

Abstract
We report the design of side-chain-to-tail linked organo-peptide hybrids incorporating an α-helical protein-binding motif. Using this strategy, macrocyclic inhibitors of the p53:HDM2 interaction displaying dual specificity against the HDMX homolog as well as increased proteolytic stability could be obtained.
The development of agents for selective modulation of protein-protein interactions (PPIs) constitutes a prominent goal in drug discovery and chemical biology.1 Since PPIs are often mediated by well defined secondary structural elements, a promising strategy in this area has involved the stabilization or mimicry of these motifs via compact molecular scaffolds.2 Reflecting its abundance in protein structures, α-helices are often encountered at the interface of protein-protein complexes.3 Accordingly, a number of strategies have been developed for stabilization of α-helical peptides4, which include the use of hydrogen bond surrogates5 as well as of a variety of inter-side-chain linkages such as disulfide,6 lactam,7, thioether8 or triazole9 bridges, ‘hydrocarbon staples’10, and cysteine cross-linking moieties.11
We recently reported strategies for the synthesis of macrocyclic organo-peptide hybrids (MOrPHs) via the chemo- and regioselective ligation of bifunctional synthetic precursors to genetically encoded precursor polypeptides (e.g. Figure 1A).12 A key feature of this new class of peptide-based macrocycles is their modular architecture, as given by the diverse non-peptidic and peptidic moieties amenable to incorporation into these scaffolds.12a,12c As part of ongoing studies directed at evaluating MOrPHs as disruptors of biomedically relevant PPIs, we were interested in assessing the potential of these macrocyclic scaffolds to accommodate, and possibly, stabilize a functional α-helical motif. In this work, we describe the successful implementation of this idea through the design and development of α-helical MOrPHs that can effectively disrupt the interaction between the tumor suppressor p53 and the oncoproteins HDM2 and HDMX.
Figure 1.

A) MOrPH macrocyclization strategy and chemical structure of synthetic precursors (box) investigated in this study. B) Crystal structure of HDM2:PMI complex (pdb 3EQS) and model of representative example of designer α-helical MOrPH (i / i+10 peptide cyclization with SP8).
HDM2/X are implicated in the negative regulation of p53 activity and overexpression of these proteins has been linked to several malignancies.13 While dual inhibition of HDM2/X has emerged as a most promising strategy for anticancer therapy,14 small-molecule inhibitors of HDM2 typically fail to potently interfere with p53:HDMX interaction due to subtle differences in the p53 binding clefts of these protein homologs.15 These limitations make the development of dual HDM2/X inhibitors a topic of current interest.10c,16 HDM2 and HMDX bind to the N-terminal transactivation domain of p53 (p5315–29), which upon complex formation adopts a well defined α-helix.17 Thus, in addition to its biomedical relevance, these structural features have made the p53:HDM2 interaction an ideal test bed to probe strategies for α-helix stabilization and mimicry.10c,11b,18
The starting point for the design of our MOrPH-based HDM2/X-targeting inhibitors was a linear 12-mer peptide isolated via phage display by Patzgier et al. (PMI: T1SFAEYWNLLSP12).19 PMI carries the triad of cofacial i / i+4 / i+7 amino acid residues known to be critical for p53 interaction with HDM2/X17 (i.e. Phe3, Trp7, and Leu10 corresponding to Phe19, Trp23, and Leu26 in p53, respectively), but inhibits these proteins with greater potency than a p53-derived peptide (IC50: 30–40 nM vs. 200–300 nM, respectively).19 Upon inspection of the PMI/HDM2 complex structure (Figure 1B),19 two solvent exposed residues, namely Thr1 and Glu5, were identified as two equally viable side-chain attachment points for MOrPH formation via substitution with p-acetyl-phenylalanine (pAcF) according to our oxime/AMA-mediated cyclization method (Figure 1A).12c
The C-terminal attachment site was chosen to lie after Ser11 (changed to Ala to promote α-helix formation), as Pro12 did not appear to establish significant contacts with the HDM2 surface.19 Analysis of models of the resulting peptide sequences, namely PMI-2 (GTSFA(pAcF)YWNLLA) and PMI-3 (G(pAcF)SFAEYWNLLA), revealed that the distances between pAcF side-chain keto group and the C-terminal carbonyl group were about 13 and 16 Å, respectively. These distances matched the spacer distance (~13 Å) furnished by one of our previously described synthetic linkers, called SP612c (Figure 1B), based on an energy-minimized model of the compound (Figure S1). SP6 was thus selected as a first candidate for macrocyclization of the target peptide sequences PMI-2 and PMI-3. To examine the influence of the non-peptidic linker structure on the functional properties of the resulting MOrPHs, a second linker reagent, SP8 (Figure 1A), was prepared (see ESI for details). SP8 satisfies the aforementioned distance requirements (Figure S1), but has higher flexibility compared to SP6 due to replacement of the triazole unit with an alkyl chain.
