

Abstract
Oligomers incorporating the tetrapeptide MSH4, the minimum active sequence of melanocyte stimulating hormone, were synthesized by an A2 + B2 strategy involving microwave-assisted copper-catalyzed azide-alkyne cycloaddition. A2 contained an MSH4 core while B2 contained a (Pro-Gly)3 spacer. Soluble mixtures containing compounds with up to eight MSH4 units were obtained from oligomerizations at high monomer concentrations. The avidities of several oligomeric mixtures were evaluated by means of a competitive binding assay using HEK293 cells engineered to overexpress the melanocortin 4 receptor. When based on total MSH4 concentrations, avidities were only minimally enhanced compared with a monovalent control. The lack of variation in the effect of ligands on probe binding is consistent with high off rates for MSH4 in both monovalent and oligomeric constructs relative to that of the competing probe.
Keywords: Oligomerization, Multivalent ligand, Melanocortin 4 receptor, Time-resolved fluorescence, Competitive binding assay
1. Introduction
Melanocortin receptors (MCRs) belong to class A of the GPCR super family and consist of five subtypes, including MC1R which is overexpressed by a majority of melanoma cancer cells.1,2 Endogenous agonists for the MCRs are the melanocortin peptides, all of which contain the sequence His-Phe-Arg-Trp, the minimum structure required for MCR activation.3 Thus, differential binding of melanocortin peptides bearing reporter groups or therapeutic agents to cells overexpressing MC1R could provide a strategy for early detection and treatment of this disease.4
Nature often uses multivalent binding to increase avidity by means of simultaneous low affinity ligand/receptor pairings.5 This strategy has also been employed to engineer biologically useful compounds.6 In designing synthetic multivalent molecules, the distances between the binding domains of the targeted receptors are key parameters. Evidence suggests that melanocortin receptors, including MC1R and MC4R, form constitutive dimers on cell surfaces.7–11 A homology model based on the crystal structure of rhodopsin estimated the distance between the binding sites of abutted receptors at 20–50 Å, depending on the relative orientations of the receptors.12 If true, melanocortin ligand/receptor systems offer excellent opportunities for application of multivalent binding to the preparation of selective imaging and therapeutic agents.
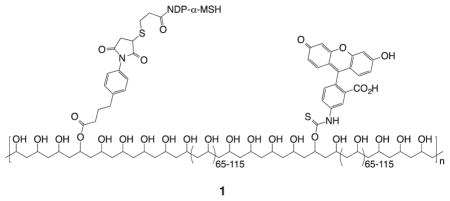
Targeting of melanocortin receptors for multivalent binding was first reported in 1981 when tobacco mosaic virus was decorated with about 500 molecules of an α-melanotropin analogue.13,14 In 1994 poly(vinyl alcohol) scaffold 1 bearing multiple copies of a fluorescent marker and copies of the highly potent tridecapeptide Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2 (NDP-α-MSH) was observed to bind specifically and irreversibly to melanocortin receptors on mouse and human melanoma cells.15 Although the distances between adjacent ligands were probably quite large (coverage was approximately one ligand per 200 monomer units), the observed specificity and irreversibility were attributed to multivalent binding. A substantial loss of entropy should disfavor binding of ligands separated by a long tether to adjacent sites on receptor dimers.16,17 However, the residence half-life of NDP-α-MSH bound to hMC1R is greater than eight hours,18 time enough for additional ligands on the surface-attached polymer to find and bind to more remote melanocortin receptors.
