

Abstract
Troglitazone (TGZ) caused delayed, life-threatening drug-induced liver injury (DILI) in some patients, but was not hepatotoxic in rats. This study investigated altered bile acid (BA) homeostasis as a mechanism of TGZ hepatotoxicity using a systems pharmacology model incorporating drug/metabolite disposition, BA physiology/pathophysiology, hepatocyte life cycle, and liver injury biomarkers. In the simulated human population, TGZ (200–600mg/day×6months) resulted in delayed increases in serum ALT>3× ULN in 0.3–5.1% of the population with concomitant bilirubin elevations>2× ULN in 0.3–3.6%. In contrast, pioglitazone (15–45mg/day×6months) did not elicit hepatotoxicity, consistent with clinical data. TGZ was not hepatotoxic in the simulated rat population. In summary, mechanistic modeling based only on BA effects accurately predicted the incidence, delayed presentation, and species differences in TGZ hepatotoxicity, and the relative liver safety of pioglitazone. Systems pharmacology models integrating physiology and experimental data can evaluate DILI mechanisms and may be useful to predict hepatotoxic potential of drug candidates.
Keywords: Drug-induced liver injury, bile acid, troglitazone, systems pharmacology modeling
INTRODUCTION
Drug-induced liver injury (DILI) is one of the primary reasons for the failure of pharmaceutical agents during drug development as well as withdrawal of approved drugs from the market.1 Unfortunately, current in vitro screening approaches or in vivo preclinical studies do not adequately predict the DILI liability of new chemical entities. Rare incidences of severe drug-related hepatotoxicity typically are not detected in the Phase III clinical trials that involve a few thousand patients, and may not be detected until the drug has been approved and administered to tens or hundreds of thousands of patients. These unexpected findings have led to black box warnings (e.g., bosentan, diclofenac, ketoconazole, isoniazid), or in severe cases, withdrawal of the drug from the market [e.g., troglitazone (TGZ), lumiracoxib, ximelagatran, bromfenac].
TGZ was the first of the thiazolidinedione drugs approved in worldwide markets for the treatment of type 2 diabetes. During clinical trials, alanine transaminase (ALT) elevations>3× upper limit of normal (ULN) in about 2% of patients, and 2 cases of jaundice, were reported.2 All of these patients recovered without permanent clinical complications, and TGZ was approved for marketing. However, after the broader diabetic population was exposed to TGZ, cases of liver failure associated with TGZ treatment were reported,3 and the drug was given a black box warning status with requirement for monthly monitoring of liver chemistries. TGZ was withdrawn from the market after rosiglitazone and pioglitazone, drugs from the same therapeutic class that demonstrated less concern about hepatotoxicity, were approved.4
Fourteen years have passed since the withdrawal of TGZ, but the mechanism(s) of TGZ-mediated hepatotoxicity have not been fully elucidated. Numerous mechanisms have been postulated including inhibition of bile acid (BA) transport by TGZ and its major metabolite, TGZ sulfate (TS),5,6 which may cause hepatic accumulation of toxic BAs and subsequent liver injury (Figure 1).7,8 The bile salt export pump (BSEP) is a canalicular transporter that is predominantly responsible for biliary excretion of BAs. Impaired BSEP function due to genetic polymorphisms induces liver injury,9,10 and BSEP inhibition mediated by drugs has been associated with DILI.11–13 In vitro vesicular transport assays revealed that TGZ and TS are potent inhibitors of BSEP and multidrug resistance-associated protein 4 (MRP4), hepatic transporters that mediate biliary and basolateral efflux of BAs, respectively.11,14,15 However, TGZ also has been shown to inhibit sodium-taurocholate cotransporting polypeptide (NTCP)-mediated BA uptake, which would reduce hepatic concentrations of BAs.16 Also, hepatotoxicity signals were not detected during preclinical testing of TGZ, even though TGZ and TS are potent inhibitors of rat Bsep.14 Thus, the role that alteration in BA homeostasis plays in TGZ-mediated hepatotoxicity remains speculative. While it is challenging to translate the results from isolated in vitro studies to in vivo, and preclinical studies to humans, systems pharmacology modeling is a useful approach to integrate data from different experimental systems and species, and biological knowledge, to predict human DILI.
Figure 1. Mechanism of troglitazone (TGZ) hepatotoxicity.
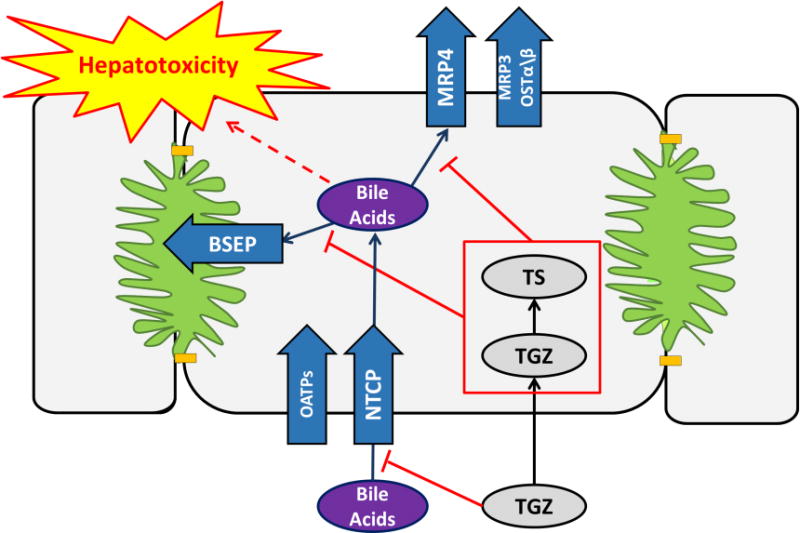
Bile acids are taken up into the hepatocytes primarily by sodium-taurocholate cotransporting polypeptide (NTCP) and also by organic anion transporting polypeptides (OATPs). Hepatocellular bile acids are excreted into bile primarily via the bile salt export pump (BSEP). Bile acids also can be transported across the basolateral membrane to sinusoidal blood via basolateral efflux transporters such as multidrug resistance-associated protein (MRP)4, MRP3, and/or organic solute transporter (OST)α/β. TGZ and its major metabolite, TGZ sulfate (TS), are potent inhibitors of hepatic bile acid transporters, which might lead to hepatic bile acid accumulation and subsequent toxicity.
In the current study, a mechanistic model of DILI (DILIsym, http://www.dilisym.com, Supplementary Figure S1) was used to investigate the role of BA transport inhibition in TGZ-mediated hepatotoxicity and underlying mechanisms for species differences. DILIsym includes sub-models representing disposition of drugs and metabolites, physiology and pathophysiology of BAs, the hepatocyte life cycle, and liver injury biomarkers (e.g., serum ALT, bilirubin) (Figure 2).17–20 TGZ-mediated DILI responses were simulated in the human and rat virtual populations (SimPops), which included variability in key model parameters. Potential risk factors for TGZ-mediated hepatotoxicity in humans in the context of BA inhibition also were assessed in human SimPops. The hepatotoxic potential of pioglitazone, a known BSEP inhibitor that is rarely associated with DILI, also was investigated as a negative control.
