

Abstract
Deuterium-labeling studies carried out in conjunction with investigations into the directed catalytic asymmetric hydroboration of unsaturated oxime ethers reveal a surprisingly facile ortho-metallation or σ-bond metathesis pathway that that diverts the expected course of CAHB to a tandem C-H activation/hydroboration reaction pathway.
Keywords: directed metallation, C-H activation, rhodium-catalyzed borylation, rhodium-catalyzed hydroboration
Introduction
The catalytic asymmetric hydroboration (CAHB) of alkenes has enjoyed a long history largely focused on the successful reactions of vinyl arene substrates and a recent resurgence in interest for the preparation of chiral organoboronates.1,2 We have developed efficient carbonyl-directed CAHBs of unsaturated amides and esters capable of chelating to Rh(I) through a two-point binding mechanism3 and shown that the presence of an aryl substituent on the alkene is not required to obtain high levels of enantioselectivity.4,5 For example, rhodium-catalyzed CAHBs of the β,γ-unsaturated amides and esters illustrated in Figure 1 lead, in the case of (E)- or (Z)-1,2-disubstituted and -trisubstituted alkenes 1, to enantioselective borylation at the β-carbon yielding the β-hydroxycarbonyl compounds 2 after oxidation. In the case of β,γ-methylidene derivatives 3, CAHB affords a γ-borylated intermediate which upon oxidation affords the γ–hydroxycarbonyl derivative 4. The successful catalyst system employs a cationic Rh(I) complex with a chiral BINOL- or TADDOL-derived phosphite or phosphoramidite (e.g., L1 and L2); enantioselectivities up to 99% ee have been obtained. Encouraged by the successful results in carbonyl-directed CAHB, we looked to explore the effectiveness of other potential directing groups. Inspired by the recent report of the oxime ether-directed asymmetric dioxygenation of alkenes6 and the novel structural features of oximes, the benzophenone-derived oxime 5a was prepared.
Figure 1.

Recent examples of carbonyl-directed CAHB serve as precedent for the anticipated oxime-directed CAHB.
Results and Discussion
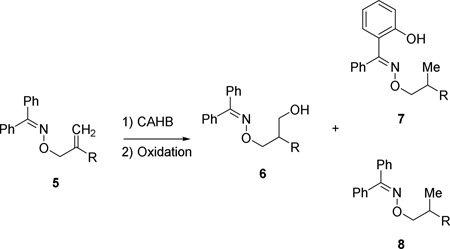
The β,γ-methylidene oxime ether 5a was subjected to rhodium-catalyzed CAHB under reaction conditions identical to those used for the structurally similar 1,1-disubstituted-β,γ-unsaturated N-phenyl amide 3 (R = Me, X = N(H)Ph) (2% [(L1)2Rh(nbd)]BF4, 2 equiv. tmdBH (B1), THF, rt). The starting material was completely consumed within 16 h, but after oxidative workup with NaBO3•monhydrate (1:3 water in THF), the expected γ-alcohol 6a was isolated in only 6% yield. Two reduced byproducts are formed in a roughly 2:1 ratio and account for approximately 90% of the mass recovered from the reaction. The minor of the two products, compound 8a, results from apparent hydrogenation of the alkene moiety. Its presence is not particularly surprising; a small amount of reduced alkene is a common byproduct in metal-catalyzed hydroborations. The 1H NMR spectrum of the major byproduct shows only nine aromatic hydrogens accompanied by a singlet at δ 11.2 ppm (integrates to one hydrogen); the latter is readily exchangeable upon treatment with D2O. High-resolution mass spectrometry and ultimately independent synthesis confirm the structure is the ortho-hydroxylated/alkene reduction product 7a. While we were unable to cleanly isolate its corresponding ortho-borylated intermediate, we hypothesize that 7a is formed via oxime-directed ortho-CH activation followed by borylation and conversion to the phenol upon oxidative workup. Alkene reduction must also occur at some point. We find no evidence for any doubly or poly-borylated products; for example, the ortho-and γ-borylated product compound 9 is not found.
