. The density and relative viscosity of the buffers were calculated as 1.004700 g/mL and 1.01418 mPa·s, respectively (14). Based on the sequence of the C-P lyase complexes, the partial specific volume (vbar) of the protein complexes were calculated as 0.7272 mL g−1. The absorbance of the samples was measured at 231 nm with a 50 second delay.
ESI-MS Measurements
ESI MS measurements were acquired with a ThermoScientific Exactive Plus EMR. In general, protein complexes were buffered with 50 mM ammonium acetate and loaded into an in-house pulled tip containing a platinum wire. All mass spectra were calibrated externally using a 1.0 μg/μL solution of cesium iodide. The protein complexes were buffered exchanged into 50 mM ammonium acetate (pH 6.8) prior to analysis using Bio-Rad Micro Bio-Spin® columns. A potential of 1500-1800 V was applied to the spray tip and m/z was scanned from 900-16000. Important instrument parameters used were: FT resolution was set to 17,500, S-Lens RF = 200, S-Lens = 42 V, Skimmer = 15 V, ion injection time was limited to 200 ms, AGC target set to 1,000,000, and 40 microscans were acquired. Typical acquisition times were 4 minutes. Source CID and HCD voltage were varied to either limit fragmentation or to promote it (see figure legends for details). The protein complex compositions and stoichiometry was determined via mass differences observed in the intact MS analysis of the protein complex. Obtained data were deconvoluted with Thermo Scientific Protein Deconvolution software. Typical settings for the deconvolution were an m/z range of 2000-13,000, output mass range 20,000-300,000, 75 ppm mass tolerance, charge state range of 4-50, 4 iterations, minimum adjacent charges 4-7, and a resolution of 12374 at 400m/z. Due to sample heterogeneity and the nature of the native spray analysis, it is estimated that all deconvoluted masses have an error of 400 ppm.
Chemical Crosslinking
The chemical crosslinking agent BS3 was added to a solution of 20 μM his-PhnG-H-I-J-K (in Buffer C) to a final concentration of 2.5 mM and then incubated on ice for two hours. The reaction was quenched by Laemmli sample buffer (2% SDS, 10% glycerol, 120 mM Tris-HCl, pH 6.8) and subsequently analyzed by SDS-PAGE. The gel was stained with 3 mg/mL Coomassie brilliant blue R250 and then destained with a solution containing 30% methanol and 10% acetic acid. The sample was proteolyzed via the addition of trypsin using a previously described protocol (15). The final samples were analyzed by LC-MS (Dionex nanoRSLC and an Orbitrap Fusion Tribrid Mass Spectrometer from Thermo Scientific). Briefly, 1.0 μL of the digested protein sample was loaded onto a 75 μm × 15 cm Acclaim Pepmap C18 column and eluted over 30 minutes with a total run time of one hour. The eluted peptides were introduced into the mass spectrometer and analyzed using the Orbitrap (OT) mass spectrometer in positive ion mode with the following settings: resolution 120K, scan range 400-1500 m/z, maximum injection time 50 ms, AGC target 2 × 104, and S-lens RF level = 60. Charge states from 2-7 were accepted and targeting ions were excluded for 60 seconds. MS/MS data were obtained using the HCD cell and fragment masses determined using the OT mass spectrometer at a resolution of 30 K. The data were analyzed using the crosslinking identification software StavroX (Version 3.4.5) (16). For the analysis of peptide masses from crosslinking experiments, a maximum mass deviation of 7 ppm and a minimal signal-to-noise ratio (S/N) of three were accepted. The mass range was set as 200 to 5000 Da. Decoy searches were also conducted with the same parameters using the reverse protein sequences to determine the false positive rate. The results were evaluated by the StavroX software (16). For scores larger than 76, the false-positive rate was less than 5%. Candidate peptides with scores higher than 76 were selected and then manually checked before being assigned as positives. Additionally, carbamidomethylation of cysteine was taken into account as a fixed modification and oxidation of methionine was set as a varied modification. Up to three missed-cleavage sites were utilized during data processing.
Hydrogen/Deuterium Exchange
The his-PhnG, his-PhnH, and his-PhnG-I were concentrated to 2.5 mg/mL and the his-PhnG-H-I-J-K complex was concentrated to 4.5 mg/mL. Deuterated buffers were prepared by removing water from the aqueous buffer containing 50 mM HEPES (pH 8.5), 200 mM NaCl, 2.0 mM TCEP using a centrifugal vacuum concentrator and then adding an equivalent volume of 99.9 % D2O (Cambridge Isotope Laboratories, Inc.). A total of 4.0 μL of the concentrated protein sample was diluted into 36 μL of aqueous buffer for the undeuterated sample. The deuterated samples were obtained by diluting 4 μL of protein into 36 μL of the deuterated buffer (90% deuteration) for various amounts of time (4 and 10 seconds, 1.0, 2.0, 5.0, 15 and 30 minutes, and 1.0 hour) at 0 °C. The deuterium exchange reaction was quenched with 60 μL ice cold buffer containing 3.0 M urea, 50 mM TCEP at pH 2.1 to bring the final pH of the resultant mixture to pH 2.3-2.4 (17, 18). A total of 50 μL of the quenched sample was added to 50 μL of immobilized pepsin resin (Thermo Scientific, USA). The pepsin beads were pre-activated by washing with 1% (v/v) TFA in water, pH 2.0. The digestion was carried out at ~6 °C for 5 minutes with frequent vortexing. The solution was then transferred to a 0.22 micron Ultrafree-MC GV centrifugal filter (EMD Millipore) and the pepsin beads were removed by centrifugation at 8,000 rpm for 30 seconds. The supernatant solution was divided into aliquots and immediately flash-frozen in liquid nitrogen and stored at -80 °C.