Figure 2. Schematic overview of the bile acid transport inhibition module in DILIsym.
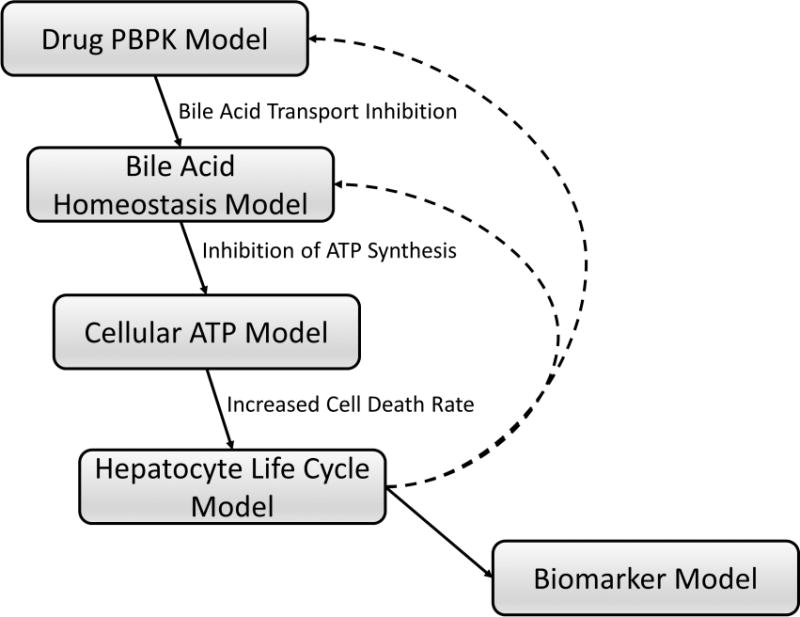
Hepatic and systemic disposition of drugs/metabolites are simulated using a physiologically-based pharmacokinetic (PBPK) model (Drug PBPK Model). The Bile Acid Homeostasis Model represents hepatobiliary disposition and enterohepatic recirculation of lithocholic acid (LCA) and chenodeoxycholic acid (CDCA) species, and all other (bulk) bile acids.20 Using bile acid transport inhibition constants of drugs/metabolites (e.g., Ki, IC50), altered bile acid disposition is simulated. Increased hepatocellular accumulation of bile acids inhibits hepatic ATP synthesis and decreases intracellular ATP concentrations (Cellular ATP Model), leading to necrotic cell death (Hepatocyte Life Cycle Model) and elevations in serum biomarkers of hepatocellular injury and function (e.g., ALT, AST, bilirubin) (Biomarker Model). Loss of hepatocytes will subsequently influence drug and bile acid disposition (dashed lines), allowing dynamic interaction between kinetics and toxicity mechanisms. Details regarding the construction and structures of sub-models can be found in the supplementary materials.
RESULTS
Physiologically-based pharmacokinetic (PBPK) modeling
A PBPK model was developed to describe the systemic disposition and hepatic concentrations of TGZ and TS in humans and male rats (Supplementary Figure S2a). Simulated TGZ and TS plasma concentration-time profiles were within 2-fold of the mean observed concentrations in humans following a single oral dose of 400mg TGZ (Supplementary Figure S3a), and within 3-fold of those in male rats following a single intravenous dose of 5mg/kg TGZ (Supplementary Figure S3b).21–23 In male rats administered a single oral dose of 5mg/kg TGZ, the simulated TGZ plasma concentration-time profile was within 2.1-fold of the mean observed concentrations (Supplementary Figure S3b).22
Simulations of TGZ hepatotoxicity in human and rat virtual populations (SimPops)
To explore TGZ hepatotoxicity at the population level, previously constructed human and rat SimPops that incorporate variability in BA disposition were employed20; variability was added to six additional parameters that describe TGZ/TS disposition, body weight, and sensitivity of ATP synthesis to hepatic BA accumulation. Several TGZ dose levels were simulated for the human SimPops (once daily oral doses of 200, 400, or 600mg for 6months) and rat SimPops (once daily oral doses of 5 or 25mg/kg for 6 months). TGZ-mediated perturbations in BA disposition and DILI responses in the human and rat SimPops are presented in Figure 3. The simulated median (range) values of maximum hepatic concentrations of chenodeoxycholic acid (CDCA) and lithocholic acid (LCA) species (sum of LCA, CDCA, and their conjugates) post-dose were 204μM (72–975), 273μM (99–1539), and 314μM (118–2610) at TGZ doses of 200, 400, and 600mg/day, respectively, compared to a baseline value of 14μM (2–127). The baseline human hepatic ATP concentration in the current model was 4.2mM. Hepatic BA accumulation led to a decrease in hepatic ATP and a decrease in viable liver mass in a subset of the human SimPops; simulated median (range) values of minimum hepatic ATP concentrations post-dose were 4.13mM (3.26–4.19), 4.10mM (2.43–4.18), and 4.07mM (2.07–4.18) at TGZ doses of 200, 400, and 600mg/day, respectively. Corresponding values for fractional viable liver mass were 1.00 (0.63–1.00), 1.00 (0.15–1.00), and 1.00 (0.15–1.00). The incidence of elevated serum ALT, serum total bilirubin, and Hy’s Law cases (serum ALT>3× ULN and serum bilirubin>2× ULN) in the human SimPops are summarized in Table 1; the reported incidence of ALT elevations and jaundice in the clinical trials also are listed.2,3 In the human SimPops, 200–600mg/day TGZ induced elevations in serum ALT>3× ULN in 0.3–5.1% of the population; Hy’s Law cases were observed in 0.3–3.6% of the human SimPops. The incidence of ALT elevations was similar to observations from the clinical trials where 200–600mg/day TGZ induced serum ALT elevations>3× ULN in 1.9% of treated patients.2 The time to peak ALT in the human SimPops with ALT elevations>3× ULN were 118±61 and 111±61 days at TGZ doses of 400 and 600mg/day, respectively; these are comparable to 147±86 days observed in the clinical trials.3 Simulated time-course dynamics of serum ALT and viable liver mass in susceptible individuals (serum ALT>3× ULN) are presented in Figure 4.
Figure 3. Simulated DILI responses in human and rat virtual populations (SimPops) at specified troglitazone (TGZ) dose levels.