Although we had not anticipated ortho-metallation under the conditions of rhodium-catalyzed CAHB, oximes are increasingly finding use as direction groups for ortho-metallation reactions in other contexts; reactions leading to ortho-acetoxylation, ortho-amidation and carbon-carbon bond formation have been recently reported (Figure 2). For example, Sanford and Dong reported the palladium-catalyzed oxime-directed oxidation of unactivated sp3 and sp2 C-H bonds.7 The reaction uses Pd(OAc)2 (5 mol%) as the catalyst precursor in an HOAc/Ac2O reaction medium at elevated temperatures (80–100 °C). Several groups have recently reported the use of (Cp*)Rh(III) complexes to catalyze the directed ortho-metallation of oxime ethers for the formation of C−N bonds.8 For example, Li and coworkers reported the oxime ether-directed ortho-amidation by N-hydroxycarbamates.8a
Figure 2.

Recent examples of the use of oximes and oxime ethers as directing groups for ortho-metallation; note Cp* = Me5Cp.
Oxime ether-directed ortho-metallation has also been utilized as a C-C bond forming cross-coupling strategy by a number of research groups. For example, as illustrated in Figure 2, Yu and coworkers reported the oxime-directed (Cp*)Rh(III)-catalyzed ortho-C−H cross-coupling with diazomalonates.9 In other work, Li10 and Cheng11 reported the oxime-directed alkynylation of arenes. In Cheng’s work the presence of both ortho-hydroxy and ortho-oxime groups lead to dual-directed C-H activation followed by cyclization and formation of highly substituted benzofurans. Organopalladium intermediates, generated by oxime-directed C-H activation, have also been intercepted by with aryl or acyl radicals to effect C−C bond formation.12
In spite of the ample precedents for the use of oxime ethers as directing groups for ortho-metallation, the formation of 7 under the conditions of CAHB is unusual in some respects. For example, in each of the rhodium-catalyzed ortho-metallations cited above, the reactions are run at elevated temperatures under mildly oxidizing conditions, and the catalyst precursor is a (Cp*)Rh(III) complex; Rh(I)-catalysts are relatively rarely used.13 In contrast, the formation of 7a arises from a Rh(I) precatalyst under reducing conditions.
The yield of 7a varies widely as a function of the ligand (e.g., L1 and L2) and borane (e.g., (±)-B1 (tmdBH), pinacolborane (pinBH, B2), or catecholborane (catBH, B3)) employed (Table 1). For example, the reaction using [(L1)2Rh(nbd)]BF4 with tmdBH (B1) afforded ortho-hydroxylated 7a as the major product (Table 1, entry 1). In contrast, the reaction using [(L2)2Rh(nbd)]BF4 under the same conditions gave γ-alcohol 6 as the major product (58%) although 7a is also formed in significant amounts (31%) (Table 1, entry 2). Electronically and sterically similar boranes tmdBH (B1) and pinBH (B2) give substantially different product distributions. Although the ratio of 7a:8a is again about 2:1, pinBH affords in addition, an isomer of 6, the β-hydroxylated product (16%) (Table 1, entry 3). The more electron deficient and relatively more reactive borane, catBH (B3), again gives a very different product mixture. Little alkene reduction is observed (approximately 7% in total) and product 7a is isolated in only trace amounts (ca 1% yield). Overall, γ-alcohol 6a (63%) is the predominant product using catBH, although the product obtained is nearly racemic (Table 1, entry 4).
Table 1.
The nature of the ligand and borane affect the distribution of borylated products and their derived alcohols.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | 5 R = | L | borane | 6 (er) | 7 | 8 |
| 1 | a Me | L1 | tmdBH | 6 (56:44) | 56 | 30 |
| 2 | a Me | L2 | tmdBH | 58 (67:34) | 31 | 10 |
| 3b | a Me | L2 | pinBH | 30 (56:44) | 16 | 24 |
| 4 | a Me | L2 | catBH | 63 (53:47) | 1 | 6 |
| 5 | b iBu | L2 | tmdBH | 52 (80:20) | 29 | 16 |
| 6 | b iBu | L2 | catBH | 16 (50:50) | 0 | 0 |
| 7 | c cC6H11 | L2 | tmdBH | 60 (70:30) | 11 | 28 |
| 8 | c cC6H11 | L2 | catBH | 52 (85:15) | 2 | 5 |
Reaction conditions: 2% [(L)2Rh(nbd)]BF4, 2 equiv. borane, THF, rt.
16% of the isomeric β-hydroxy isomer also formed.