The subsequent analysis by mass spectrometry was performed using a Thermo Scientific Q Exactive instrument within two to three days of sample preparation. The samples were thawed on ice prior to injection to minimize back-exchange. The peptic digests of his-PhnG, his-PhnH, his-PhnG-I, and his-PhnG-H-I-J-K were applied to a 2.1 × 100 mm Symmetry C18 reverse-phased column (Waters Corporation, Milford, MA) using 0.1% (v/v) formic acid in water at a flow rate of 200 μL/minute. The peptides were eluted using a gradient of 8–55% acetonitrile over the course of eight minutes. The solvents and column were placed in an ice-bath throughout the experiment to reduce the back-exchange of deuterons during analysis. A blank solution using acetonitrile was utilized between each sample to avoid peak carry-over between runs. The mass spectrometer was calibrated using an LTQ ESI positive ion calibration solution from Thermo Scientific, USA. The peptides were identified using the software Peptide Mapping (Thermo Scientific) with a mass tolerance of 10 ppm for the theoretical mass-to-charge (m/z) ratios. Oxidation of methionine was set as a possible modification. The peptides were further confirmed by LC-MS/MS analysis on an Orbitrap Fusion Tribrid Mass Spectrometer from Thermo Scientific as described earlier. The pepsin digestion provided sequence coverages of 88%, 83%, and 86% for the PhnG, PhnH, and PhnG-I complexes, respectively. The PhnG-H-I-J-K complex had a sequence coverage of 89% for PhnG and PhnI, and 83% for PhnH.
The deuterium content and centroid values of each peptide at various time points were calculated using the software HDExaminer (Sierra Analytics, Modesto, CA). Each identified peptide was manually examined and validated. It was assumed that the maximum number of exchangeable amides in a peptide is two less than the total number of amino acids in the peptide minus any proline residues (17-19). The shifts in the centroid values for the deuterated samples were determined using the undeuterated sample as a control. The various charge states of an individual peptide were used to calculate the deuterium level and the values between them did not show much change. For regions of the proteins containing multiple overlapping peptides, a maximum of three overlaps encompassing the wider region were used in the analysis. The peptide exchange plots were generated using MS-Excel. The deuterated samples were not corrected for back-exchange and all of the reactions were carried out under identical conditions.
Homology Modeling and Secondary Structure Predictions
The homology model for PhnK was created using the Intensive Mode of the web-based structure prediction software Phyre2 (20) with the nucleotide-binding domain of a dipeptide ABC transporter (PDB id: 4FWI), which has a 32% sequence identity with PhnK. For PhnG, PhnI and PhnJ, the homology modeling was conducted using Swiss Model (21) and the secondary structure prediction was carried out using SABLE protein structure prediction server (22). All protein structure figures were generated using Pymol (http://www.pymol.org).
RESULTS
Characterization of PhnG-I
Two different contructions were prepared for the expression of a complex that contained only PhnG and PhnI. The first of these had a polyhistidine purification tag at the N-terminus of PhnG and the second had the polyhistidine tag at the C-terminus of PhnI. The calculated molecular masses for the subunits within these two complexes are as follows: PhnG (16,524 Da); his-PhnG (18,688 Da); PhnI (38,852 Da); and PhnI-his (39,917 Da). Both of these complexes migrated as a single peak upon elution from a calibrated Superdex 200 10/300 GL gel filtration column (GE Heathcare) (data not shown) and the approximate molecular weight was determined to be 140 ± 26 kDa.
The molecular weight for the his-PhnG-I complex was also determined using analytical ultracentrifugation and ESI mass spectrometry. For the his-PhnG-I complex, the sedimentation coefficient () of 5.89 S corresponds to an apparent molecular weight of 115.8 ± 1.7 kDa at a protein concentration of 0.25 mg/mL. The sedimentation velocity experiment was also conducted at a protein concentration of 0.75 mg/mL and a van Holde-Weischet analysis indicated that the oligomeric state of his-PhnG-I did not change at the higher protein concentration. Under native ESI conditions, the his-PhnG-I complex exhibited charge states in the gas phase with a range of +21 to +25 and a mass of 115,300 Da (Figure 1). CID fragmentation of the his-PhnG-I complex resulted in the loss of a PhnG monomer and a mass corresponding to PhnGI2 (96,600 Da), as well as the appearance of a mass at 56,000 Da that corresponds to PhnGI, indicating that PhnG and PhnI form a heterodimer (Supplemental Figure S1).
Figure 1.

Native ESI of the PhnG2I2 complex. The upper figure is the raw data obtained using 75V of energy in the HCD cell and 50V of energy in the source region. The bottom figure is the deconvoluted data using the isotope clusters from the upper mass spectrum of the complex, which represents the +19 to +23 charge states of the complex.
The stoichiometry of the PhnG-I-his complex was addressed by N-terminal amino acid sequence analysis. After seven rounds of N-terminal amino acid sequencing, the ratio of the amino acids specific for PhnG to that for PhnI averaged 0.9 ± 0.1. Therefore, the apparent stoichiometry of the two subunits contained within the PhnG-I-his complex is ~1:1. Given the mass of the his-PhnG-I complex determined from analytical ultracentrifugation (115,800 Da) and ESI mass spectrometry (115,770 Da) it is concluded that the stoichiometry of the complex containing only PhnG and PhnI is PhnG2I2.
SDS Electrophoresis of PhnG2I2
The subunit composition of the PhnG2I2 complex was confirmed by SDS-PAGE and the identity of each band was verified by in-gel digestion with trypsin and mass spectroscopy (Figure 2). The intensity of the Coomassie Blue staining for each of the two separated subunits of PhnG2I2 was measured using a Gel Doc EZ System densitometer (BioRad). For the PhnG2I2-his complex, the relative staining intensity for subunits PhnG and PhnI-his was 1.00:1.22 and 1.00:1.07 for his-PhnG2I2. When the his-tag was removed by proteolysis with thrombin the relative staining intensity for these two subunits was similar (1.00:1.17). Since the molecular weight ratios for the two subunits are 1.00:2.42 (for PhnG and PhnI-his) and 1.00:2.09 (for his-PhnG and PhnI), PhnG stains approximately twice as dense per amino acid than does PhnI under these conditions
Figure 2.