In the subsequent twenty years, multivalent molecules with relatively low molecular weights that incorporate melanocortin peptides have been reported.19 We have described multivalent constructs based on linear,20–23 spherical,24 and trigonal25 scaffolds, some bearing variable ligand numbers and with variable ligand spacing. Much of this work (and the work of others26–28) has focused on incorporation of the weakly binding tetrapeptide His-DPhe-Arg-Trp (MSH4) since cooperative binding leading to enhanced avidity is more evident when weakly binding ligands are utilized.29,30
In the present work, we describe a method for rapid assembly of multiple copies of the MSH4 ligand to produce mixtures of oligomeric multivalent compounds. Multivalent oligomers are less well studied than multivalent small molecules and polymers, but may offer advantages with respect to in vivo distribution and metabolism.31 Because modifications of both the N- and C-termini of MSH4 are generally possible without loss of activity,3 the sequence His-DPhe-Arg-Trp was incorporated in the main chain of our construct. Copper-catalyzed azide-alkyne cycloaddition (CuACC)32 was chosen for oligomerization because of the orthogonality of CuACC chemistry with typical peptide functional and protecting groups.33–36 Finally, an A2 + B2 assembly strategy was selected over an AB strategy in order that the oligomer structure might be more easily altered (by changing out the B2 co-monomer) and to limit macrocyclization.34,37
2. Synthesis and Oligomerization of the A2 and B2 Co-monomers
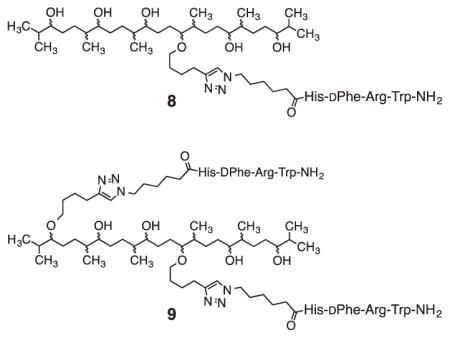
The A2 and B2 co-monomers were prepared by solution-phase peptide synthesis as described in the Supplementary Data that accompanies this article. Solution-phase synthesis was used so that multigram quantities of the co-monomers might be produced. The A2 co-monomer 2 (see Scheme 1) incorporates the MSH4 tetrapeptide, His-DPhe-Arg-Trp,38 with the side chains unprotected. The A2 co-monomer 3 (not shown) incorporates MSH4 with the side chains protected.39 The B2 co-monomer 4 incorporates the semi-rigid hexapeptide (Pro-Gly)3, which has proven useful in previous multivalent constructs involving melanocortin ligands.12,40,41
Scheme 1.

CuAAC Reaction of 2 with 4 to Produce Oligomers 5.
Oligomerization of 2 with 4 by microwave-assisted CuAAC is depicted in Scheme 1. All reactions were carried out using tetrakis(acetonitrile)copper(I) hexafluorophosphate (TACP) and tris(benzyltriazolylmethyl)amine (TBTA) in degassed DMF in a microwave reactor. Oligomerization of 3 with 4 was conducted similarly, with subsequent side chain deprotection under standard conditions.39 Results from oligomerization runs are given in Table 1. Specific reaction and workup procedures for each run are described in the Supplementary Data. Product oligomeric mixtures 5 were analyzed by MALDI-TOF mass spectrometry. Mass plots and peak identities for the product mixtures from runs 1, 5, and 10 appear in Figure 1. Mass plots and peak identities for the remaining runs are given in the Supplementary Data.
Table 1.
Summary of Oligomerization Runs.
| Run | Monomers | Monomer Conc.a (mM) | Reaction | Yieldb (%) | |
|---|---|---|---|---|---|
| Time (min) | Temp. (°C) | ||||
| 1 | 3 + 4 | 18 | 120 | 100 | 71 |
| 2 | 3 + 4 | 36 | 120 | 100 | 29 |
| 3 | 3 + 4 | 180 | 120 | 100 | 0c |
| 4 | 3 + 4 | 180 | 60 | 100 | 47d |
| 5 | 3 + 4 | 180 | 30 | 100 | 36 |
| 6 | 3 + 4 | 180 | 30 | 100 | 43 |
| 7 | 2 + 4 | 180 | 20 | 100 | 71 |
| 8 | 2 + 4 | 180 | 30 | 100 | 95 |
| 9 | 2 + 4 | 180 | 30 | 150e | 70 |
| 10 | 2 + 4 | 180 | 40 | 100 | 71 |
| 11 | 2 + 4 | 180 | 80 | 100 | 71 |
Concentration of each monomer.