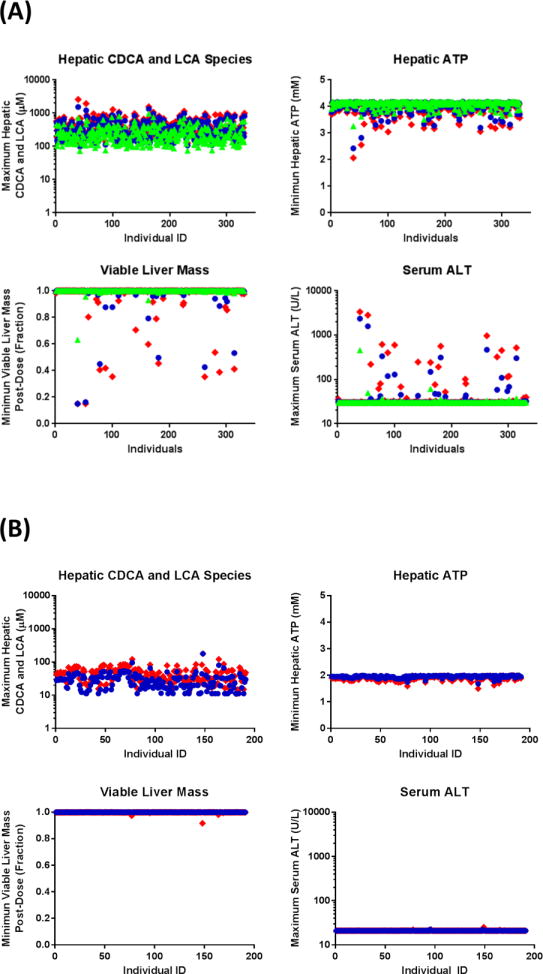
Predicted maximum hepatic accumulation of CDCA and LCA species and DILI responses (i.e., minimum hepatic ATP, minimum viable liver mass, maximum serum ALT) post-dose in human SimPops at oral doses of 200 (green triangle), 400 (blue circle), or 600 (red diamond) mg/day TGZ for 6months (A), and rat SimPops at oral doses of 5 (blue circle) or 25 (red diamond) mg/kg/day for 6months (B).
Table 1.
Summary of troglitazone (TGZ)-mediated hepatotoxicity in human SimPops and clinical trials.
| Simulationsa | Clinical Trials2,3 | ||||
|---|---|---|---|---|---|
|
| |||||
| TGZ 200 mg (n=331) |
TGZ 400 mg (n=331) |
TGZ 600 mg (n=331) |
TGZ 200 – 600 mg (n=2510) |
Placebo (n=475) |
|
| ALT > 3× ULN (%)b | 0.3 | 3.0 | 5.1 | 1.9 | 0.6 |
| ALT > 5× ULN (%)b | 0.3 | 1.8 | 4.2 | 1.7 | N/A |
| ALT > 8× ULN (%)b | 0.3 | 1.8 | 3.6 | 0.9 | 0 |
| ALT > 30× ULN (%)b | 0 | 0.6 | 0.9 | 0.2 | 0 |
| Time to peak ALT (Days)c | 180d | 118 ± 61 | 111 ± 61 | 147 ± 86 | N/A |
| Total Bilirubin > 2× (%)e | 0.3 | 1.8 | 3.6 | N/A | N/A |
| Hy’s Law cases (%) | 0.3 | 1.8 | 3.6 | N/A | N/A |
| Jaundice (%) | N/A | N/A | N/A | 0.08 | 0 |
Each dose level was simulated for 6 months.
Upper limit of normal (ULN) was 34 U/L in the clinical trials. In the human SimPops, ULN was 30 U/L because all the individuals had the same baseline ALT (30 U/L) before troglitazone administration.
Mean ± S.D.
S.D. was not calculated because only one individual showed ALT elevation > 3× ULN
Baseline serum total bilirubin in human SimPops was 0.55 mg/dL.
N/A, not available.
Figure 4. Simulated serum ALT and viable liver mass in susceptible individuals.
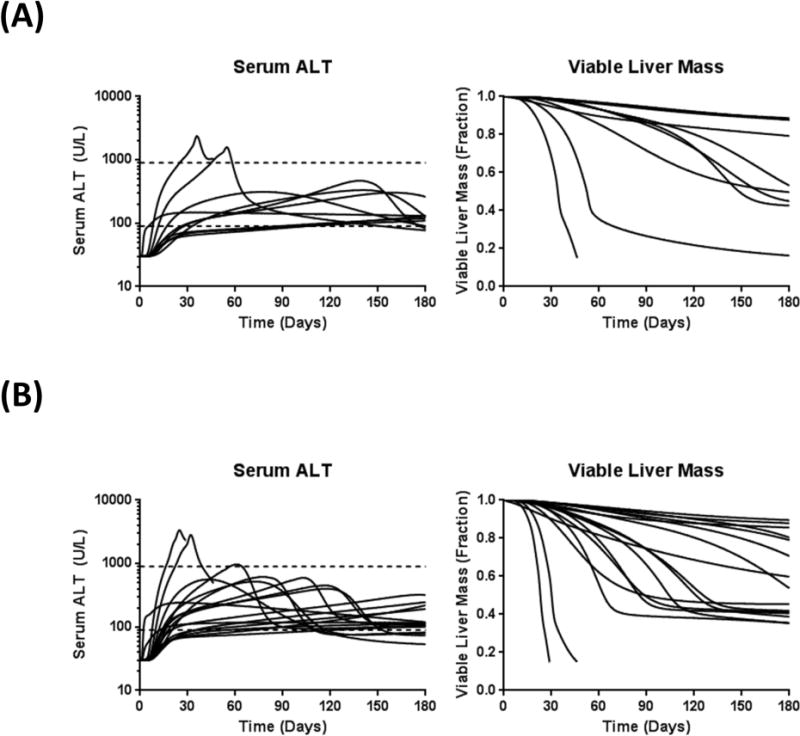
In human SimPops administered 400 (A) or 600 (B) mg/day troglitazone (TGZ) for 6months, individuals with serum ALT elevations > 3× ULN (n=10 at 400mg/day; n=17 at 600mg/day) are presented. One individual at 400mg/day and two individuals at 600mg/day lost >85% of viable liver mass and was classified as dead.
In the rat SimPops, the simulated median (range) values of maximum hepatic concentrations of CDCA and LCA species post-dose were 27μM (5–105) and 44μM (7–151) at TGZ doses of 5 and 25mg/kg/day, respectively, compared to baseline values of 13.8μM (2.4–126.6). The baseline rat hepatic ATP concentration was 2.0mM. Simulated median (range) minimum hepatic ATP concentrations after TGZ doses of 5mg/kg/day and 25mg/kg/day were 1.96mM (1.66–1.99) and 1.92mM (1.49–1.99), respectively. The corresponding values for fractional viable liver mass were 1.00 (0.99–1.00) and 1.00 (0.92–1.00), respectively. None of the rat SimPops exhibited serum ALT elevations>3× baseline (21 U/L).
Sensitivity analysis
To investigate the sensitivity of DILI responses to transporter inhibition constants, simulations were performed with 10-fold smaller and larger inhibition constants of TGZ/TS for BSEP, MRP4, and NTCP. Simulated maximum serum ALT levels in human and rat SimPops treated with TGZ (600mg/day for humans; 5mg/kg/day for rats) for 1month are presented in Figure 5. In the human SimPops, serum ALT levels were sensitive to the Ki value of TGZ/TS for BSEP inhibition; when BSEP Ki was decreased by 10-fold (assuming 10-fold more potent inhibition), 15.7% of the population exhibited serum ALT>3× ULN compared to only 3.6% of the population with the measured BSEP Ki. None of the individuals showed elevated serum ALT>3× ULN when BSEP Ki was increased 10-fold (assuming 10-fold less potent inhibition). The Ki of TGZ/TS for MRP4 inhibition also influenced serum ALT elevations, but to a smaller extent compared to BSEP Ki; the incidence of serum ALT elevations > 3× ULN ranged 2.1 – 3.6% when the MRP4 Ki was decreased or increased by 10-fold. Modulation of the Ki for NTCP inhibition by TGZ/TS led to opposite effects compared to modulation of BSEP and MRP4 Ki; a decrease in the NTCP Ki by 10-fold led to a decreased incidence of elevations in serum ALT>3× ULN (2.1% of the population), whereas an increase in the NTCP Ki by 10-fold increased the incidence of elevations in serum ALT>3× ULN (3.9% of the population), compared to an incidence of 3.6% with the measured NTCP Ki. These findings are consistent with the suggested protective role of BA uptake inhibition in hepatic BA accumulation and subsequent DILI. In the rat SimPops, simulated serum ALT levels did not exceed 3× baseline values (21 U/L) even when Ki values for BSEP or MRP4 were decreased by 10-fold, or the Ki for NTCP was increased 10-fold. These results support the hypothesis that TGZ is not expected to be hepatotoxic in rats.