SDS-PAGE separation of the subunits contained within the C-P lyase complexes from E. coli. Lane 1: his-PhnG2I2. Lane 2: his-PhnG2H2I2J2; Lane 3: his-PhnG2H2I2J2K. All protein complexes were diluted to 2mg/mL in sample buffer (65.8 mM Tris-HCl, pH 6.8, 2.1% SDS, 26.3% (w/v) glycerol, 0.01% bromophenol blue) and loaded onto a Bio-Rad mini-protean precast gel. The gel was stained by Coomassie Brilliant Blue and the relative intensity of each band quantified using a Gel Doc EZ System densitometer (BioRad).
Characterization of PhnG-H-I-J
The complex that contains subunits PhnG, PhnH, PhnI, and PhnJ was cloned with a polyhistidine tag at the C-terminus of PhnJ. This complex migrated as a single homogeneous peak after elution from a gel filtration column (data not shown). The approximate molecular weight for this complex was determined to be 243 ± 38 kDa using a calibrated Superdex 200 10/300 GL gel filtration column (GE Heathcare). Two species were identified by ultracentrifugation at a protein concentration of 0.25 mg/mL. The major component (79.9%) exhibited an value of 9.43 S and an estimated molecular weight of 218.8 ± 7.4 kDa. The minor component (19.1%) had an value of 7.90 S with an estimated molecular weight of 191.0 kDa ± 9.1 kDa. The sedimentation velocity experiments were also conducted at a higher protein concentration (0.75 mg/mL) and these experiments indicated that the PhnG-H-I-J-his complex aggregated at the higher protein concentration during the ultracentrifugation process.
The subunit composition of the PhnG-H-I-J-his complex was confirmed by SDS-PAGE and the identity of each band was verified by in-gel digestion with trypsin and mass spectroscopy (Figure 2). The intensity of the Coomassie Blue staining for each of the separated subunits was measured and the relative staining intensities for subunits PhnG, PhnH, PhnI, and PhnJ-his are listed in Table 1. From the relative staining intensities for PhnG and PhnI in this complex (1.19 to 1.28) we can conclude that these two subunits are in a ratio of 1:1 since this is essentially the same ratio as found in the PhnG2I2 complex (1.17).
Table 1.
Relative Staining Intensities in PhnG-H-I-J and PhnG-H-I-J-K
| PhnG | PhnH | PhnI | PhnJ | |
|---|---|---|---|---|
| PhnG-H-I-J | 27.6 | 11.2 | 35.4 | 25.9 |
| PhnG-H-I-J-his | 26.0 | 13.5 | 31.0 | 29.5 |
| PhnG-H-I-J-K | 25.4 | 11.8 | 35.8 | 27.0 |
| PhnG-H-I-J-K-his | 22.7 | 11.3 | 38.0 | 27.9 |
| his-PhnG-H-I-J-K | 28.5 | 9.3 | 37.5 | 24.8 |
| Average | 26.0 | 11.4 | 35.5 | 27.0 |
Under native ESI conditions, the parent mass of the PhnG-H-I-J-his complex was measured to be approximately 222,200 Da (Figure 3). In an effort to elucidate the subunit composition of this complex, the collisional energy was raised as high as 200 V and the resulting deconvoluted fragment masses of 197,900, 166,300, 144,000, 111,100, 94,500 and 55,600 were obtained (Supplemental Figure S2). These fragments correspond to the sequential loss of PhnH (21.0 kDa), PhnJ-his (32.8 kDa), PhnH (21.0 kDa), PhnJ-his (31.7 kDa), PhnG (16.5 kDa), and PhnI (38.9 kDa). PhnGI is the smallest fragment this is observed. These results are consistent with the formation of PhnHJ and PhnGI heterodimers in the complex and that the overall composition for this complex is PhnG2H2I2J2.
Figure 3.

Native ESI of the PhnG2H2I2J2 complex. The upper figure is the raw data obtained using 10 V of energy in the HCD cell and 10 V of energy in the source region. The bottom figure is the deconvoluted data utilizing the isotope clusters from the upper mass spectrum of the complex, which represents the +28 to +33 charge states of the complex.
Characterization of PhnG-H-I-J-K
The complex composed of PhnG through PhnK was cloned and subsequently expressed with a polyhistidine tag at either the N-terminus of PhnG or the C-terminus of PhnK. Both of these complexes migrated through a calibrated gel filtration column as a single peak (data not shown) with an estimated molecular weight of 285 ± 46 kDa. The subunit compositions of the two PhnG-H-I-J-K complexes were confirmed by SDS-PAGE and the identity of each band was verified by in-gel digestion with trypsin and mass spectroscopy (Figure 2). To estimate the stoichiometry of the subunits contained within these two complexes, the relative Coomassie Blue staining intensities for PhnG, PhnH, PhnI, and PhnJ were compared to the relative staining intensities for these same subunits in the complex that contains only four subunits (Table 1). Within experimental error, the relative staining intensities for PhnG, PhnH, PhnI, and PhnJ contained within the PhnG2H2I2J2 complexes matched those contained within either of the complexes that contained all five subunits. Therefore, the PhnG2H2I2J2 core is retained within the two complexes that now contain PhnK.