Yield after deprotection (Runs 1–6), workup, and lyophylization (see the Supplementary Data for details).
Intractable product.
Partially soluble product.
Reaction mixture turned brown.
Figure 1.

Mass plots and peak identities from MALDI-TOF analyses of oligomeric mixtures 5 from run 1 (top), run 5 (middle), and run 10 (bottom). The A2 co-monomer incorporates the MSH4 tetrapeptide ligand and is represented by A in the oligomer structures in this figure. The B2 co-monomer incorporates the (Pro-Gly)3 spacer and is represented by B.
Oligomerization runs 1–6 used co-monomers 3 and 4. Run 1 employed relatively low monomer concentrations (18 mM) and, following deprotection, workup, and lyophilization, gave a good yield of a mixture of soluble oligomeric products 5. Run 3, with ten-fold higher monomer concentrations (180 mM), produced an insoluble amorphous gel. Run 4 also employed 180 mM monomer concentrations, but only half the reaction time (60 minutes), and gave products of limited solubility. Halving the reaction time again to 30 minutes in run 5 gave a mixure of soluble oligomeric products 5. These results suggest that increasing the monomer concentrations and/or the reaction times increases the degree of oligomerization. The mass plots and peak identities from the MALDI-TOF analyses of the oligomer mixtures 5 from runs 1 and 5 are depicted in Figure 1. In each case oligomers are evident, with higher masses present in the mass plot from run 5. A peak for residual co-monomer A2 is observed in both runs in spite of the fact that (A2B2)n oligomers are present. Interestingly, peaks with two terminal A2 subunits appear, but no peaks with two terminal B2 subunits are evident. In the case of run 1, peaks for the A2B2 and A2B2A2B2 oligomers are most intense. These facts are suggestive of macrocyclic structures for the (A2B2)n oligomers. Macrocyclization is favored in AB and A2 + B2 polymerizations when monomer concentrations are low.34
Oligomerization runs 7–11 employed 180 mM concentrations of co-monomers 2 and 4. The yields of product were generally higher than those observed with co-monomers 3 and 4 in runs 1–6. Runs 7, 8, 10, and 11 were conducted at 100 °C for 20, 30, 40, and 80 minutes, respectively. The longer the time, the more viscous the reaction mixture became. MALDI-TOF analyses of the products from these runs confirmed that increasing the time of reaction increases the degree of oligomerization (see Supplementary Data). Run 9 was conducted at 150 °C for 30 minutes, during which time the reaction mixture became brown (the others had remained green). MALDI-TOF analysis of the product from run 9 indicated a lower level of oligomerization, possibly due to decomposition at the elevated temperature of the reaction.
The differences between the mass plots from runs 1–6 and 7–11 are noteworthy. The mass plot and peak identities from the MALDI-TOF analysis of the oligomeric mixture 5 from run 10 is also depicted in Figure 1. Peaks from (A2B2)n, (A2B2)nA2, and B2(A2B2)n oligomers are all in evidence (the latter were absent in runs 1–6). While the (A2B2)n oligomers may be linear, macrocyclic, or a mixture of both, the existence of (A2B2)nA2 and B2(A2B2)n oligomers guarantees that linear molecules are included in the mxture, and that further oligomerization was possible at the time this run was halted.
Oligomer mixtures 5 from runs 1, 5, and 10 were subjected to analysis by infrared spectroscopy (see Supplementary Data for spectra). The A2 co-monomer 2 exhibits very weak terminal alkyne stretching at 2115 cm−1, while the B2 co-monomer 4 exhibits strong azide stretching at 2098 cm−1. In the IR spectra of product 5 from runs 1 and 5, where alkyne end groups must exist and azide end groups will exist if the (A2B2)n oligomers are linear, peaks for terminal alkyne and azide stretching were absent. However, in the IR spectrum of product 5 from run 10, where both azide and alkyne end groups must exist, a peak for azide stretching was observed. Assuming that absorption due to terminal alkyne stretching was simply too weak to appear, these results are consistent with macrocyclic structures for the (A2B2)n oligomers in runs 1 and 5, linear and/or macrocyclic structures for the (A2B2)n oligomers in run 10, and the presence of B2(A2B2)n oligomers in the mixture of products in run 10.