Figure 5. Sensitivity analysis of transporter inhibition constants.
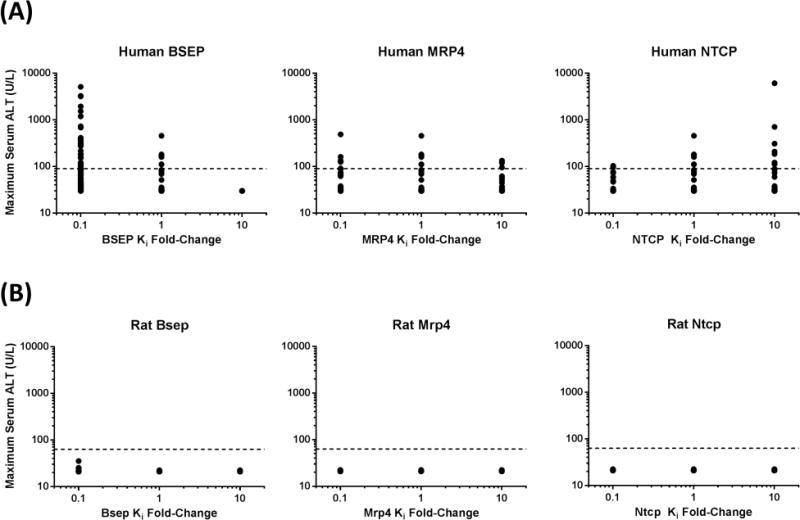
Inhibition constants for BSEP, MRP4, and NTCP were altered 10-fold in either direction of the values measured in isolated transport systems (Supplementary Table S2). Predicted maximum serum ALT concentrations in human and rat SimPops after an oral dose of 600mg/day and 5 mg/kg/day troglitazone (TGZ), respectively, for 1month are presented. Dashed lines represent 3× baseline ALT in human (90 U/L) and rat (63 U/L) SimPops.
Multiple regression analysis
Hepatotoxicity was not predicted in the baseline human simulation, which did not include population variability (data not shown), whereas simulations with human SimPops revealed a subset of individuals susceptible to TGZ-mediated hepatotoxicity. To identify the most important parameters in our model in the context of BA-mediated DILI, multiple regression analysis was performed with the lowest hepatic ATP post-dose as the dependent variable and the 16 parameters used to develop the human SimPops as independent variables. Table 2 lists the statistical significance (p-values) and standardized coefficients of parameters varied in human SimPops. Among the 16 parameters varied in human SimPops, seven parameters were statistically significant predictors of hepatic ATP levels; the maximum rate of LCA-sulfate biliary excretion was the most important variable influencing hepatic ATP decline followed by the maximum rate of LCA synthesis in the intestinal lumen, the canalicular efflux regulation scaling factor, biliary clearance of TS, body weight, the toxicity Km for CDCA and LCA species, and the maximum rate of CDCA-amide biliary excretion.
Table 2. List of parameters varied in the human and rat SimPops and results of multiple regression analysis in human SimPops administered 600 mg/day troglitazone (TGZ) for 6 months.
Human and rat population samples incorporating variability in parameters governing bile acid homeostasis (Bile Acid SimPops) have been constructed previously.20 Four parameters in the Drug PBPK Sub-model and two system-specific parameters also were varied. (See supplementary material for methods and data used for construction of SimPops). In the human SimPops administered 600 mg/day TGZ for 6 months, a multiple regression analysis was performed to identify the most important parameters in TGZ-mediated hepatotoxicity using 16 varied parameters as independent variables and minimum hepatic ATP as the dependent variable. Statistical significance and standardized coefficients were calculated using JMP 10.
| Parameter Name | Parameter Description | Significance | Standardized Coefficienta |
|---|---|---|---|
|
| |||
| Bile Acid Homeostasis Sub-model | |||
| LCA-sulfate uptake Vmax | Maximum velocity of hepatic uptake of LCA-sulfate | N/S | −0.08 |
| LCA-sulfate canalicular efflux Vmax | Maximum velocity of biliary excretion of LCA-sulfate | P< 0.001 | 0.41 |
| CDCA-amide uptake Vmax | Maximum velocity of hepatic uptake of CDCA-amide | N/S | −0.01 |
| CDCA-amide canalicular efflux Vmax | Maximum velocity of biliary excretion of CDCA-amide | P< 0.01 | 0.14 |
| CDCA-amide basolateral efflux Vmax | Maximum velocity of hepatic basolateral efflux of CDCA-amide | N/S (P=0.06) | 0.08 |
| CDCA amidation Vmax | Maximum velocity of CDCA amidation in hepatocytes | N/S | 0.06 |
| LCA-amide sulfation Vmax | Maximum velocity of LCA-amide sulfation in hepatocytes | N/S | −0.06 |
| LCA synthesis Vmax | Maximum velocity of LCA synthesis by the gut microbiome | P< 0.001 | −0.21 |
| Uptake regulation scaling factor | Scaling factor governing the magnitude of feedback regulation of hepatic uptake transporter function by hepatic bile acid accumulation | N/S | 0.02 |
| Canalicular efflux regulation scaling factor | Scaling factor governing the magnitude of FXR-mediated feedback regulation of hepatic canalicular transporter function by hepatic bile acid accumulation | P< 0.001 | 0.2 |
| LCA hydroxylation Vmaxb | Maximum velocity of LCA hydroxylation in hepatocytes | N/A | N/A |
|
| |||
|
Drug PBPK Sub-model
| |||
| TGZ intestinal absorption Kab | First-order rate constant for TGZ absorption from intestine | N/S | −0.05 |
| TGZ hepatic uptake Vmax | Maximum velocity of TGZ hepatic uptake | N/S | −0.07 |
| TGZ sulfation Vmax | Formation rate of TGZ-sulfate (TS) | N/S | −0.06 |
| TS biliary clearance | Biliary clearance of TS | P< 0.001 | 0.15 |
|
| |||
|
Other system-specific parameters
| |||
| Body weightc | Body weight | P< 0.001 | 0.15 |
| Toxicity Km for CDCA and LCA speciesc | Intracellular bile acid concentrations that induce half-maximal inhibition of ATP synthesis | P< 0.001 | 0.15 |
Parameter estimates that would have resulted from the regression if all of the variables had been standardized to a mean of 0 and a variance of 1. The greater the absolute value of the standardized coefficient, the greater the effects of the independent variable on the model output.