The molecular weight of the PhnG-H-I-J-K complex was estimated using analytical ultracentrifugation. At a protein concentration of 0.25 mg/mL, the value was determined to be 8.94 S for the PhnG-H-I-J-K-his complex with a calculated molecular weight of 236.9 ± 8.3 kDa. The sedimentation velocity experiments were also conducted at a protein concentration of 0.75 mg/mL and a van Holde-Weischet analysis indicated that the oligomeric state of PhnG-H-I-J-K-his was constant at the two protein concentrations. The mass of the PhnG-H-I-J-K-his complex was also determined using ESI mass spectrometry. Under native ESI conditions, the PhnG-H-I-J-K-his complex enters the gas phase with charge states of +30 to +36, corresponding to a mass of 249,700 (Figure 4), a value that is consistent with that determined by analytical ultracentrifugation. Using 75 V of source fragmentation energy and 45 V in the HCD cell, the mass at 249,200 most likely represents the PhnG2H2I2J2K-his complex (calculated mass = 245,144) and that at 221,300 represents the PhnG2H2I2J2 complex (calculated mass = 216,234) and the loss of PhnK-his (calculated mass = 28,910). The mass at 196,234 represents the loss of PhnHJ (calculated mass = 52,741) to give PhnG2HI2JK (calculated mass = 192,403). The mass at 168,598 represents the PhnG2HI2J complex with the loss of PhnK (Supplemental Figure S3).
Figure 4.

Native ESI of the PhnG2H2I2J2K complex. The upper figure is the raw data obtained using 13 V of collision energy in the HCD cell and 10 V of energy in the source region. The bottom figure is the deconvoluted data utilizing the isotope clusters from the upper mass spectrum of the complex, which represents the +30 to +36 charge states of the complex.
Structural Analysis by Chemical Crosslinking
The contact regions for interaction of the subunits contained within the PhnG2H2I2J2K complex were probed by chemical crosslinking. Crosslinking was initiated with the homo-bifunctional reagent bis-sulfosuccinimidyl suberate (BS3) that can form a linkage of up to 11.4 Å between the ε-amino groups of lysine residues or the free amino group of the N-terminus (23). The products of the crosslinking reactions with PhnG2H2I2J2K-his were separated by SDS-PAGE and the results are presented in Figure 5. Four prominent high molecular weight cross-linked protein complexes of approximately 47 (band 1), 70 (band 2), 87 (band 3) and 110 kDa (band 4) were formed after incubation with BS3. Two bands of higher molecular weight at approximately 125 and 165 kDa were also formed, but are less distinct. The crosslinked proteins were excised from the gel and subjected to in-gel digestion with trypsin and characterized by LC-ESI/MS. Based on the mass spectrometry data, the 47 kDa band contains one copy each of PhnG (17 kDa) and PhnJ (31 kDa); the 70 kDa band contains one copy each of PhnI (39 kDa) and PhnJ and the 87 kDa band contains one copy each of PhnG, PhnI, and PhnJ. The bands at 110, 125, and 165 kDa contain mixtures of PhnG, PhnI, and PhnJ. No crosslinked proteins were identified to contain PhnH or PhnK.
Figure 5.
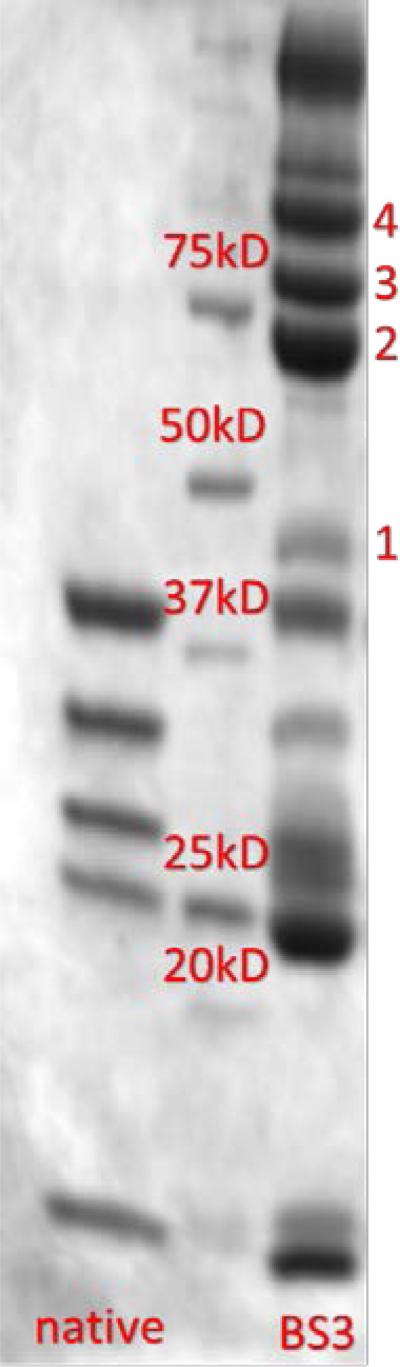
SDS-PAGE of the BS3-initiated crosslinking of those proteins contained within the various carbon-phosphorus lyase complexes. The left lane represents the separation of the subunits contained within the PhnG2H2I2J2K complex prior to the addition of BS3. The middle lane is a standard molecular weight protein ladder (Bio-Rad). The rightmost lane represents the separation of crosslinked proteins after the addition of BS3 to the PhnG2H2I2J2K complex. Four prominent crosslinked bands were labeled as 1, 2, 3 and 4 from the bottom to the top.
The crosslinked proteins were excised, proteolyzed with trypsin, and then analyzed by LC-ESI-MS/MS to identify the specific crosslinking sites. The purified peptides were subjected to high-energy collision dissociation (HCD). A representative example of an MS/MS spectrum of a triply charged ion at m/z 1004.16 (corresponding to an M+H+ = 3010.46) is provided in Figure 6. The daughter ion spectrum exhibited most of the y-ion series for two peptides from PhnG (β-peptide, residues 89-95) and PhnJ (α-peptide, residues 2-18). The y16β and y5α ions indicate that Lys-89 of PhnG and the N-terminus of PhnJ are covalently linked to one another. A similar analysis was conducted to identify all of the other crosslinked peptides and the identity of the crosslinked peptide pairs are presented in Table 2.
Figure 6.

ESI-MS/MS analysis of the crosslinked peptides contained within the PhnG2H2I2J2K complex. The peptide 88DKHAER94- from PhnG and the peptide 2ANLSGNYFAYLDEQTKR18 were crosslinked through Lys-89 from DKHAER and the N-terminus of ANLSGNYFAYLDEQTKR.
Table 2.