To further delineate the structures of the oligomers in the product mixture from run 10, a method for end-group tagging was employed. Alkyne 6 (Scheme 2) containing the 7-nitrobenzo[c][1,2,5]oxadiazol-4-yl (NBD) moiety was synthesized as described in the Supplementary Data. Initially, a CuAAC reaction between 6 and 4 established suitable reaction conditions, which were then used for the reaction of the product mixture 5 from run 10 with an excess of 6 (Scheme 2). The product mixture 7 so obtained was analyzed by MALDI-TOF (see Supplementary Data). As expected, all the B2(A2B2)n peaks disappeared and instead the corresponding doubly tagged NBD-B2(A2B2)n-NBD peaks were observed. While (A2B2)n peaks remained in evidence, peaks corresponding to tagged A2B2-NBD, A2B2A2B2-NBD, and A2B2A2B2A2B2-NBD were also observed. These results confirm that (A2B2)n are not non-covalently bound dimers or trimers of A2B2, and indicate that at least some of the (A2B2)n products in run 10 were acyclic.
Scheme 2.
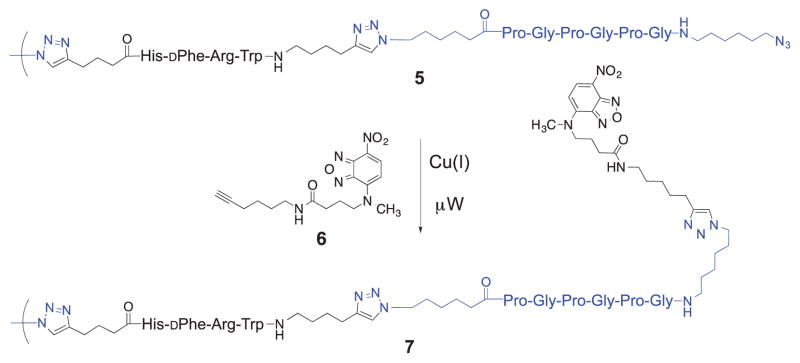
Azide end-group tagging with alkyne 6.
MALDI-TOF analyses can determine qualitatively the oligomers present in a given mixture, but we were concerned that they might be less useful in quantifying the relative amounts of these oligomers.42,43 To test this point, squalene-derived compounds 8 and 944 were mixed in a 1:1 molar ratio and the mixture subjected to MALDI-TOF analysis. As can be seen in Figure 2, the intensity of the higher mass peak due to compound 9 was greatly attenuated relative to the lower mass peak due to 8, evidence that the distributions of the oligomers 5 in mixtures from runs 1–11 are not well represented by the peak intensities in the MALDI-TOF mass spectra we had obtained. Since the oligomer size distributions and thus concentrations are imperfectly known, bioassay results are reported based on the total concentrations of MSH4 ligand in solution.
Figure 2.

Mass plot and peak identities from MALDI-TOF analysis of a 1:1 mixture of compounds 8 and 9.