According to these design principles, macrocycles 3–5 and 7–9 were prepared via cyclization of PMI-2 or PM-3 target sequences with SP6 or SP8 (see ESI for details). 7–9 thus feature an i / i+10 side-chain-to-backbone connectivity, whereas in 3–5 the non-peptidic moiety bridges the i and i+6 residue. As controls, the same two peptide sequences were cyclized in the presence of the shorter reagent SP4 (Figure 1B). Since the spacing distance provided by SP4 (~8 Å, Figure S1) represents a mismatch with the target ones (13–16 Å), the resulting macrocycles (5 and 9) were intended to serve as negative control designs.
The ability of the designed macrocycles to disrupt the p53:HDM2/X interaction was assessed using a surface plasmon resonance (SPR) inhibition assay (Figure S2). Herein, biotinylated p53(15–29) was immobilized on a streptavidin-coated biosensor chip and increasing concentrations of inhibitors were added to a fixed concentration of HDM2 or HDMX. Using this assay, half-maximal inhibitory concentrations (IC50) were determined for the i / i + 6 macrocycles 3–5 and compared to those obtained for the corresponding acyclic 2 and for a linear peptide (1) corresponding to the Hdm2/X-binding domain in p53. Gratifyingly, these studies revealed that both 3 and 4 possess improved inhibitory activity as compared to the acyclic counterpart 2 (Figure S3), exhibiting an approximately 2-fold lower IC50 for HDMX (4) or for both HMD2 and HDMX (3) (Table 1). In contrast, SP4-based macrocycle 5 showed very weak inhibition (IC50 ≈ 10 μM). Thus, these initial data supported the ability of designer MOrPHs 3 and 4 to accommodate the target α-helical motif, a conclusion supported also by the poor activity of 5. The latter indeed highlighted the deleterious effect of a mismatch between the length of the synthetic linker and the target side-chain···C-terminus bridging distance as anticipated. To our disappointment, however, both SP6- and SP8-based macrocycles were weaker inhibitors of HDM2/X compared to the wild-type p53 sequence (Table 1). This result can be rationalized based on the negative effect of replacing Glu5 with pAcF as required for macrocyclization. Indeed, in the crystal structure of the HDM2:PMI complex, Glu5 is found to form a hydrogen bond network with the neighboring Ser2 (Figure S4), which is likely to contribute to α-helix stabilization.16 This conclusion is supported also by the much higher inhibitory activity of the linear peptide 6 (vs. 2), in which the Ser2/Glu5 pair is preserved (Table 1).
Table 1.