In related studies of the carbonyl-directed CAHB, the nature of the alkene substituent proved important. Increasing the size of the substituent generally increased the yield of the desired γ-borylated products. In contrast, the bulkier substituents have little effect on the observed product distribution in the present case (Table 1, entries 5–8). Increasing the size of the substituent does lead to a modest increase in the enantiomeric ratio (er) for the CAHB reactions with L2 and tmdBH (B1). However, this may simply be the consequence of more easily differentiating the prochiral faces of the reacting alkene due the greater difference in bulk of the substituents.
Following the distribution of products obtained over the course of the reaction using the [(L2)2Rh(nbd)]BF4 catalyst precursor with tmdBH (B1) gives useful insight into the reaction (Figure 2). Employing slightly modified reaction conditions for this experiment, the reaction mixture was stirred at 0 °C for 30 minutes after complete addition of borane, and then allowed to warm to room temperature; aliquots were removed, quenched by oxidative workup and the product distributions shown graphically in Figure 3. The majority of reduced product 8a is formed within the first 30 minutes at 0 °C, and then only slowly increases thereafter. Meanwhile, the γ-borylated product 6a and ortho-borylated product 7 are formed at a relatively constant rate and ratio over the course of the reaction. These results suggest that non-borylated product 8a is formed by an independent pathway while the γ-borylated and ortho-borylated products, 6a and 7, diverge from a common intermediate. Furthermore, resubjecting isolated 8a to the standard reaction conditions does not lead to the formation of 7a; 8a is recovered unchanged. In light of the concurrent formation of γ-borylated product 6a and ortho-borylated product 7a, we prepared deuterium-labelled d10-10 to follow the fate of the hydrogen involved in apparent ortho-metallation. The reaction of d10-10 under conditions used previously (2 mol% [(L2)2Rh(nbd)]BF4 and 2 equiv tmdBH (B1)) yields surprising results. Ignoring for the moment the precise isotope distribution, the labeled and unlabeled substrates give virtually identical product distributions (see Figure 4 vs Table 1, entry 2). However, it was surprising to find evidence for ortho-metallation and H/D-exchange not only for the ortho-hydroxylated product but for the products of all three reaction modes, 11–13!
Figure 3.

a) Following the product distribution over time indicates that the non-borylated product 8a is formed in the early stages of reaction while 6a and 7a are formed more or less in parallel. b) Viewing the data another way, the percent composition of the isolated reaction products over the allotted time normalized to 100 percent highlights the rapid formation of 8 at the initial stages of the reaction.
Figure 4.

The redistribution of deuterium shows that ortho-C-H activation is occurring in all reaction modes during the course rhodium-catalyzed CAHB.
Deuterated product 12, derived from ortho-borylation and alkene reduction, shows clean redistribution of one ortho-deuterium to the terminus of what was the methylidene group (i.e., the methyl group in the product) with apparent incorporation of boron in the ortho-position; the latter replaced by OH upon oxidative workup. We see evidence for partial H/D exchange at the other ortho-position of the same phenyl substituent resulting in approximately 60% proton incorporation at that position. However, we see no evidence for H/D-exchange on the other oxime phenyl substituent. For reduced product 13, one ortho-deuterium is replaced by hydrogen virtually quantitatively, and the deuterium is again cleanly incorporated into a methyl group in 13.
One ortho-deuterium atom is also redistributed in the course CAHB leading to the major product 11. Quantitative substitution of one deuterium by hydrogen at the ortho-position of the aromatic ring is accompanied by its complete transfer to the propenyl moiety affording a 50:50 mixture of 11a and 11b. One possible explanation that accounts for distribution of deuterium to both carbons is a tandem two-step dehydrogenative borylation and reduction.14
The nature of the borane has a significant impact on the propensity to ortho-C–H activation. Treating d10-10 with 2 mol% [((L2)2Rh(nbd)]BF4 and 2 equivalents catBH (B3) in place of tmdBH (B1) affords γ-alcohol d10-14. While the yield is low (20%), no H/D exchange is observed in either d10-14 or the recovered starting material (Figure 5). Given the relatively low conversion when using catBH in lieu of tmdBH, we considered that the lack of ortho-H/D exchange could be simply due to the background reaction of catBH in a non-catalyzed hydroboration of the alkene rather than rhodium-catalyzed hydroboration. However, this proved not to be the case. Attempted hydroboration using catBH in the absence of a rhodium catalyst resulted in the isolation of only trace amounts of d10-14 after oxidative workup.