3. Bioassays
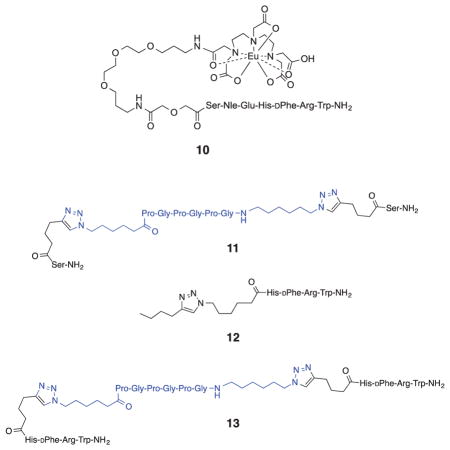
A time-resolved fluorescence (TRF) binding assay in which the MSH4 oligomers 5 from runs 1, 5, and 10 were competed against the probe Eu-DTPA-PEGO-MSH7-NH2 (10)44 for binding and uptake by genetically engineered45 HEK293 cells was used to characterize the bioactivity of these multivalent constructs. The HEK293 cells stably overexpress the human melanocortin 4 receptor (hMC4R) at 6.4×105 receptors per cell and the human cholecyctokinin 2 receptor (hCCK2R) at 1.1×106 receptors per cell.45 While this assay targets hMC4R, the melanoma-related receptor hMC1R has a similar pharmacophore.46 Compounds 11-13 were used as controls. Compounds 11 and 13 were prepared as described in the Supplementary Data. The competitive binding assays are described in detail in the Supplementary Data. Representative competitive binding curves are shown in Figure 3. Ki values are listed in Table 2 and, for oligomers 5 and control 13, were calculated based on the total MSH4 concentration. A Ki value for the monovalent MSH4 control compound 12 was previously reported.44
Figure 3.

Competitive binding curves for oligomers 5 from runs 1 (top), 5 (middle), and 10 (bottom).
Table 2.
Competitive binding of compounds 11, 12, 13, and oligomers 5 to hMC4R.
| Compound | Run | Kia (nM) | nb |
|---|---|---|---|
| 11 | nbc | 1 | |
| 12d | 350±28 | 4 | |
| 13e | 560±110 | 5 | |
| 5e | 1 | 190±24 | 6 |
| 5e | 5 | 270±31 | 7 |
| 5e | 10 | 200±32 | 5 |
The Ki was calculated using the equation Ki = IC50/(1+([ligand]/Kd)) where [ligand] = [10] = 20 nM and Kd = 27 nM.
The value of Ki given represents the average of n independent competition binding experiments, each done in quadruplicate.
Compound 11 was unable to inhibit the binding and uptake of probe 10 in the concentration range tested.
Ki calculated based on [12], taken from ref 44.
Ki calculated based on [MSH4].
4. Discussion
The solution-phase synthesis of co-monomer 3 required five steps starting from 6-amino-1-hexyne and was accomplished in 30% overall yield. Deprotection of 3 produced 2 in 41% yield after purification by preparative reversed phase HPLC. The solution-phase synthesis of co-monomer 4 required eight steps in the longest linear sequence starting from 6-amino-1-hexanol and was accomplished in 8% overall yield. All reactions are amenable to scale-up with the potential for multi-gram production of the co-monomers.
Oligomerizations of 3 and 4 under conditions of high dilution appear to give mostly macrocyclic product mixtures of relatively low molecular weight. Oligomerizations of 3 and 4 under conditions of high monomer concentration appear to give both macrocyclic and linear products of comparatively high molecular weight. Similar results were observed for oligomerizations of 2 and 4, although additional linear products with the structure B2(A2B2)n were observed. Qualitative analysis of the product mixtures by MALDI-TOF is possible, but quantitative analysis by this method is not possible as peaks of higher mass are much less responsive than peaks of lower mass. For this reason, the oligomeric mixtures produced here are likely to be of much higher average molecular weight than the MALDI-TOF mass plots suggest.
The oligomeric construct 5 was designed with an inter-ligand spacing of 45 atoms, a distance estimated at ~55 Å assuming full extension of all chain segments.47 While this is longer than the estimated distance between binding sites of abutted receptors,12 we expected that the oligomers would have sufficient conformational freedom to allow multiple ligands to span shorter distances to bind to hMC4 receptors simultaneously.48,49
A standard high throughput TRF assay was used to screen for functional binding.50,51 This assay provides an indirect readout of the ability of oligomers 5 to bind to hMC4 receptors on the surface of HEK293 cells through measurement of the inhibition of the binding and uptake52 of a competed TRF probe, 10.