Sequence and inhibitory activity of peptides
| Name | Sequence | HDM2 IC50 (nM) | HDMX IC50 (nM) |
|---|---|---|---|
| 1(p5315–29) | SQETFSDLWKLLPEN | 920 ± 65 | 1,200 ± 110 |
| 2 | GTSFAYYWNLLA | 1,510 ± 95 | 7,500 ± 250 |
| 3 |
|
870 ± 53 | 4,100 ± 190 |
| 4 |
|
1,500 ± 115 | 3,500 ± 95 |
| 5 |
|
10,000 ± 400 | ND |
| 6 | GYSFAEYWNLLA | 65 ± 9 | 355 ± 31 |
| 7 |
|
475 ± 37 | 910 ± 105 |
| 8 |
|
110 ± 15 | 340 ± 44 |
| 9 |
|
>50,000 | >50,000 |
To our delight, the i / i + 10 macrocycles 7 and 8 exhibited significantly improved ability to disrupt p53 interaction with HMD2/X as compared to 3 and 4 (Figures 2A and S3). A notable effect of the type of synthetic linker on the binding properties of the corresponding MOrPH was also apparent. Notably, the SP4-containing 9 was found to possess negligible inhibitory activity against HDM2 or HDMX (IC50 > 50 μM), confirming that cyclization via the ‘mismatching’ SP4 strongly disfavored adoption of the bioactive α-helical conformation by the embedded PMI-3 peptide sequence. In stark contrast, much higher inhibitory activity was observed in the presence of the ‘distance-matching’ SP6, leading to a compound with sub-micromolar IC50 values for both protein homologues (Table 1). Interestingly, the simple replacement of the triazole unit in 7 with the alkyl chain in 8 led to a significant further improvement of inhibitory activity (3- to 4-fold) against both HDM2 (IC50 : 110 vs. 475 nM) and HDMX (IC50 : 340 vs. 910 nM). Intriguingly, the nature of the linker was found to have an effect also on the selectivity of the compounds against the two protein homologs. Indeed, while the unconstrained peptide 6 has stronger preference for HDM2 over HDMX, the macrocyclic counterparts, and in particular 7, behave more as dual, equipotent inhibitors (IC50(HDMX)/IC50(HDM2) = 5.5 vs.1.9). Overall, these studies led to the identification of macrocyclic inhibitors of the p53:HDM2/X interaction with much improved inhibitory activity compared to the wild-type p53 sequence, with the best compound, 8, exhibiting a 8- and 3.5-fold lower IC50 value in the presence of HDM2 and HDMX, respectively.
Figure 2.
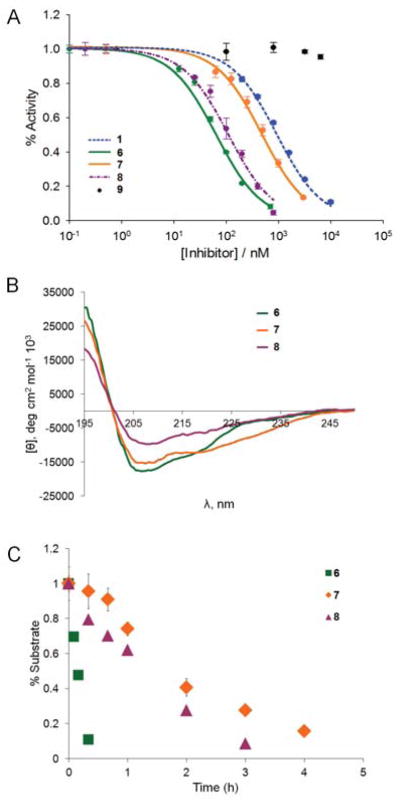
Characterization data for representative compounds of Table 1. (A) SPR-based inhibition curves corresponding to disruption of HDM2/p53 interaction; (B) Circular dichroism spectra measured in phosphate buffer (pH 7.0) with 40% TFE; (C) Proteolytic stability tests in the presence of chymotrypsin (1.0 μg / mL) at room temperature.
To examine the impact of macrocyclization on the peptide conformational properties, circular dichroism (CD) analyses were performed on the most potent compounds 7 and 8 as well as on the linear peptide 6 as a control (Figure 2B). Peptide 6 was found to display minima at 222 nm and 208 nm, which is consistent with the presence of an α-helical conformation. The α-helical content of the peptide was estimated to be about 31%. Cyclization of this sequence with SP6 (7) produced an increase in α-helicity (40%), whereas 8 showed a reduction in the α-helical content of the embedded peptide sequence (21%). The lack of a strict correlation between α-helicity and in vitro inhibitory activity has been observed for other types of p53:HDM2 inhibitors10c,11b and it is not entirely unexpected considering that additional factors can affect the binding properties of these compounds, including potential interactions of the linker moiety with the protein surface.16 Nevertheless, these experiments proved that a functional α-helical motif can be accommodated, and to some extent stabilized within the MOrPH scaffolds, thereby providing a proof-of-principle validation of the design strategy outlined in Figure 1B.