Used in rat SimPops only.
Used in human SimPops only.
N/S, not significant.
N/A, not available.
DISCUSSION
BA transport inhibition by TGZ and its major metabolite, TS, is one proposed mechanism of TGZ-mediated hepatotoxicity. Although TGZ and TS are potent inhibitors of BA transporters in isolated membrane vesicle systems, the relationship between BA transport inhibition and in vivo hepatotoxicity has not been evaluated. In the current study, a mechanistic model of DILI was used to investigate the hepatotoxic potential of TGZ via BA transport inhibition in humans and rats. It is important to consider population variability when predicting BA-mediated hepatotoxicity due to large variability in BA exposure and the low incidence of hepatotoxicity.2,24 Nonexistence of TGZ-mediated DILI in the baseline human model in the current study also supports the necessity of population-based analysis. Thus, human and rat SimPops that included variability in key parameters in BA disposition, TGZ and TS disposition, body weight, and sensitivity of ATP synthesis to hepatic BA accumulation, were employed to investigate the hepatotoxic potential of TGZ at the population level.
At common clinical doses (200–600mg/day), the simulated incidence of elevated serum ALT>3× ULN was 0.3–5.1%, which was similar to that observed in clinical trials (1.9%) (Table 1). Hy’s Law cases were observed in 0.3–3.6% of human SimPops, whereas 2 cases of jaundice (0.08%, both Hy’s Law cases) relevant to TGZ treatment were reported in clinical trials (Table 1). The incidence of serum bilirubin elevations might have been overestimated in the simulations because TGZ was not discontinued even when serum ALT was increased, in contrast to the usual situation in a clinical trial. Simulations also adequately predicted the delayed time to peak ALT observed in the clinical trials (Table 1). The delayed ALT elevations in the present mechanistic model were driven by a delayed build-up of toxic BAs in hepatocytes, which resulted from two factors: 1) farnesoid X receptor-mediated feedback regulation of BA synthesis/transport initially delayed BA accumulation until it could no longer compensate and 2) competitive inhibition of biliary BA excretion by TGZ/TS. The impact of a competitive inhibitor on BA transport decreases as hepatic BA concentrations increase and begin to outcompete the inhibitor, which slows down the rate of accumulation.20
It should be noted that a delay in DILI presentation for weeks to months is characteristic of idiosyncratic hepatotoxicity produced by multiple drugs.25 With some drugs, the risk of DILI has been associated with specific human leukocyte antigen alleles suggesting that latency in onset may in part reflect the time required to mount an adaptive immune response. However, it should not take several months to mount an adaptive immune response, suggesting that evolution of non-immunological events precede initiation of adaptive immunity in these cases. Moreover, the largest genome-wide association analysis of all-cause DILI to date did not find evidence for HLA associations once DILI cases attributed to flucloxacillin and amoxicillin-clavulanate were excluded.26 This observation, together with our accurate modeling of the latency associated with TGZ-mediated DILI based on altered BA homeostasis alone, support the conclusion that adaptive immunity may not underlie most cases of idiosyncratic DILI.
Preclinical animals are less sensitive to BA-mediated DILI compared to humans, and thus, do not reliably predict human hepatotoxicity that involves BA transport inhibition.25,27 Toxicity signals for TGZ were not detected during the standard preclinical toxicity testing before approval, and minimal hepatotoxicity was observed in 104 weeks of long-term toxicity studies.28 A unifying hypothesis is that differential hepatotoxicity of TGZ could be attributed to species differences in toxic BA profiles. Rats have a hydrophilic, and thus, less toxic BA pool; CDCA, the most widely implicated BA in cholestatic liver injury,29 is one of the dominant BAs in humans, whereas it contributes a smaller proportion of the BA pool in rats and mice.24,30 Less toxic, tri-hydroxy BAs such as cholic acid and muricholic acid are more abundant in rodents.30 LCA, the most hydrophobic and potentially toxic BA, is predominantly sulfated in humans, whereas LCA primarily undergoes 6β-hydroxylation to form murideoxycholic acid in rats.31 In DILIsym, CDCA, LCA, and their conjugates were exclusively modeled as the toxic BAs.20 Simulated maximum hepatic concentrations of CDCA and LCA species in the human SimPops administered 200–600mg/day TGZ were 72–2610μM (Figure 3A). Although hepatic BA concentrations after administration of BA transport inhibitors to humans have not been reported, several investigations revealed that concentrations of hepatic BAs increased up to 215±39μM and 1961μM in patients with end-stage chronic cholestatic liver disease and hepatolithiasis, respectively,32,33 suggesting that simulated hepatic BA concentrations are not physiologically unrealistic. In rat SimPops administered 5–25mg/kg/day TGZ, simulated maximum hepatic concentrations of CDCA and LCA species ranged from 5–151μM (Figure 3B). This is much lower compared to humans due to the hydrophobic BA pool and detoxification of LCA by hydroxylation20; hepatotoxicity was not predicted based on the rat SimPops. Sensitivity analysis also revealed that rat SimPops did not exhibit hepatotoxicity even with 10-fold lower (more potent) inhibition constants for BA efflux transporters (Figure 5). These results demonstrated that a mechanistic model that incorporates species differences in BA homeostasis correctly predicted differential hepatotoxicity of TGZ in humans versus rats.
Only a small subset of patients treated with TGZ experienced elevated serum ALT, indicating that certain patients are more susceptible to TGZ-mediated toxicity. Multiple linear regression analysis identified potential risk factors for TGZ-mediated hepatotoxicity associated with BA transport inhibition (Table 2). Decreased expression and/or function of hepatic canalicular transporters may decrease biliary excretion of BAs (via BSEP) and TS [via breast cancer resistance protein (BCRP) and MRP2], resulting in increased hepatic exposure to toxic BAs as well as perpetrator drugs/metabolites. Decreased function of basolateral efflux transporters [e.g., MRP4, MRP3, organic solute transporter (OST)α/β], which are important compensatory pathways for BA excretion when biliary excretion is impaired,34 could potentiate hepatic accumulation of toxic BAs. LCA is synthesized from CDCA in the intestine by the gut microbiome, but the rate and variability of LCA synthesis have not been well-characterized. One approach to test these susceptibility factors that were identified by modeling would be to interrogate genetic polymorphisms with known functional changes.35–37 However, genomic DNA was not archived from the TGZ clinical trials and no patient has received TGZ treatment in almost two decades. Therefore, cases of TGZ-mediated hepatotoxicity are not present in the current DILI registries or DNA banks. An alternative approach would be to retrospectively recruit subjects who experienced and did not experience TGZ-mediated hepatotoxicity for phenotyping studies using selected transporter substrates as in vivo probes38,39; next generation sequencing also could be used to profile the gut microbiome. However, the DILI event and the passage of time could alter these phenotypes. Although it would be challenging to test the hypothesis regarding risk factors for TGZ-mediated hepatotoxicity, an important message is that these genetic and phenotyping approaches could be used to test modeling results with drugs in current and future clinical development. Body weight was identified as a significant predictor of hepatotoxicity because a fixed dose was employed in SimPops, which led to different dosage per kg body weight. Because liver weight, blood flow, and hepatic enzyme/transporter expression were proportional to body weight in the current model, individuals with higher body weight were able to clear drugs faster, resulting in lower hepatic exposure to TGZ and TS. In the clinic, however, body weight may correlate with many other factors, and rarely has been identified as a risk factor for DILI.
