

Abstract
Connexin 43 (Cx43) is a gap junction protein that plays an integral role in the skeletal response to mechanical loading and unloading. In a previous study, we demonstrated preservation of trabecular bone mass and cortical bone formation rate in mice with an osteoblast/osteocyte-selective deficiency of Cx43 (cKO) following mechanical unloading via hindlimb suspension (HLS). In the present study, we sought to define the potential mechanisms underlying this response. Following three weeks of HLS, mRNA levels of Sost were significantly greater in wild-type (WT)-Suspended mice vs. WT-Control, while there was no difference between cKO control and cKO-Suspended. Unloading-induced decreases in P1NP, a serum marker of bone formation, were also attenuated in cKO-Suspended. The proportion of sclerostin-positive osteocytes was significantly lower in cKO-Control vs. WT-Control (−72%, p < 0.05), a difference accounted for by the presence of numerous empty lacunae in the cortical bone of cKO vs. WT. Abundant TUNEL staining was present throughout the cortical bone of the tibia and femur, suggesting an apoptotic process. There was no difference in empty lacunae in the trabecular bone of the tibia or femur. Trabecular and cortical osteoclast indices were lower in cKO-Suspended vs. WT-Suspended; however, mRNA levels of the gene encoding RANKL increased similarly in both genotypes. Connexin 43 deficient mice experience attenuated sclerostin-mediated suppression of cortical bone formation and lower cortical osteoclast activity during unloading. Preservation of trabecular bone mass and attenuated osteoclast activity during unloading, despite an apparent lack of effect on osteocyte viability at this site, suggests that an additional mechanism independent of osteocyte apoptosis may also be important. These findings indicate that Cx43 is able to modulate both arms of bone remodeling during unloading.
Keywords: Connexin 43, Unloading, Sclerostin, RANKL, Osteocyte apoptosis
Introduction
Connexin 43 (Cx43)is the predominant gap junction protein in bone [1]. Gap junctions facilitate the movement of molecules <1 kD between adjacent cells [2] or between the cell and extracellular milieu when unopposed as hemichannels. Cx43 also interacts with signaling molecules and structural proteins [3–7], indicating a role beyond that of intercellular transmission of small molecules. Cx43 has also been shown to play a critical role in skeletal development [8], fracture repair [9], and bone response to mechanical forces [5].
Mechanical loading is required to maintain bone mass and strength [10]. Bone loss due to mechanical unloading is characterized by an uncoupling of bone turnover: bone formation decreases, while bone resorption increases [11]. The net result is deterioration of bone micro-structure, particularly in the trabecular bone compartment, which is more sensitive to unloading-induced bone loss [12,13]. The effects of mechanical unloading are most vividly demonstrated by the profound bone loss experienced by astronauts following long duration stays on the International Space Station [14]. A more clinically-relevant example of unloading-induced bone loss would be prolonged bed rest as a result of neurological injury or trauma [15].
Previous work in our laboratory and others has demonstrated the integral role that gap junctions play in the response to both mechanical loading [16–18] and unloading [19,20]. In a recent study, we subjected mice with an osteoblast/osteocyte-selective deficiency of Cx43 (cKO) to mechanical unloading via hindlimb suspension (HLS) [20]. Following three weeks of HLS, we detected attenuation of trabecular bone loss in Cx43 deficient mice [20]. In addition, histomorphometric bone formation rate at the endocortical and periosteal surface was maintained at baseline levels in cKO mice, in contrast to characteristic unloading-induced suppression of bone formation in WT mice [11]. Despite these findings, we were unable to detect a difference in cortical bone loss between WT and cKO. However, a separate study by Grimston et al. found attenuated cortical bone loss in younger Cx43 deficient mice subjected to botox immobilization of the hindlimbs [19].
One of the primary mediators of bone loss associated with unloading is the production of sclerostin (product of the Sost gene) by osteocytes [21]. No previous studies have investigated the effect of Cx43 deficiency on Sost expression during unloading. Sclerostin is an antagonist of the Wnt/β-catenin pathway that inhibits bone formation and osteogenesis [21]. Osteocyte-derived sclerostin may also support osteoclast activity by increasing the levels of receptor activator of nuclear factor kappa B ligand (RANKL) [22]. Bivi et al. demonstrated increased osteocyte apoptosis in Cx43 deficient mice [23], while Watkins et al. showed decreased baseline Sost mRNA levels [24]. Osteocyte apoptosis associated with Cx43 deficiency, and its effects on bone signaling pathways, may be a crucial mediator of the desensitization of these mice to certain effects of mechanical unloading.