Identification of specific crosslinking site of PhnG-H-I-J-K C-P lyase complex.
| Band | M+H+ | Deviation (ppm) | Sequence 1 | Protein 1 | Sequence 2 | Protein 2 |
|---|---|---|---|---|---|---|
| Band 1 | 3010.45 | −2.65 | 88DKQHAER94 | PhnG | 2ANLSGYNFAYLDEQTKR18 | PhnJ |
| 2838.42 | −0.88 | 84NFFKR88 | PhnJ | 2ANLSGYNFAYLDEQTKR18 | PhnJ | |
| Band 2 | 2501.29 | 2.34 | 84NFFKR88 | PhnJ | 7GGEKAIDAAHALQESR22 | PhnI |
| 3214.42 | −1.95 | 1MYVAVKGGEK10 | PhnI | 274QQSEAKNQ281 | PhnJ | |
| 2857.42 | −3.37 | 1mYVAVK6 | PhnI | 2ANLSGYNFAYLDEQTKR18 | PhnJ | |
| 1930.02 | 1.53 | 84NFFKR88 | PhnJ | 1MYVAVKGGEK10 | PhnI | |
| 2838.42 | 1.09 | 84NFFKR88 | PhnJ | 2ANLSGYNFAYLDEQTKR18 | PhnJ | |
| Band 3 | 2501.29 | 0.65 | 84NFFKR88 | PhnJ | 7GGEKAIDAAHALQESR22 | PhnI |
| 3010.47 | 1.68 | 88DKQHAER94 | PhnG | 2ANLSGYNFAYLDEQTKR18 | PhnJ | |
| 3137.47 | −3.92 | −1MMHADTATR8 | PhnG | 196MDMmPALQLFGAGREKR212 | PhnJ | |
| 2167.06 | −0.97 | 274QQSEAKNQ281 | PhnJ | 1mYVAVKGGEK10 | PhnI | |
| 2838.42 | 1.09 | 84NFFKR88 | PhnJ | 2ANLSGYNFAYLDEQTKR18 | PhnJ | |
| Band 4 | 3010.47 | 1.68 | 88DKQHAER94 | PhnG | 2ANLSGYNFAYLDEQTKR18 | PhnJ |
| 7691.66 | −6.61 | −1mMHADTATRQHWMSVLAHSQPAELAAR27 | PhnG | 41EMPMPYGWGTGGIQLTASVIGESDVLKVIDQGADDTTNAVSIR81 | PhnJ | |
| 2501.28 | 0.65 | 84NFFKR88 | PhnJ | 7GGEKAIDAAHALQESR22 | PhnI | |
| 1930.02 | −2.17 | 84NFFKR88 | PhnJ | 1MYVAVKGGEK10 | PhnI | |
| 2682.31 | −2.83 | 84NFFKR88 | PhnJ | 2ANLSGYNFAYLDEQTK17 | PhnJ |
m: oxidized form of methionine.
In addition to the intermolecular crosslinks between different protein subunits there were several intramolecular crosslinks contained within PhnI and PhnJ. In all of the 3010.46) is provided in . The daughter ion spectrum exhibited most of the y-ion series for two peptides from PhnG (β-peptide, residues 89-95) and PhnJ (α-peptide, residues 2-18). The y16β and y5α ions indicate that Lys-89 of PhnG and the crosslinking experiments, only peptides from PhnG, PhnI or PhnJ were identified. There was no trace of PhnH or PhnK in the crosslinked proteins. However, further analysis of the ESI-MS data identified several internally crosslinked peptides and individual lysine residues within PhnH and PhnK that were labeled with the crosslinking reagent. For example, Lys-38 from PhnH was derivatized with BS3. In PhnK, the N-terminus, Lys-127, and Lys-220 were labeled with BS3 and an internal crosslink was identified between Lys-19 and Lys-49.
Structural Analysis by Hydrogen/Deuterium Exchange
Hydrogen/deuterium exchange experiments were carried out on PhnG, PhnG2I2, and PhnG2H2I2J2K from the C-P lyase complex in an attempt to determine those regions of PhnG that are most affected by the association with PhnI. It was assumed that those portions of PhnG that interact directly with PhnI will exhibit a change in the rate of H/D exchange in either PhnG2I2 or PhnG2H2I2J2K, relative to the H/D exchange profile for PhnG alone. Reduced rates of H/D exchange may also be observed for those portions of PhnG that become more structured in either of these two complexes than in the isolated subunit alone. A difference of 10% or more in the exchange rate between any two protein complexes is considered significant. The overall coverage of peptides from PhnG that were detected by mass spectrometry in all three complexes was approximately 87%. The time course for the exchange of deuterium from solvent incorporated into the peptide 68TRAAVRLTDGTLGYS82 from PhnG is presented in Figure 7. The time courses for H/D exchange within PhnG alone indicate that many regions of the protein undergo H/D exchange very rapidly. This is especially true for those regions of PhnG that extend from residues 28-79 and from residues 116-124 (Figure 8). All of the peptides identified for PhnG, PhnG2I2 and PhnG2H2I2J2K exhibit EX2 kinetics for deuterium uptake (24-27). Bimodal isotopic distribution spectra, which indicate the presence of local or global unfolding through EX1 kinetic, were not observed for any of the peptides of PhnG or PhnI (24-26)
Figure 7.

Time course for the uptake of deuterium by the peptide 68TRAAVRLTDGTLGYS 82 from PhnG as measured by ESI-MS after 0, 4 seconds, 5 minutes and 1 hour.
Figure 8.

Differential deuterium exchange for the peptides contained within PhnG alone and the same peptide contained within PhnG22. The incubation times are as follows: 4 seconds (◆); 10 seconds (■); 1 minute (▲); 2 minutes (□) 5 minutes (•) 15 minutes (•); 30 minutes (■); 60 minutes (◆).