Pioglitazone, another thiazolidinedione drug that is still used in diabetic patients, is rarely associated with hepatotoxicity.40 Pioglitazone also was identified as a potent inhibitor of BSEP and MRP4 in vesicular transport assays; steady-state plasma concentrations of TGZ and pioglitazone were comparable.11 These data suggest that the hepatotoxic potential of these compounds cannot be differentiated using in vitro transporter inhibition and systemic exposure data alone. Simulations using DILIsym revealed that no hepatotoxicity was predicted in the human SimPops at clinical doses of pioglitazone due mainly to the low hepatic exposure of pioglitazone as a result of extensive hepatic metabolism (see supplementary material for pioglitazone modeling results). These results re-emphasize that drug/metabolite concentrations at the site of toxicity (hepatocellular concentrations in this case) need to be considered when predicting toxicity.41 TGZ also is metabolized extensively in the liver, but its major metabolite, TS, is a more potent BSEP inhibitor contributing to BA accumulation and subsequent hepatotoxicity. Hepatic exposure and the inhibitory effects on hepatic transporters of pioglitazone metabolites have not been investigated experimentally, and thus, were not included in the current simulation. Systemic exposure of the major metabolite of pioglitazone, M-IV, is more than 10-fold smaller compared to TS at respective clinical doses.42,43 Pioglitazone metabolites may be less likely to cause hepatotoxicity, but further studies are warranted to confirm this. The current study demonstrated that systems pharmacology modeling that integrates physiological information and experimental data was able to predict differential hepatotoxicity between TGZ and pioglitazone.
Increasing evidence supports the hypothesis that drug-mediated functional disturbances in hepatic BA transporters leads to intracellular accumulation of potentially harmful BAs and subsequent hepatic injury. A systematic investigation of a panel of drugs for their inhibitory effects on BA efflux transporters using isolated membrane vesicles and hepatotoxic potential demonstrated that inhibition of BA efflux transporters is associated with DILI.11–13,44 More sophisticated model systems such as sandwich-cultured hepatocytes have been used to assess effects of drugs and generated metabolites on hepatic accumulation of BAs.13,45–47 However, results from in vitro systems may not always translate directly to in vivo hepatotoxicity risk due to the complexity of BA homeostasis (i.e., vectorial transport, enterohepatic recirculation), dynamic changes in the systemic and hepatic exposure of drugs/metabolites, and feedback regulation of BA synthesis and transport as an adaptive response to hepatic BA accumulation.48,49 It is of note that knowledge gaps exist in BA homeostasis as discussed above and elsewhere,20 which may limit accurate, quantitative prediction of BA-mediated DILI. Nonetheless, systems pharmacology modeling incorporating 1) physiology/pathophysiology of BAs in humans and rats, 2) systemic and hepatic disposition of drugs/metabolites, and 3) in vitro inhibition potency data reasonably predicted altered BA disposition in rats administered glibenclamide,20 and also adequately predicted delayed presentation and species differences in TGZ hepatotoxicity. Although effects of pioglitazone metabolites were not incorporated due to lack of data, differential hepatotoxicity between TGZ and pioglitazone was predicted correctly in the present study. It is also important to note that use of the pre-existing human SimPops, with no modification except for adding variability to the parameters describing drug disposition, body weight, and sensitivity of ATP synthesis to hepatic BA accumulation, accurately predicted the incidence of TGZ hepatotoxicity; the current mechanistic model was truly predictive. These findings suggest that systems pharmacology modeling combined with population analysis may provide a useful tool to integrate our current knowledge about physiological and experimental data obtained during the drug development process, and prospectively predict hepatotoxic potential of new chemical entities that are in the drug development pipeline.
METHODS
PBPK model development
A PBPK model was developed to describe the disposition of TGZ/TS in humans and male rats, and pioglitazone in humans (Figure 2, Supplementary Figure S3). Details regarding the construction and final structure of the PBPK model are provided in the supplementary material (Supplementary Figure S2a, Supplementary Table S1).
Construction of human and rat population samples (SimPops)
Human (n=331) and rat (n=191) population samples with variability in 10 (human) or 11 (rat) parameters in the BA homeostasis sub-model (Bile Acid SimPops) were constructed previously within DILIsym using the genetic algorithm in MATLAB20; these Bile Acid SimPops are system-specific, and thus the same Bile Acid SimPops are used to simulate hepatotoxic effects of different compounds. In the current study, parameters governing TGZ and TS disposition (human and rat), pioglitazone disposition (human), body weight (human), and sensitivity of hepatic ATP decline to hepatic BA accumulation (human) also were varied using the probability distribution of each parameter obtained from the literature. Parameters varied in SimPops and literature data employed to construct human and rat SimPops are listed in Table 2 and Supplementary Table S3. Details related to construction of the SimPops can be found in the supplementary material.
Simulation of DILI responses
Perturbation of BA disposition and DILI responses after TGZ administration in human [200, 400, or 600mg/day (common clinical doses) for 6months] and rat [5 (equivalent to the clinical dose) or 25mg/kg/day for 6month] SimPops, and pioglitazone administration in human SimPops [15, 30, or 45mg/day (common clinical doses) for 6months] were simulated using PBPK model predictions of TGZ/TS or pioglitazone disposition, a previously developed BA homeostasis sub-model,20 and BA transport inhibition constants for TGZ/TS or pioglitazone (i.e., Ki, IC50) measured in isolated membrane vesicle transport systems (Supplementary Table S2). To assess the sensitivity of DILI responses to inhibition constants, simulations were performed with 10-fold smaller or greater inhibition constants of TGZ/TS or pioglitazone for BSEP, MRP4, and NTCP in human (600mg/day TGZ or 45mg/pioglitazone) and rat SimPops (5mg/kg/day TGZ). Simulations for the sensitivity analysis studies were performed for 1month due to the extensive computational time required for long-term simulations, and also because even for the individuals with delayed presentation of hepatotoxicity, slight increases (<3× ULN) in serum ALT could be detected within 1month of simulation. To identify the most important parameters in the context of BA-mediated DILI in humans administered TGZ, a multiple regression analysis was performed with minimum hepatic ATP as the dependent variable. Hepatic ATP was selected because perturbations in cellular ATP synthesis is a key step in the development of BA-mediated DILI in the current model (Figure 2), and thus is the most sensitive and variable model output compared to other DILI responses (i.e., serum ALT, fractional viable liver mass). Sixteen parameters used to develop the human SimPops were utilized as independent variables. Because the units of independent variables were different by orders of magnitude, standardized coefficients were calculated to determine which of the independent variables have a greater effect on the minimum hepatic ATP. Statistical analyses were performed using JMP 10 (SAS, Cary, NC).
Supplementary Material
{kind=link}
Study Highlights.
What is the current knowledge on the topic?