A potential benefit deriving from peptide macrocyclization is an enhancement in proteolytic stability. Despite its high potency in vitro, the linear peptide PMI was indeed found to be ineffective in cell-based assays in part due to rapid proteolysis.19 To assess this aspect, macrocycles 7 and 8, along with the linear peptide 6, were incubated in the presence of chymotrypsin (Figure 2C). Not surprisingly, 6 was found to undergo rapid proteolytic degradation, with the original peptide becoming undetectable after only 30 minutes. In contrast, the macrocyclic peptides 7 and 8 survived up to 3 and 4 hour incubation with the protease, respectively, exhibiting a 10- to 15-fold longer half-life compared to the acyclic counterpart. These data clearly showed the beneficial effect of the intramolecular linkage in imparting these compounds with increased resistance against proteolysis. It was also interesting to note how the linker SP6 provided superior performance in term of both α-helix stabilization and proteolytic resistance as compared to SP8, which may be linked to the reduced conformational flexibility of the former over the latter.
In summary, we have described the rational design of macrocyclic organo-peptide hybrids that can effectively accommodate and, to a certain extent, stabilize an α-helical protein binding motif. While a common approach in the area of α-helix stabilization has involved the use of inter-side-chain covalent linkages,4 this work represents, to the best of our knowledge, the first example of exploiting side-chain-to-tail peptide cyclization for this purpose. Using this strategy, submicromolar inhibitors of the p53:HDM2 interaction which display dual specificity against the HDMX isoform as well as increased proteolytic stability were obtained. Another intriguing aspect concerns the influence of the non-peptidic moiety in modulating the functional, conformational, and stability properties of these α-helical MOrPHs. These findings lay the ground for future efforts directed at leveraging this feature to further optimize these compounds and exploring the potential of the present approach toward disrupting other α-helix-mediated protein-protein interactions.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Science Foundation grant CHE-1112342. J.M.S. acknowledges the NSF GRF program for financial support. MS instrumentation was supported by the U.S. National Science Foundation grants CHE-0840410 and CHE-0946653.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental and synthetic procedures, additional inhibition curves. See DOI: 10.1039/b000000x/
Notes and references
- 1.a) Wells JA, McClendon CL. Nature. 2007;450:1001. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]; b) Smith MC, Gestwicki JE. Expert Rev Mol Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Robinson JA, Demarco S, Gombert F, Moehle K, Obrecht D. Drug Discov Today. 2008;13:944. doi: 10.1016/j.drudis.2008.07.008. [DOI] [PubMed] [Google Scholar]; b) Whitby LR, Boger DL. Acc Chem Res. 2012;45:1698. doi: 10.1021/ar300025n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jochim AL, Arora PS. Mol Biosyst. 2009;5:924. doi: 10.1039/b903202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henchey LK, Jochim AL, Arora PS. Curr Opin Chem Biol. 2008;12:692. doi: 10.1016/j.cbpa.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Liao W, Arora PS. Angew Chem Int Ed. 2005;44:6525. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG. J Am Chem Soc. 1991;113:9391. [Google Scholar]
- 7.Osapay G, Taylor JW. J Am Chem Soc. 1992;114:6966. [Google Scholar]
- 8.Brunel FM, Dawson PE. Chem Commun. 2005:2552. doi: 10.1039/b419015g. [DOI] [PubMed] [Google Scholar]