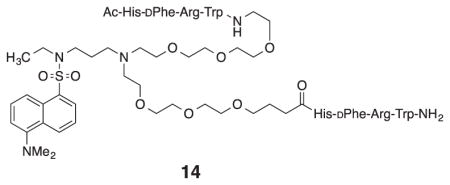
As expected, bis-serinamide control 11 did not inhibit the binding and uptake of probe 10 over the concentration range tested. The divalent MSH4 control 13 was 1.6 times less potent as an inhibitor when compared to monovalent MSH4 control 12. A similar result was previously reported for the divalent compound 14.21 Presumably, entropy disfavors the simultaneous binding of a second ligand at the end of a flexible tether if the ligand spacing is too great, effectively reducing by as much as one half the concentration of ligands available for binding.
Given this premise, oligomers 5 present a conundrum. Despite the fact that the ligand spacing in the oligomers is the same as in 13, the oligomers 5 from runs 1, 5, and 10 were of comparable inhibitory potency to the monovalent MSH4 control 12. Thus, the tethered ligands in 5 were just as effective as the untethered ligands in 12 at inhibiting the binding and uptake of probe 10. Ki values for 5 based on oligomer concentrations, were they accurately known, would be lower than the ~200 nM values calculated based on the total concentrations of MSH4, and quite possibly much lower, suggestive of multivalent binding. If multivalent binding does occur with oligomers 5, why then is there no greater effect on the binding and uptake of 10? One possible answer may lie in the off-rate of the MSH4 ligand. The apparent off-rate of the highly potent ligand NDP-α-MSH when bound to hMC1R was determined to be 1.3 × 10−3 min−1.18 To the best of our knowledge, the off-rate of MSH4, which should be much higher, has not been reported. If the off-rates of tethered and untethered MSH4 ligands are both similar and high, this could render comparable their effects on the binding and uptake of the probe 10, even as some ligands of an oligomer construct remain bound to the cell surface. To resolve this conundrum, we plan to measure the off-rates of MSH4 and of multivalent MSH4 constructs, and to study by direct methods the binding and uptake of fluorescently tagged MSH4 constructs such as 7.
Supplementary Material
Acknowledgments
This work was supported by grants R33 CA95944, RO1 CA97360, RO1 CA123547, and P30 CA23074 from the National Cancer Institute.
Footnotes
Details of the syntheses of compounds 2-4, 6, 11, 13, of the oligomerizations of 2 and 3 with 4, MALDI-TOF spectra of oligomers 5 and 7, IR spectra of compounds 2, 4, and 5, and details of the binding assays. Supplementary Material associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bagutti C, Stolz B, Albert R, Bruns C, Pless J, Eberle AN. Int J Cancer. 1994;58:749–755. doi: 10.1002/ijc.2910580521. [DOI] [PubMed] [Google Scholar]
- 2.Miao Y, Whitener D, Feng W, Owen NK, Chen J, Quinn TP. Bioconjugate Chem. 2003;14:1177–1184. doi: 10.1021/bc034069i. [DOI] [PubMed] [Google Scholar]
- 3.Singh A, Haslach EM, Haskell-Luevano C. Structure-Activity Relationships (SAR) of Melanocortin and Agouti-Related (AGRP) Peptides. In: Catania A, editor. Melanocortins: Multiple Actions and Therapeutic Potential. Chapter 1. Landes Bioscience/Springer Science+Business Media; New York: 2010. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 4.Newton JR, Miao Y, Deutscher SL, Quinn TP. J Nuc Med. 2007;48:429–436. [PubMed] [Google Scholar]
- 5.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 6.Choi S-K. Synthetic Multivalent Molecules Concepts and Biomedical Applications. Wiley-Interscience; Hoboken, NJ: 2004. [Google Scholar]