-
✓
Troglitazone (TGZ) induced hepatotoxicity in humans, but not in preclinical species. Inhibition of bile acid (BA) transport is one of many proposed mechanisms of TGZ-mediated hepatotoxicity. Pioglitazone also inhibits BA transport, but is rarely hepatotoxic in humans.
What question this study addressed?
-
✓
Can a mechanistic model based on BA transport inhibition correctly predict susceptibility to TGZ- and pioglitazone-mediated hepatotoxicity?
What this study adds to our knowledge?
-
✓
Inhibition of BA transport by TGZ and its major metabolite, TGZ sulfate, was predicted to induce delayed hepatotoxicity in humans due to hepatocellular accumulation of toxic BAs. A systems pharmacology model incorporating species-specific physiology/pathophysiology of BAs and drug disposition correctly predicted species differences in TGZ hepatotoxicity and the relative liver safety of pioglitazone; toxic BA profiles and hepatic drug exposure were important contributors.
How this might change clinical pharmacology and therapeutics?
-
✓
Systems pharmacology models incorporating data generated from human-derived in vitro systems provide a framework for more accurate prediction of BA-mediated DILI risk in humans.
Acknowledgments
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01 GM041935 (K.L.R.B), and by members of the DILI-sim Initiative. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. For more information on the DILI-sim Initiative, see www.DILIsym.com. K. Yang was supported, in part, by a fellowship from Amgen, Inc.
Footnotes
Conflict of Interest/Disclosure
The authors declare no conflicts of interest.
Author Contributions
Wrote Manuscript: K.Y., J.L.W., P.B.W., B.A.H., K.L.R.B
Designed Research: K.Y., J.L.W., P.B.W., B.A.H., K.L.R.B
Performed Research: K.Y.
Analyzed Data: K.Y., J.L.W., P.B.W., B.A.H., K.L.R.B
Contributor Information
Kyunghee Yang, Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599.
Jeffrey L Woodhead, The Hamner-UNC Institute for Drug Safety Sciences, The Hamner Institutes for Health Sciences, Research Triangle Park, NC 27709.
Paul B Watkins, Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; The Hamner-UNC Institute for Drug Safety Sciences, The Hamner Institutes for Health Sciences, Research Triangle Park, NC 27709.
Brett A Howell, The Hamner-UNC Institute for Drug Safety Sciences, The Hamner Institutes for Health Sciences, Research Triangle Park, NC 27709.
Kim LR Brouwer, Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599.
References
- 1.Temple RJ, Himmel MH. Safety of newly approved drugs: implications for prescribing. JAMA. 2002;287:2273–5. doi: 10.1001/jama.287.17.2273. [DOI] [PubMed] [Google Scholar]
- 2.Watkins PB, Whitcomb RW. Hepatic dysfunction associated with troglitazone. N Engl J Med. 1998;338:916–7. doi: 10.1056/NEJM199803263381314. [DOI] [PubMed] [Google Scholar]
- 3.Presented at the Endocrinologic and Metabolic Drugs Advisory Committee Meeting; 1999. Aprial 26. Available from the US Food and Drug Administration ( http://www.fda.gov/ohrms/dockets/ac/99/transcpt/3499t1a.pdf). Accessed January 25, 2014. [Google Scholar]
- 4.Isley WL. Hepatotoxicity of thiazolidinediones. Expert Opin Drug Saf. 2003;2:581–6. doi: 10.1517/14740338.2.6.581. [DOI] [PubMed] [Google Scholar]
- 5.Smith MT. Mechanisms of troglitazone hepatotoxicity. Chem Res Toxicol. 2003;16:679–87. doi: 10.1021/tx034033e. [DOI] [PubMed] [Google Scholar]
- 6.Chojkier M. Troglitazone and liver injury: in search of answers. Hepatology. 2005;41:237–46. doi: 10.1002/hep.20567. [DOI] [PubMed] [Google Scholar]
- 7.Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–89. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maillette de Buy Wenniger L, Beuers U. Bile salts and cholestasis. Dig Liver Dis. 2010;42:409–18. doi: 10.1016/j.dld.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Jansen PL, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–9. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 10.van Mil SW, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379–84. doi: 10.1053/j.gastro.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 11.Morgan RE, et al. A Multifactorial Approach to Hepatobiliary Transporter Assessment Enables Improved Therapeutic Compound Development. Toxicol Sci. 2013 doi: 10.1093/toxsci/kft176. [DOI] [PubMed] [Google Scholar]
- 12.Dawson S, Stahl S, Paul N, Barber J, Kenna JG. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab Dispos. 2012;40:130–8. doi: 10.1124/dmd.111.040758. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen JM, et al. Early Identification of Clinically Relevant Drug Interactions With the Human Bile Salt Export Pump (BSEP/ABCB11) Toxicol Sci. 2013;136:328–43. doi: 10.1093/toxsci/kft197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funk C, Ponelle C, Scheuermann G, Pantze M. Cholestatic potential of troglitazone as a possible factor contributing to troglitazone-induced hepatotoxicity: in vivo and in vitro interaction at the canalicular bile salt export pump (Bsep) in the rat. Mol Pharmacol. 2001;59:627–35. [PubMed] [Google Scholar]
- 15.Yang K, Yue W, Koeck K, Brouwer KLR. Interaction of troglitazone sulfate with hepatic basolateral and canalicular transport proteins. 2011 AAPS Annual Meeting and Exposition [Google Scholar]
- 16.Marion TL, Leslie EM, Brouwer KL. Use of sandwich-cultured hepatocytes to evaluate impaired bile acid transport as a mechanism of drug-induced hepatotoxicity. Mol Pharm. 2007;4:911–8. doi: 10.1021/mp0700357. [DOI] [PubMed] [Google Scholar]
- 17.Shoda LK, Woodhead JL, Siler SQ, Watkins PB, Howell BA. Linking physiology to toxicity using DILIsym((R)), a mechanistic mathematical model of drug-induced liver injury. Biopharm Drug Dispos. 2014;35:33–49. doi: 10.1002/bdd.1878. [DOI] [PubMed] [Google Scholar]
- 18.Howell BA, et al. In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsym: a mechanistic, mathematical model of DILI. J Pharmacokinet Pharmacodyn. 2012;39:527–41. doi: 10.1007/s10928-012-9266-0. [DOI] [PubMed] [Google Scholar]
- 19.Woodhead JL, et al. An analysis of N-acetylcysteine treatment for acetaminophen overdose using a systems model of drug-induced liver injury. J Pharmacol Exp Ther. 2012;342:529–40. doi: 10.1124/jpet.112.192930. [DOI] [PubMed] [Google Scholar]
- 20.Woodhead JL, et al. Mechanistic modeling reveals the critical knowledge gaps in bile acid-mediated DILI. CPT Pharmacometrics Syst Pharmacol. 2014;3:e123. doi: 10.1038/psp.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loi CM, Randinitis EJ, Vassos AB, Kazierad DJ, Koup JR, Sedman AJ. Lack of effect of type II diabetes on the pharmacokinetics of troglitazone in a multiple-dose study. J Clin Pharmacol. 1997;37:1114–20. doi: 10.1002/j.1552-4604.1997.tb04295.x. [DOI] [PubMed] [Google Scholar]