Figure 5.

catBH (B3) does not promote H/D exchange under the conditions of CAHB, and the labeled reduced product d10-13 does not undergo ortho-metallation-H/D exchange or conversion to 11 or 12.
It is important to note that the redistribution of the ortho-deuterium, now inferred to occur in 5 as well, is completely transparent in the proteo substrate. A simple explanation that could potentially account for the quantitative exchange of the ortho-deuterium is that of an unexpectedly facile oxime-directed ortho-metallation and H/D-exchange. To further explore this possibility, d10-13 was independently synthesized and subjected to standard CAHB conditions. No H/D exchange is observed and d10-13 is not converted to either 11 or 12 under the reaction conditions (Figure 5). Since ortho-H/D exchange was essentially quantitative in all the observed CAHB products (Figure 4) and yet does not occur in reduced byproduct d10-13 (Figure 5), H/D exchange must occur only from intermediates generated in the course of the alkene reacting.
While the mechanistic details are not yet clear, based on our previous mechanistic studies,3 we propose the model in Figure 6 to account for the ligand and borane dependent formation of the ortho-borylated/alkene reduction product 12. The substrate presumable chelates rhodium forming a complex such as 15. Oxidative addition of borane would generate a Rh(III) intermediate such as 16 suitably disposed for alkene insertion into the Rh-H bond affording a coordinately unsaturated intermediate such as 17. Reductive elimination to form the C-B bond is the expected CAHB pathway that would lead, and in the case of catBH does lead (Figure 5), to a γ-borylated intermediate and ultimately to alcohol d10-14 after oxidation.
Figure 6.

A model involving σ-bond metathesis to account for facile ortho-borylation and deuterium scrambling in the course of CAHB of d10-10.
We speculate that if reductive elimination from 17 is slow, the interchange of H/D with boron, whether by formal Rh(III) to Rh(V) oxidative addition (i.e., ortho-metallation),15 σ-bond metathesis, or a σ-cam mechanism,16 may compete favorably to form an intermediate such as 18. While the analogy is somewhat indirect, this type of “exchange” during Rh-CAHB has been previously reported for the Rh-catalyzed addition of CCl4 to alkenes using Wilkinson’s catalyst and pinBH (B2).17 Reductive elimination from 18 would release 19 that upon oxidative workup would account for the formation of the ortho-borylated/alkene reduction product 12. A similar sequence, perhaps involving diastereomers of 16 and/or competing alkene insertion into the Rh-B bond, can be envisioned to account for ortho-H/D exchange en route to the formation of 11. While the formation of a mixture of deuterated isomers 11a and 11b is an additional puzzling aspect, a well-documented dehydrogenative borylation mechanism may be involved to account for the deuterium scrambling.14
Conclusions
In summary, oxime ether 5 undergoes rhodium-catalyzed CAHB with tmdBH, pinBH and catBH to afford the expected products of hydroboration and, in the case of tmdBH and pinBH, an unexpected ortho-borylated product (Table 1, entries 1–4). Deuterium-labeling studies suggest that H/D-exchange at the ortho-position must be associated with hydroboration of the β,γ-alkene, and that ortho-metallation or σ-bond metathesis is exceptionally facile leading to a tandem C-H activation/hydroboration reaction. Studies exploring the use of other directing groups for directed CAHB are currently underway.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support for these studies from the NIH (GM100101) and from the NSF (CHE-0091975, MRI-0079750) and NIH (SIG-1-510-RR-06307) for the NMR spectrometers used in these studies carried out in facilities renovated under NIH RR016544. We thank M. P. Takacs for carrying out some early experiments synthesizing the oxime ether substrates and S. M. Smith for some early exploratory investigations into the CAHB of oxime ether substrates.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Experimental procedures and spectral characterization are available free of charge via the Internet at http://____________.
References and notes
- 1.For review articles, please see: Carroll AM, O’Sullivan TP, Guiry PJ. Adv. Synth. Catal. 2005;347:609–631. Crudden CM, Edwards D. Eur. J. Org. Chem. 2003;24:4695–4712.