There are significant differences in the rates of deuterium exchange for those peptides from PhnG when this protein is contained within the PhnG2I2 or PhnG2H2I2J2K complexes. These differences are graphically depicted in the representative times courses for comparable peptides in Figure 8. For the peptides that encompass residues 16-23, 28-36, and 98-111, there are essentially no differences in the time courses for the uptake of deuterium by PhnG alone and PhnG contained within the PhnG2I2 complex. These results are in stark contrast to the significant differences in the exchange profiles for peptides that encompass residues 47-79 and 68-97 (Figure 8).
The H/D exchange profiles for the PhnG and PhnI subunits contained within the PhnG2I2 and PhnG2H2I2J2K complexes are quite similar to one another (Figure 9). For PhnG, there are virtually no differences in the exchange rates for portions of the protein when this complex is embedded in either PhnG2I2 or PhnG2H2I2J2K. For PhnI, the only regions exhibiting differential deuterium exchange profiles between the two complexes are the peptides contained within PhnI that encompass residues from 33-41, 81-85, and 109-130 (Figure 9). These peptides show slightly more protection in the PhnG2H2I2J2K complex than in the PhnG2I2 complex.
Figure 9.

Differential deuterium exchange for the peptides contained within PhnG and PhnI for the complexes PhnG2I2 and PhnG2H2I2J2K. The peptides differing in exchange pattern between the two complexes are highlighted in red. The incubation times are as follows: 4 seconds (◆); 10 seconds (■); 1 minute (▲); 2 minutes (□); 5 minutes (•) 15 minutes (•); 30 minutes (■); 60 minutes (◆).
H/D exchange analysis of PhnH alone was carried out and compared with the peptides from PhnH obtained from the PhnG2H2I2J2K complex to help identify those regions of PhnH that undergo structural variations upon complex formation. A total of 23 common peptides, spanning 83% of PhnH, were identified. From the deuterium uptake plots for the PhnH peptides in the individual subunit and the PhnG2H2I2J2K complex, many regions along the entire length of PhnH are more protected in the PhnG2H2I2J2K complex (Supplemental Figure S4). The N-terminal region of PhnH, which comprises the dimerization interface in the crystal structure of PhnH, is represented by the peptides encompassing residues 6-18, 7-18, 19-24 and 19-31. This region exhibits significant reduction in deuterium exchange in the PhnG2H2I2J2K complex as compared to the individual subunit of PhnH. A major difference in deuterium exchange profile is also observed in the region connecting α-helix D with a disordered loop and stretches up to β-strand β5 for the peptides spanning residues 110 to 123 (peptides 107-122 and 110-123) (Supplemental Figures S4 and S5). A high degree of differential exchange is also observed in the region extending from residues 136-146 as illustrated by the peptides 136LRLTGAG142, 136LRLTGAGIAE145 and 136LRLTGAGIAEE146 (Supplemental Figure S4). This region extends from β-strand β6 and connects through a disordered loop region to β-strand β7 (Supplemental Figure S5). Finally, the C-terminal region of PhnH, illustrated by the peptides encompassing residues 157-173, 159-173 and 183-194 are more protected in the complex compared to the PhnH alone (Supplemental Figure S4).
DISCUSSION
Stoichiometry of the C-P Lyase Complexes
Three different protein complexes from the carbon-phosphorus lyase enzyme were expressed, purified and characterized by gel electrophoresis, analytical ultra-centrifugation, N-terminal protein sequencing, and high-resolution mass spectrometry. For the complex that contains only PhnG and PhnI, the results are consistent with an equal mixture of the two subunits and an overall stoichiometry of PhnG2I2. In the complex that contains these two subunits, in addition to PhnH and PhnJ, the results are consistent with a stoichiometry of PhnG2H2I2J2. For the complex that contains five different subunits, the complex contains two copies of PhnG, PhnH, PhnI, and PhnJ, plus a single copy of PhnK (PhnG2H2I2J2K). These results suggest that PhnG and PhnI associate to form a central heterodimeric core (PhnG2I2) that is required prior to the binding of two additional copies each of PhnH and PhnJ (PhnHJ) to facilitate the assembly of PhnG2H2I2J2. The largest complex is subsequently formed by the binding of a single copy of PhnK. The proposed assembly pathway for the formation of the PhnG2H2I2J2 and the PhnG2H2I2J2K complexes is further supported by the mass spectrometry results where individual subunits are progressively dissociated from the central core complex. With the PhnG2H2I2J2 complex, we observed the sequential dissociation of PhnH, PhnJ, PhnH, PhnJ, PhnG and PhnI.
Chemical Crosslinking
Chemical crosslinking experiments were conducted to identify those subunits that are physically able to contact one another and these results are graphically illustrated in Figure 10. Using PhnG2H2I2J2K we demonstrated that subunit contacts occur between PhnG and PhnJ, and between PhnJ and PhnI. PhnG and PhnI must further interact with one another since a PhnG2I2 complex can be isolated. Unfortunately, no crosslinks could be identified between PhnH or PhnK and any other subunit. This result must reflect the fact that the N-terminus and the available lysine residues within these two proteins are insufficiently close to the appropriate residues contained within PhnG, PhnI, or PhnJ. We did, however, identify an internal crosslink between two lysine residues within PhnK and labeling of single lysine residues in both PhnH (Lys-38) and PhnK (Lys-127 and Lys-220).
Figure 10.

The crosslinking map of the PhnG2H2I2J2K complex. All of the crosslinked residues are highlighted in red while all of the residues that participate in a mono-linkage are highlighted in blue. Two residues that were reported to be key residues within the active site of PhnJ (Gly-32 and Cys-272) are highlighted in green. The crosslinking bonds are depicted as straight lines between different crosslinking sites. The internal crosslinked bonds are presented as a curve connecting two lysine residues.