- 9.a) Scrima M, Le Chevalier-Isaad A, Rovero P, Papini AM, Chorev M, D’Ursi AM. Eur J Org Chem. 2010:446. [Google Scholar]; b) Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang S. J Med Chem. 2012;55:1137. doi: 10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Blackwell HE, Grubbs RH. Angew Chem Int Ed. 1998;37:3281. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; b) Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891. [Google Scholar]; c) Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. Cancer Cell. 2010;18:411. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Zhang FZ, Sadovski O, Xin SJ, Woolley GA. J Am Chem Soc. 2007;129:14154. doi: 10.1021/ja075829t. [DOI] [PubMed] [Google Scholar]; b) Muppidi A, Wang Z, Li X, Chen J, Lin Q. Chem Commun. 2011;47:9396. doi: 10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jo H, Meinhardt N, Wu YB, Kulkarni S, Hu XZ, Low KE, Davies PL, DeGrado WF, Greenbaum DC. J Am Chem Soc. 2012;134:17704. doi: 10.1021/ja307599z. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Spokoyny AM, Zou Y, Ling JJ, Yu H, Lin YS, Pentelute BL. J Am Chem Soc. 2013;135:5946. doi: 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Smith JM, Vitali F, Archer SA, Fasan R. Angew Chem Int Ed. 2011;50:5075. doi: 10.1002/anie.201101331. [DOI] [PubMed] [Google Scholar]; b) Satyanarayana M, Vitali F, Frost JR, Fasan R. Chem Commun. 2012;48:1461. doi: 10.1039/c1cc13533c. [DOI] [PubMed] [Google Scholar]; c) Frost JR, Vitali F, Jacob NT, Brown MD, Fasan R. Chembiochem. 2013;14:147. doi: 10.1002/cbic.201200579. [DOI] [PubMed] [Google Scholar]
- 13.a) Marine JC, Dyer MA, Jochemsen AG. J Cell Sci. 2007;120:371. doi: 10.1242/jcs.03362. [DOI] [PubMed] [Google Scholar]; b) Wahl GM, Wade M. Mol Cancer Res. 2009;7:1. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. J Biol Chem. 2006;281:33030. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]; b) Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. J Biol Chem. 2006;281:33036. doi: 10.1074/jbc.M605405200. [DOI] [PubMed] [Google Scholar]
- 15.Popowicz GM, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, Holak TA. Cell Cycle. 2007;6:2386. doi: 10.4161/cc.6.19.4740. [DOI] [PubMed] [Google Scholar]
- 16.Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, Khoo KH, Choong ML, Lee MA, Yurlova L, Zolghadr K, Joseph TL, Verma CS, Lane DP. ACS Chem Biol. 2013;8:506. doi: 10.1021/cb3005148. [DOI] [PubMed] [Google Scholar]
- 17.a) Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]; b) Popowicz GM, Czarna A, Holak TA. Cell Cycle. 2008;7:2441. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 18.a) Fasan R, Dias RL, Moehle K, Zerbe O, Vrijbloed JW, Obrecht D, Robinson JA. Angew Chem Int Ed Engl. 2004;43:2109. doi: 10.1002/anie.200353242. [DOI] [PubMed] [Google Scholar]; b) Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J Am Chem Soc. 2004;126:9468. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]; c) Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew Chem Int Ed. 2005;44:2704. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]; d) Fasan R, Dias RL, Moehle K, Zerbe O, Obrecht D, Mittl PR, Grutter MG, Robinson JA. Chembiochem. 2006;7:515. doi: 10.1002/cbic.200500452. [DOI] [PubMed] [Google Scholar]; e) Hara T, Durell SR, Myers MC, Appella DH. J Am Chem Soc. 2006;128:1995. doi: 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]; f) Murray JK, Gellman SH. Biopolymers. 2007;88:657. doi: 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]; g) Li C, Liu M, Monbo J, Zou G, Yuan W, Zella D, Lu WY, Lu W. J Am Chem Soc. 2008;130:13546. doi: 10.1021/ja8042036. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Henchey LK, Porter JR, Ghosh I, Arora PS. Chembiochem. 2010;11:2104. doi: 10.1002/cbic.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pazgier M, Liu M, Zou G, Yuan W, Li C, Li J, Monbo J, Zella D, Tarasov SG, Lu W. Proc Natl Acad Sci USA. 2009;106:4665. doi: 10.1073/pnas.0900947106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.