- 2.(a) He Z, Zhao Y, Tian P, Wang C, Dong H, Lin G. Org. Lett. 2014;16:1426–1429. doi: 10.1021/ol500219e. [DOI] [PubMed] [Google Scholar]; (b) Feng X, Jeon H, Yun J. Angew. Chem. Int. Ed. 2013;52:3989–3992. doi: 10.1002/anie.201208610. [DOI] [PubMed] [Google Scholar]; (c) Moteki SA, Toyama K, Liu Z, Ma J, Holmes AE, Takacs JM. Chem. Commun. 2012;48:263–265. doi: 10.1039/c1cc16146f. [DOI] [PubMed] [Google Scholar]; (d) Noh D, Yoon SK, Won J, Lee JY, Yun J. Chem. Asian J. 2011;6:1967–1969. doi: 10.1002/asia.201100146. [DOI] [PubMed] [Google Scholar]; (e) Noh D, Chea H, Ju J, Yun J. Angew. Chem. Int. Ed. 2009;48:6062–6064. doi: 10.1002/anie.200902015. [DOI] [PubMed] [Google Scholar]; (f) Moteki SA, Takacs JM. Angew. Chem. Int. Ed. 2008;47:894–897. doi: 10.1002/anie.200703127. [DOI] [PubMed] [Google Scholar]; (g) Moteki SA, Wu D, Chandra KL, Reddy DS, Takacs JM. Org. Lett. 2006;8:3097–3100. doi: 10.1021/ol061117g. [DOI] [PubMed] [Google Scholar]; h) Kwong FY, Yang QT, Mak CW, Chan ASC, Chan KS. J. Org. Chem. 2002;67:2769–2777. doi: 10.1021/jo0159542. [DOI] [PubMed] [Google Scholar]; (i) Köllner C, Togni A. Can. J. Chem. 2001;79:1762–1774. [Google Scholar]; (j) Schnyder A, Hintermann L, Togni A. Angew. Chem. Int. Ed. 1995;34:931–933. [Google Scholar]; (k) Togni A, Breutel C, Schnyder A, Spindler F, Landert H, Tijani A. J. Am. Chem. Soc. 1994;116:4062–4066. [Google Scholar]; (l) Hayashi T, Matsumoto Y, Ito Y. J. Am. Chem. Soc. . 1989;111:3426–3428. [Google Scholar]
- 3.Yang Z, Pal R, Hoang GL, Zeng XC, Takacs JM. ACS Catal. 2014;4:763–773. doi: 10.1021/cs401023j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Smith SM, Thacker NC, Takacs JM. J. Am. Chem. Soc. 2008;130:3734–3735. doi: 10.1021/ja710492q. [DOI] [PubMed] [Google Scholar]; (b) Smith SM, Takacs JM. J. Am. Chem. Soc. 2010;132:1740–1741. doi: 10.1021/ja908257x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith SM, Uteuliyev M, Takacs JM. Chem. Commun. 2011;47:7812–7814. doi: 10.1039/c1cc11746g. [DOI] [PubMed] [Google Scholar]
- 5.Smith SM, Hoang GL, Pal R, Khaled MOB, Pelter LSW, Zeng XC, Takacs JM. Chem. Commun. 2012;48:12180–12182. doi: 10.1039/c2cc36199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neufeldt SR, Sanford MS. Org. Lett. 2013;15:46–49. doi: 10.1021/ol303003g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ren Z, Mo F, Dong G. J. Am. Chem. Soc. 2012;134:16991–16994. doi: 10.1021/ja3082186. [DOI] [PubMed] [Google Scholar]; (b) Kubota A, Sanford MS. Synthesis. 2011:2579–2589. doi: 10.1055/s-0030-1260087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Neufeldt SR, Sanford MS. Org. Lett. 2010;12:532–535. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Desai LV, Stowers KJ, Sanford MS. J. Am. Chem. Soc. 2008;130:13285–13293. doi: 10.1021/ja8045519. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Desai LV, Malik HA, Sanford MS. Org. Lett. 2006;8:1141–1144. doi: 10.1021/ol0530272. [DOI] [PubMed] [Google Scholar]; (f) Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 8.(a) Zhou B, Du J, Yang Y, Feng H, Li Y. Org. Lett. 2014;16:592–595. doi: 10.1021/ol403477w. [DOI] [PubMed] [Google Scholar]; (b) Yu S, Wan B, Li X. Org. Lett. 2013;15:3706–3709. doi: 10.1021/ol401569u. [DOI] [PubMed] [Google Scholar]; (c) Ng K, Zhou Z, Yu W. Org. Lett. 2012;14:272–275. doi: 10.1021/ol203046n. [DOI] [PubMed] [Google Scholar]