We investigated the response of Cx43 cKO mice to three weeks of HLS. Compared to WT mice, unloaded cKO mice were found to have increased baseline osteocyte apoptosis, decreased Sost expression, and preserved serum P1NP levels. Indices of bone resorption, including osteoclast number and surface were reduced in Cx43 deficient mice subjected to unloading. Taken together, these results demonstrate that Cx43 deficiency reduces the sensitivity of bone to the deleterious cellular and molecular changes induced by mechanical unloading.
Methods
Mice
Given that global knockout of the Cx43 gene is embryonic lethal [25], we utilized mice with conditional deletion of Gja1 (encodes connexin 43) specifically in osteoblasts/osteocytes. Our strategy for generation of transgenic mice, confirmation of conditional knockout in osteocytes/osteoblasts, and determination of the specificity of the Cx43 deletion was described previously [18,20]. Briefly, mice expressing Cre recombinase under the control of the human osteocalcin promoter (OC-Cre; Cx43+/+) [26] were bred with mice in which Gja1 is flanked by two loxP sites (Cx43flx/flx) [27] to generate OC-Cre; Cx43flx/+ mice. We then crossed OC-Cre; Cx43flx/+ mice with Cx43flx/flx mice to generate OC-Cre; Cx43flx/flx mice. We then back bred OC-Cre; Cx43flx/flx mice with Cx43flx/flx mice to generate an equal number of OC-Cre; Cx43flx/flx (conditional Cx43 deficient equivalent; cKO) and Cx43flx/flx (wild-type; WT). Mice derived from this breeding strategy were bred with C57BL/6 mice for three generations, resulting in mice with a C57BL/6 background. Genotyping was performed by PCR using genomic DNA isolated from mouse earpieces and appropriate primers [18]. The mice used in this study were separate from those used in our previous unloading study [20].
Hindlimb suspension
The present study utilized six-month-old male Cx43 cKO and WT mice. Mice of this age and sex have been used successfully in our previous studies of mechanical loading [18] and unloading [20]. Mice were housed in standard vivarium enclosures until one-week prior to experimentation when they were moved to hindlimb suspension (HLS) enclosures (2 mice per cage) to acclimate while ambulating normally. WT and cKO mice were then divided into normally loaded (i.e., Control) and unloaded (i.e., Suspended) groups (n = 5/group). The HLS protocol was a modified version of that described by Morey-Holton and colleagues [28] and described in detail previously [20]. Our HLS enclosures consisted of a modified rat cage with standard bedding placed below a wire mesh insert. Two metal crossbars were located at either end of the cage, along with water bottles. Under isoflurane anesthesia (2%), two strips of bandage tape were braided around the tail, with loose ends fixed to a swivel hook attached to a string. The string was wound around the cross bar at the top of the cage. The crossbar could be rotated, raising or lowering the hindquarters of the animal to achieve a 30° elevation. This angle of suspension has been previously demonstrated to keep the forelimbs normally-loaded, while minimizing tail tension and animal stress [43]. Two mice were suspended per cage in this manner, although their placement at opposite ends prevented physical contact. Control mice were housed in this same cage environment, albeit without attachment of the HLS apparatus. Based on previous studies, animals were subjected to three weeks of HLS [20]. The Institutional Animal Care and Use Committee at the Penn State College of Medicine approved of all animal procedures (Protocol #2010-117).