Hydrogen/Deuterium Exchange
The assembly of multi-protein complexes was further addressed by measuring the rates of exchange of hydrogen and deuterium into the backbone of PhnG and PhnI from solvent. For PhnG alone, about 40% of the protein backbone undergoes deuterium exchange in a very short time (< 10 sec). This result demonstrates that significant portions of PhnG alone are highly dynamic and likely to be unstructured. Some of these regions become more structured upon interaction with PhnI. This is particularly true for that portion of the protein that extends from Val-47 through Leu-67 (Figure 8). However, further differences are not observed for PhnG within the PhnG2H2I2J2K complex but significant differences in the H/D exchange rates are observed for PhnI in certain regions when the PhnG2I2 complex is compared with the PhnG2H2I2J2K complex (Figure 9). Similar structural transitions are often found in multi-protein complexes and in intrinsically disordered proteins that undergo synergistic folding in the presence of their binding partners (28, 29).
The N-terminal dimerization interface of PhnH is altered in the PhnG2H2I2J2K complex. This may indicate a loss in the interaction of the PhnH monomer with another PhnH. The structural change near the dimer interface region may also imply the binding of PhnH with another subunit in the PhnG2H2I2J2K complex. The crystal structure of PhnH shows the formation of a large hydrophobic pocket on the surface of each monomer as the result of dimerization by the N-terminal ends of β-strands β4 (91-94), β5 (110-121) and β8 (166-173) (30). Interestingly almost all of the regions other than the N-termini of β-strand β4 become more protected in the complex. This clearly indicates the change in the hydrophobic pocket region within the PhnG2H2I2J2K complex. The C-terminal region spanning residues 185-194 also become more protected in the complex. This indicates the possible role of this region involved in subunit interactions in the complex.
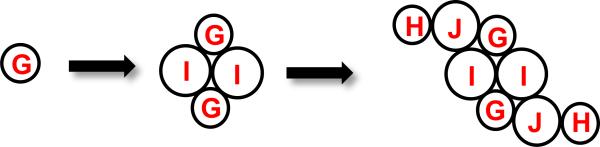
Low Resolution Structural Model
The results obtained from the mass spectrometry and chemical crosslinking experiments can be used to propose a low-resolution model for the assembly of the C-P Lyase complex (Scheme 2). In this model a heterodimeric core complex is formed from the association of two copies each of PhnI and PhnG. This complex serves as a platform for the further addition of two copies each of PhnH and PhnJ. Crosslinking experiments demonstrate the physical interaction between PhnI and PhnJ but we were unable to obtain evidence for the association of PhnH with any other protein. However, we have assumed that PhnH must interact with PhnJ since both proteins are apparently required for elaboration of the PhnG2I2 core.
Scheme 2.
The closest sequence homologues for PhnG, PhnI and PhnJ were found to be the putative Trp repressor protein (PDB id: 3KOR), carboxymethyl-2-hydroxymuconate semialdehyde dehydrogenase (PDB id: 2D4E) and PhnH (PDB id: 2FSU), respectively. However, the sequence identities of these proteins to their closest identified homologues are less than 20% and so a reliable homology model could not be built for any of these proteins. However, it is quite interesting that the closest structural model for PhnJ is PhnH. The secondary structural predications for PhnG, PhnI and PhnJ are presented in Supplemental Figure S6. The predicted secondary structures are consistent with the results of H/D exchange. With PhnG, the region with the most rapid H/D exchange rate (residues 28-61) is predicted to be mostly unstructured or β-strands. For PhnI contained within PhnG2I2 and PhnG2H2I2J2K, most of the dynamic regions are also predicted to be unstructured or beta strands.
Lys-89 from PhnG was shown to be a crosslinking site between PhnG and PhnJ. The H/D exchange results showed a dramatic change of the exchange rate on Lys-89 when PhnG is in the PhnG2H2I2J2K complex. Unfortunately, the crosslinking sites for PhnG and PhnI are all located close the N-terminus, which is not covered by the H/D exchange experiments. It is possible that the N- and C-terminal regions of PhnH are involved in the interaction with PhnJ, based on the differential deuterium exchange analysis of PhnH. The peptides which contain Lys-23 (19RRLLKA24 and 19RRLLKAMSEPGVI31) exchange at a much slower rate in the PhnG2H2I2J2K complex than in PhnH itself and the region containing Lys-38 does not show a significant difference between the two protein complexes. Since there are no crosslinked peptides for PhnH, we are not able to identify the regions involved in interaction with PhnJ or any other subunit in the complex.
The homology model of PhnK indicates that residues Lys-19 and Lys-49, which are internally crosslinked are separated by a distance of about 20 Å (Supplemental Figure S7). The crosslinking agent is ~11 Å in length. This indicates that the region containing these two lysine residues must come closer together in the complex. Although the lysine residues in the homology model of PhnK appear to be on the protein surface, no peptides of PhnK were crosslinked to any other subunit. It is currently unclear how PhnK binds to the PhnG2H2I2J2 complex or why only one copy of PhnK binds to an apparently dimeric complex. What is obviously required is the crystallization and three-dimensional structure determination of these complexes.
Supplementary Material
Acknowledgements
We thank John Patrick (Texas A&M University) for valuable assistance with the ESI experiments, and Professor Vicki Wysocki and Royston Quintyn (Ohio State University) for valuable discussions and assistance with ESI-MS of protein complexes. We also gratefully acknowledge Jamie K. Humphries and ThermoScientific for providing access to the Exactive mass spectrometer used in these studies.
This work was supported in part by the National Institutes of Health (GM 103917) and (1S10DO10407).