- 22.Izumi T, et al. Prediction of the human pharmacokinetics of troglitazone, a new and extensively metabolized antidiabetic agent, after oral administration, with an animal scale-up approach. J Pharmacol Exp Ther. 1996;277:1630–41. [PubMed] [Google Scholar]
- 23.Izumi T, Hosiyama K, Enomoto S, Sasahara K, Sugiyama Y. Pharmacokinetics of troglitazone, an antidiabetic agent: prediction of in vivo stereoselective sulfation and glucuronidation from in vitro data. J Pharmacol Exp Ther. 1997;280:1392–400. [PubMed] [Google Scholar]
- 24.Trottier J, Caron P, Straka RJ, Barbier O. Profile of serum bile acids in noncholestatic volunteers: gender-related differences in response to fenofibrate. Clin Pharmacol Ther. 2011;90:279–86. doi: 10.1038/clpt.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watkins PB. Idiosyncratic liver injury: challenges and approaches. Toxicologic pathology. 2005;33:1–5. doi: 10.1080/01926230590888306. [DOI] [PubMed] [Google Scholar]
- 26.Urban TJ, Goldstein DB, Watkins PB. Genetic basis of susceptibility to drug-induced liver injury: what have we learned and where do we go from here? Pharmacogenomics. 2012;13:735–8. doi: 10.2217/pgs.12.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leslie EM, Watkins PB, Kim RB, Brouwer KL. Differential inhibition of rat and human Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1)by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther. 2007;321:1170–8. doi: 10.1124/jpet.106.119073. [DOI] [PubMed] [Google Scholar]
- 28.Herman JR, et al. Rodent carcinogenicity with the thiazolidinedione antidiabetic agent troglitazone. Toxicol Sci. 2002;68:226–36. doi: 10.1093/toxsci/68.1.226. [DOI] [PubMed] [Google Scholar]
- 29.Greim H, et al. Mechanism of cholestasis. 6. Bile acids in human livers with or without biliary obstruction. Gastroenterology. 1972;63:846–50. [PubMed] [Google Scholar]
- 30.Garcia-Canaveras JC, Donato MT, Castell JV, Lahoz A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J Lipid Res. 2012;53:2231–41. doi: 10.1194/jlr.D028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev. 2004;36:703–22. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 32.Shoda J, Tanaka N, He BF, Matsuzaki Y, Osuga T, Miyazaki H. Alterations of bile acid composition in gallstones, bile, and liver of patients with hepatolithiasis, and their etiological significance. Dig Dis Sci. 1993;38:2130–41. doi: 10.1007/BF01297095. [DOI] [PubMed] [Google Scholar]
- 33.Fischer S, Beuers U, Spengler U, Zwiebel FM, Koebe HG. Hepatic levels of bile acids in end-stage chronic cholestatic liver disease. Clin Chim Acta. 1996;251:173–86. doi: 10.1016/0009-8981(96)06305-x. [DOI] [PubMed] [Google Scholar]
- 34.Trauner M, Wagner M, Fickert P, Zollner G. Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroenterol. 2005;39:S111–24. doi: 10.1097/01.mcg.0000155551.37266.26. [DOI] [PubMed] [Google Scholar]
- 35.Ieiri I, Higuchi S, Sugiyama Y. Genetic polymorphisms of uptake (OATP1B1, 1B3) and efflux (MRP2, BCRP) transporters: implications for inter-individual differences in the pharmacokinetics and pharmacodynamics of statins and other clinically relevant drugs. Expert opinion on drug metabolism & toxicology. 2009;5:703–29. doi: 10.1517/17425250902976854. [DOI] [PubMed] [Google Scholar]
- 36.Lang C, et al. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenetics and genomics. 2007;17:47–60. doi: 10.1097/01.fpc.0000230418.28091.76. [DOI] [PubMed] [Google Scholar]
- 37.Meier Y, et al. Increased susceptibility for intrahepatic cholestasis of pregnancy and contraceptive-induced cholestasis in carriers of the 1331T>C polymorphism in the bile salt export pump. World J Gastroenterol. 2008;14:38–45. doi: 10.3748/wjg.14.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfeifer ND, et al. Effect of Ritonavir on (99m)Technetium-Mebrofenin Disposition in Humans: A Semi-PBPK Modeling and In Vitro Approach to Predict Transporter-Mediated DDIs. CPT Pharmacometrics Syst Pharmacol. 2013;2:e20. doi: 10.1038/psp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kusuhara H. Imaging in the study of membrane transporters. Clin Pharmacol Ther. 2013;94:33–6. doi: 10.1038/clpt.2013.85. [DOI] [PubMed] [Google Scholar]
- 40.Livertox Database: pioglitazone. ( http://livertox.nlm.nih.gov/Pioglitazone.htm). Accessed April 4, 2014.
- 41.Chu X, et al. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther. 2013;94:126–41. doi: 10.1038/clpt.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loi CM, Alvey CW, Vassos AB, Randinitis EJ, Sedman AJ, Koup JR. Steady-state pharmacokinetics and dose proportionality of troglitazone and its metabolites. J Clin Pharmacol. 1999;39:920–6. doi: 10.1177/00912709922008533. [DOI] [PubMed] [Google Scholar]
- 43.Eckland DA, Danhof M. Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes. 2000;(Suppl2):S234–S42. [Google Scholar]
- 44.Kock K, et al. Risk Factors for Development of Cholestatic Drug-induced Liver Injury: Inhibition of Hepatic Basolateral Bile Acid Transporters MRP3 and MRP4. Drug Metab Dispos. 2013 doi: 10.1124/dmd.113.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marion TL, Perry CH, St Claire RL, 3rd, Brouwer KL. Endogenous bile acid disposition in rat and human sandwich-cultured hepatocytes. Toxicol Appl Pharmacol. 2012;261:1–9. doi: 10.1016/j.taap.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ansede JH, Smith WR, Perry CH, St Claire RL, 3rd, Brouwer KR. An in vitro assay to assess transporter-based cholestatic hepatotoxicity using sandwich-cultured rat hepatocytes. Drug Metab Dispos. 2010;38:276–80. doi: 10.1124/dmd.109.028407. [DOI] [PubMed] [Google Scholar]
- 47.Griffin LM, Watkins PB, Perry CH, St Claire RL, 3rd, Brouwer KL. Combination lopinavir and ritonavir alter exogenous and endogenous bile acid disposition in sandwich-cultured rat hepatocytes. Drug Metab Dispos. 2013;41:188–96. doi: 10.1124/dmd.112.047225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang K, Kock K, Sedykh A, Tropsha A, Brouwer KL. An updated review on drug-induced cholestasis: mechanisms and investigation of physicochemical properties and pharmacokinetic parameters. J Pharm Sci. 2013;102:3037–57. doi: 10.1002/jps.23584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodrigues AD, Lai Y, Cvijic ME, Elkin LL, Zvyaga T, Soars MG. Drug-induced perturbations of the bile acid pool, cholestasis, and hepatotoxicity: mechanistic considerations beyond the direct inhibition of the bile salt export pump. Drug Metab Dispos. 2014;42:566–74. doi: 10.1124/dmd.113.054205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.