- 7.Mandrika I, Petrovska R, Wikberg J. Biochem Biophys Res Commun. 2005;326:349–354. doi: 10.1016/j.bbrc.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 8.Nickolls SA, Maki RA. Peptides. 2006;27:380–387. doi: 10.1016/j.peptides.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 9.Sánchez-Laorden BL, Sánchez-Más J, Martínez-Alonso E, Martínez-Menárguez JA, García-Borrón JC, Jiménez-Cervantes C. J Invest Dermatol. 2006;126:172–181. doi: 10.1038/sj.jid.5700036. [DOI] [PubMed] [Google Scholar]
- 10.Zanna PT, Sánchez-Laorden BL, Pérez-Oliva AB, Turpín MC, Herraiz C, Jiménez-Cervantes C, García-Borrón JC. Biochem Biophys Res Commun. 2008;368:211–216. doi: 10.1016/j.bbrc.2008.01.060. [DOI] [PubMed] [Google Scholar]
- 11.Chapman KL, Findlay JB. Biochim Biophys Acta. 2013;1828:535–542. doi: 10.1016/j.bbamem.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 12.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Bioconjugate Chem. 2007;18:1101–1109. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwyzer R, Kriwaczek VM. Biopolymers. 1981;20:2011–2020. doi: 10.1002/bip.1981.360200923. [DOI] [PubMed] [Google Scholar]
- 14.Wunderlin R, Sharma SD, Minakakis P, Schwyzer R. Helv Chim Acta. 1985;68:12–22. [Google Scholar]
- 15.Sharma SD, Granberry ME, Jiang J, Leong SPL, Hadley ME, Hruby VJ. Bioconjugate Chem. 1994;5:591–601. doi: 10.1021/bc00030a015. [DOI] [PubMed] [Google Scholar]
- 16.Kitov PI, Bundle DR. J Am Chem Soc. 2003;125:16271–16284. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- 17.Shewmake TA, Solis FJ, Gillies RJ, Caplan MR. Biomacromol. 2008;9:3057–3064. doi: 10.1021/bm800529b. [DOI] [PubMed] [Google Scholar]
- 18.Haskell-Luevano C, Miwa H, Dickinson C, Hadley ME, Hruby VJ, Yamada T, Gantz I. J Med Chem. 1996;39:432–435. doi: 10.1021/jm950407s. [DOI] [PubMed] [Google Scholar]
- 19.Carrithers MD, Lerner MR. Chem Bio. 1996;3:537–542. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 20.Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Bioconjugate Chem. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu L, Jagadish B, Hruby VJ, Gillies RJ, Mash EA. J Org Chem. 2007;72:1675–1680. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Bioorg Med Chem Lett. 2007;17:3310–3313. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alleti R, Rao V, Xu L, Gillies RJ, Mash EA. J Org Chem. 2010;75:5895–5903. doi: 10.1021/jo101043m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao V, Alleti R, Xu L, Tafreshi NK, Morse DL, Gillies RJ, Mash EA. Bioorg Med Chem. 2011;19:6474–6482. doi: 10.1016/j.bmc.2011.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elshan NGRD, Jayasundera T, Anglin BL, Weber CS, Lynch RM, Mash EA. Org Biomol Chem. doi: 10.1039/c4ob02094d. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Bioorg Med Chem Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 27.Brabez N, Lynch RM, Xu L, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. J Med Chem. 2011;54:7375–7384. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brabez N, Saunders K, Nguyen KL, Jayasundera T, Weber C, Lynch RM, Chassaing G, Lavielle S, Hruby VJ. ACS Med Chem Lett. 2013;4:98–102. doi: 10.1021/ml300312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. ACS Chem Biol. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 30.Kiessling LL, Lamanna AC. Multivalency in Biological Systems. NATO Science Series, II: Mathematics, Physics and Chemistry. 2003;129:345–357. [Google Scholar]
- 31.Wester HJ, Kessler H. J Nuc Med. 2005;46:1940–1945. [PubMed] [Google Scholar]
- 32.Hein JE, Fokin VV. Chem Soc Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.TornØe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 34.van Dijk M, Mustafa K, Dechesne AC, van Nostrum CF, Hennink WE, Rijkers DTS, Liskamp RMJ. Biomacromolecules. 2007;8:327–330. doi: 10.1021/bm061010g. [DOI] [PubMed] [Google Scholar]