Footnotes
Supporting Information
Supplemental Figures S1, S2, and S3 for the ESI mass spectra for the fragmentation of complexes PhnG2I2, PhnG2H2I2J2, and PhnG2H2I2J2K are provided. Supplementary Figures S4, S5, S6, and S7 are provided for H/D exchange data for PhnH, the structure of PhnH, secondary structure predictions for PhnG, PhnI, and PhnJ, and the homology model for PhnK. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Hsieh YJ, Wanner BL. Global regulation by the seven-component Pi signaling system. Curr. Opin. Microbiol. 2010;13:198–203. doi: 10.1016/j.mib.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jia Y, Lu Z, Huang K, Herzberg O, Dunaway-Mariano D. Insight into the mechanism of phosphoenolpyruvate mutase catalysis derived from site-directed mutagenesis studies of active site residues. Biochemistry. 1999;38:14165–14173. doi: 10.1021/bi990771j. [DOI] [PubMed] [Google Scholar]
- 3.McGrath JW, Chin JP, Quinn JP. Organophosphonates revealed: new insights into the microbial metabolism of ancient molecules. Nat. Rev. Microbiol. 2013;11:412–419. doi: 10.1038/nrmicro3011. [DOI] [PubMed] [Google Scholar]
- 4.Kononova SV, Nesmeyanova MA. Phosphonates and their degradation by microorganisms. Biochemistry-Moscow. 2002;67:184–195. doi: 10.1023/a:1014409929875. [DOI] [PubMed] [Google Scholar]
- 5.Kamat SS, Raushel FM. The enzymatic conversion of phosphonates to phosphate by bacteria. Curr. Opin. Chem. Biol. 2013;17:589–596. doi: 10.1016/j.cbpa.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Chen CM, Ye QZ, Zhu ZM, Wanner BL, Walsh CT. Molecular biology of carbon-phosphorus bond cleavage. Cloning and sequencing of the phn (psiD) genes involved in alkylphosphonate uptake and C-P lyase activity in Escherichia coli. B. J. Biol. Chem. 1990;265:4461–4471. [PubMed] [Google Scholar]
- 7.Gebhard S, Cook GM. Differential regulation of high-affinity phosphate transport systems of Mycobacterium smegmatis: identification of PhnF, a repressor of the phnDCE operon. J. Bacteriol. 2008;190:1335–1343. doi: 10.1128/JB.01764-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yakovleva GM, Kim SK, Wanner BL. Phosphate-independent expression of the carbon-phosphorus lyase activity of Escherichia coli. Appl. Microbiol. Biotechnol. 1998;49:573–578. doi: 10.1007/s002530051215. [DOI] [PubMed] [Google Scholar]
- 9.Hove-Jensen B, McSorley FR, Zechel DL. Physiological role of PhnP-specified phosphoribosyl cyclic phosphodiesterase in catabolism of organophosphonic acids by the carbon-phosphorus lyase pathway. J. Am. Chem. Soc. 2011;133:3617–3624. doi: 10.1021/ja1102713. [DOI] [PubMed] [Google Scholar]
- 10.Hove-Jensen B, McSorley FR, Zechel DL. Catabolism and detoxification of 1-aminoalkylphosphonic acids: N-acetylation by the phnO gene product. Plos One. 7. 2012 doi: 10.1371/journal.pone.0046416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hove-Jensen B, Rosenkrantz TJ, Haldimann A, Wanner BL. Escherichia coli phnN, encoding ribose 1,5-bisphosphokinase activity (phosphoribosyl diphosphate forming): Dual role in phosphonate degradation and NAD biosynthesis pathways. J. Bacteriol. 2003;185:2793–2801. doi: 10.1128/JB.185.9.2793-2801.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamat SS, Williams HJ, Raushel FM. Intermediates in the transformation of phosphonates to phosphate by bacteria. Nature. 2011;480:570–573. doi: 10.1038/nature10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jochimsen B, Lolle S, McSorley FR, Nabi M, Stougaard J, Zechel DL, Hove-Jensen B. Five phosphonate operon gene products as components of a multi-subunit complex of the carbon-phosphorus lyase pathway. Proc. Nat.l Acad. Sci. U S A. 2011;108:11393–11398. doi: 10.1073/pnas.1104922108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demeler B. UltraScan: a comprehensive data analysis software package for analytical ultracentrifugation experiments. Modern analytical ultracentrifugation: techniques and methods. 2005:210–229. [Google Scholar]
- 15.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protocols. 2007;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 16.Gotze M, Pettelkau J, Schaks S, Bosse K, Ihling CH, Krauth F, Fritzsche R, Kuhn U, Sinz A. StavroX--a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass. Spectrom. 2012;23:76–87. doi: 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- 17.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 19.Walters BT, Ricciuti A, Mayne L, Englander SW. Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J. Am. Soc. Mass. Spectrom. 2012;23:2132–2139. doi: 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelley LA, Sternberg MJE. Protein structure prediction on the web: A case study using the Phyre server. Nature Protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 21.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 22.Adamczak R, Porollo A, Meller J. Combining prediction of secondary structure and solvent accessibility in proteins. Proteins: Strcuture, Function and Bioinformatics. 2005;59:467–475. doi: 10.1002/prot.20441. [DOI] [PubMed] [Google Scholar]
- 23.Sinz A. Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J. Mass Spectrom. 2003;38:1225–1237. doi: 10.1002/jms.559. [DOI] [PubMed] [Google Scholar]
- 24.Krishna MM, Hoang L, Lin Y, Englander SW. Hydrogen exchange methods to study protein folding. Methods (San Diego, Calif.) 2004;34:51–64. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Weis DD, Wales TE, Engen JR, Hotchko M, Ten Eyck LF. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass. Spectrom. 2006;17:1498–1509. doi: 10.1016/j.jasms.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 27.Liyanage R, Devarapalli N, Puckett LM, Phan NH, Gidden J, Stites WE, Lay JO., Jr. Comparison of two ESI MS based H/D exchange methods for extracting protein folding energies. Int. J. Mass spectrom. 2009;287:96–104. doi: 10.1016/j.ijms.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacherjee A, Wallin S. Coupled folding-binding in a hydrophobic/polar protein model: impact of synergistic folding and disordered flanks. Biophys. J. 2012;102:569–578. doi: 10.1016/j.bpj.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams MA, Luo Y, Hove-Jensen B, He SM, van Staalduinen LM, Zechel DL, Jia Z. Crystal structure of PhnH: an essential component of carbon-phosphorus lyase in Escherichia coli. J. Bacteriol. 2008;190:1072–1083. doi: 10.1128/JB.01274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.