- 9.Chan W, Lo S, Zhou Z, Yu W. J. Am. Chem. Soc. 2012;134:13565–13568. doi: 10.1021/ja305771y. [DOI] [PubMed] [Google Scholar]
- 10.Xie F, Qi Z, Yu S, Li X. J. Am. Chem. Soc. 2014;136:4780–4787. doi: 10.1021/ja501910e. [DOI] [PubMed] [Google Scholar]
- 11.Yeh C, Chen W, Gandeepan P, Hong Y, Shih C, Cheng C. Org. Biomol. Chem. 2014;12:9105–9108. doi: 10.1039/c4ob01876a. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yu W, Sit WN, Zhou Z, Chan AS. Org. Lett. 2009;11:3174–3177. doi: 10.1021/ol900756g. [DOI] [PubMed] [Google Scholar]; (b) Chan C, Zhou Z, Chan AS, Yu W. Org. Lett. 2010;12:3926–3929. doi: 10.1021/ol101618u. [DOI] [PubMed] [Google Scholar]
- 13.N-Phenyl amides have been reported to direct C-H activation. Huckins JR, Bercot EA, Thiel OR, Hwang T, Bio MM. J. Am. Chem. Soc. 2013;135:14492–14495. doi: 10.1021/ja405140f. Schroeder N, Wencel-Delord J, Glorius F. J. Am. Chem. Soc. 2012;134:8298–8301. doi: 10.1021/ja302631j. Hesp KD, Bergman RG, Ellman JA. Org. Lett. 2012;14:2304–2307. doi: 10.1021/ol300723x. Quinones N, Seoane A, Garcia-Fandino R, Mascarenas JL, Gulias M. Chem. Sci. 2013;4:2874–2879.
- 14.(a) Caballero A, Sabo-Etienne S. Organometallics. 2007;26:1191–1195. [Google Scholar]; (b) Brown JM, Lloyd-Jones GC. J. Am. Chem. Soc. 1994;116:866–878. [Google Scholar]; (c) Westcott SA, Marder TB, Baker RT. Organometallics. 1993;12:975–979. [Google Scholar]; (d) Geier MJ, Vogels CM, Decken A, Westcott SA. J. Organomet. Chem. 2009;694:3154–3159. [Google Scholar]
- 15.(a) Vyboishchikov SF, Nikonov GI. Organometallics. 2007;26:4160–4169. [Google Scholar]; (b) Taw FL, Bergman RG, Brookhart M. Organometallics. 2004;23:886–890. [Google Scholar]; (c) Cook KS, Incarvito CD, Webster CE, Fan Y, Hall MB, Hartwig JF. Angew. Chem. Int. Ed. 2004;43:5474–5477. doi: 10.1002/anie.200460430. [DOI] [PubMed] [Google Scholar]; (d) Nagashima H, Tatebe K, Ishibashi T, Nakaoka A, Sakakibara J, Itoh K. Organometallics. 1995;14:2868–2879. [Google Scholar]; (e) Duckett SB, Perutz RN. J. Chem. Soc. Chem. Commun. 1991:28–31. [Google Scholar]; (f) Duckett SB, Haddleton DM, Jackson SA, Perutz RN, Poliakoff M, Upmacis RK. Organometallics. 1988;7:1526–1532. [Google Scholar]; (g) Fernandez MJ, Bailey PM, Bentz PO, Ricci JS, Koetzle TF, Maitlis PM. J. Am. Chem. Soc. 1984;106:5458–5463. [Google Scholar]
- 16.(a) Perutz RN, Sabo-Etienne S. Angew. Chem. Int. Ed. 2007;46:2578–2592. doi: 10.1002/anie.200603224. [DOI] [PubMed] [Google Scholar]; (b) Balcells D, Clot E. Chem. Rev. 2010;110:749–823. doi: 10.1021/cr900315k. [DOI] [PubMed] [Google Scholar]
- 17.Pereira S, Srebnik M. J. Am. Chem. Soc. 1996;118:909–910. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.