- 35.van Dijk M, Nollet ML, Weijers P, Dechesne AC, van Nostrum CF, Hennink WE, Rijkers DTS, Liskamp RMJ. Biomacromolecules. 2008;9:2834–2843. doi: 10.1021/bm8005984. [DOI] [PubMed] [Google Scholar]
- 36.Ahmad Fuaad AAH, Azmi F, Skwarczynski M, Toth I. Molecules. 2013;18:13148–13174. doi: 10.3390/molecules181113148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grieshaber SE, Farran AJE, Lin-Gibson S, Kiick KL, Jia X. Macromolecules. 2009;42:2532–2541. doi: 10.1021/ma802791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cody WL, Mahoney M, Knittel JJ, Hruby VJ, de L, Castrucci AM, Hadley ME. J Med Chem. 1985;28:583–588. doi: 10.1021/jm50001a008. [DOI] [PubMed] [Google Scholar]
- 39.Following oligomerization of 3 with 4, the His (Trt), Arg (Boc2), and Trp (Boc) protecting groups were removed by treatment with a mixture of trifluoroacetic acid, thioanisole, triisopropylsilane, and water (91:03:0.3:0.3).
- 40.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Bioconjug Chem. 2011;22:1270–1278. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu L, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, Gillies RJ. Proc Nat Acad Sci USA. 2012;109:21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Byrd HCM, McEwen CN. Anal Chem. 2000;72:4568–4576. doi: 10.1021/ac0002745. [DOI] [PubMed] [Google Scholar]
- 43.Goldschmidt RJ, Wetzel SJ, Blair WR, Guttman CM. J Am Soc Mass Spectrom. 2000;11:1095–1106. doi: 10.1016/S1044-0305(00)00177-X. [DOI] [PubMed] [Google Scholar]
- 44.Alleti R, Vagner J, Dehigaspitiya DC, Moberg VE, Elshan NG, Tafreshi NK, Brabez N, Weber CS, Lynch RM, Hruby VJ, Gillies RJ, Morse DL, Mash EA. Bioorg Med Chem. 2013;21:5029–5038. doi: 10.1016/j.bmc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu L, Vagner J, Josan J, Lynch RM, Morse DL, Baggett B, Han H, Mash EA, Hruby VJ, Gillies RJ. Mol Cancer Ther. 2009;8:2356–2365. doi: 10.1158/1535-7163.MCT-08-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cone RD. The Melanocortin Receptors. Humana Press; Totowa, NJ: 2000. [Google Scholar]
-
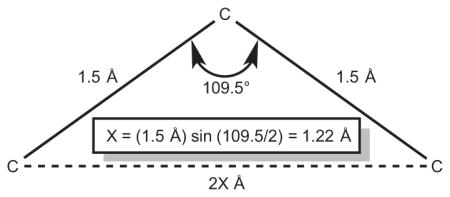
47.The sketch below illustrates the geometric basis for estimating the maximum inter-ligand distance given an extended interconnecting chain of sp3 atoms. The distance between atoms separated by n (an even number of) bonds is approximately (1.22 × n) Å.
- 48.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. J Am Chem Soc. 2007;129:1312–1320. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.CuAAC between 2 and 3 presumably occurs by four different combinations of monomer ends, leading to head-to-head and head-to-tail arrangements of adjacent MSH4 ligands in the chain. Any dependence of the directionality of adjacent ligands on binding is not addressed here.
- 50.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Anal Biochem. 2004;330:242–250. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 51.Josan JS, De Silva CR, Yoo B, Lynch RM, Pagel MD, Vagner J, Hruby VJ. Drug Design and Discovery. Humana Press; New York: 2011. Fluorescent and Lanthanide Labeling for Ligand Screens, Assays, and Imaging; pp. 89–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.As these are live cells, ligand binding leads to internalization of the receptor and the bound probe 10. Receptor recycling occurs and is responsible for the high signal-to-noise ratios obtained in these TRF assays.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.