RNA collection
Mice were removed from the suspension apparatus and immediately killed via carbon dioxide asphyxiation followed by cervical dislocation. The right femur and tibia were isolated and cleaned of all non-osseous tissue. The ends of each bone were cut off using a sterile scalpel blade and the marrow flushed from the diaphysis with ice cold phosphate buffered saline (PBS). Bones were then snap frozen in liquid nitrogen and kept frozen at −80 °C until use. The tibia and femur were pulverized using a frozen mortar and pestle on ice. Total RNA was extracted using TRIZOL (Invitrogen Corporation; Carlsbad, CA, USA). RNA was then subjected to DNase digestion using an RNase-Free DNase Kit (Qiagen; Valencia, CA, USA) followed by cleanup using an RNeasy Mini Kit (Qiagen). Single-stranded cDNA was made using a reverse transcription kit (iScript, BioRad; Hercules, CA, USA) and used as a template for realtime PCR with SYBR Green PCR Master Mix (Applied Biosystems; Foster City, CA, USA) and gene specific primers in a Rotor-Gene 2000 thermocycler (Corbett Research; Sydney, Australia). Targets of interest included Sost (encodes sclerostin), Tnsfs11b (encodes osteoprotegerin), and Tnsfs11 (encodes RANKL, receptor activator of nuclear factor kappa-B ligand). mRNA levels were standardized with the internal control Actb (encodes β-actin). All samples (n = 5/group) were tested in triplicate.
Histology, TRAP, and TUNEL staining
Left femurs and tibias were harvested immediately after sacrifice and embedded in paraffin as described previously [9]. 5 μm sections (n = 5/group) were stained with Alcian blue/hematoxylin/Orange G/Eosin (i.e., H&E) or used for tartrate resistant acid phosphatase (TRAP) staining as described previously [9]. The number of multinucleated (> 3 nuclei/cell) TRAP + osteoclasts was counted in the proximal tibia, in an area extending 500 μm distal from the epiphy-seal plate. For cortical osteoclasts, we analyzed a 2400 μm length section of the endocortical surface at the femur midshaft. BioQuant Osteo software (v12.5.60, BIOQUANT Image Analysis Corporation; Nashville, TN, USA) was used to determine osteoclast number (Oc.N; 1/mm) and surface (Oc.S/BS; %) (n = 3–5/group). A TACS 2 TdT Blu Label In Situ Apoptosis Detection Kit (Trevigen; Gaithersburg, MD, USA) was used to detect apoptotic osteocytes in sections of cortical and trabecular bone from femur and tibia. H&E sections were used to identify empty lacunae in a 2400 μm long region of tibia cortical bone (just distal of the trabecular bone at the proximal end) or femur midshaft cortical bone (n = 5/group).
Immunohistochemistry
Paraffin sections were de-waxed, and dehydrated, and underwent antigen retrieval using sodium citrate buffer (pH 6.0). Sections were probed with an anti-Cx43 (#SAB4300504, Sigma Aldrich; St. Louis, MO, USA) or an anti-sclerostin antibody (#AF1589 R&D Systems; Minneapolis, MN, USA) diluted 1:500 in normal goat serum, followed by goat anti-rabbit secondary antibody (#PK6101, Vector Laboratories; Burlingame, CA), and staining was visualized with DAB Chromogen (Invitrogen). Sections were counterstained with methyl green. BioQuant Osteo software (BIOQUANT Image Analysis Corporation) was used to quantify positive and negative cells (n = 3–5/group) as well as empty lacunae in 20× images of a 2400 μm region of cortical bone at femur midshaft relative to either the total number of lacunae (including empty lacunae) or the total number of occupied lacunae (excluding empty lacunae).
Serum markers of bone formation and resorption
At the study endpoint, approximately 0.5 mL of whole blood was collected from the posterior vena cava of all mice following an overnight fast (n = 5/group). Blood was allowed to clot for 20 min and then serum was separated via centrifugation (2000 ×g for 10 min). Type I collagen N-terminal propeptide (P1NP) and C-terminal telopeptides (CTX) were quantified with ELISA kits (Immunodiagnostic Systems Inc.; Fountain Hills, AZ, USA) according to the manufacturer's protocols and using an Epoch Microplate Spectrophotometer (BioTek Instruments, Inc.; Winooski, VT).
Statistical analyses
Statistical analysis was conducted using GraphPad Prism (v5.0f, GraphPad Software Inc.; La Jolla, CA, USA). All data are expressed as mean ± standard error (SE). Statistical evaluation of the data was performed using a two-way ANOVA with post-hoc Student-Newman–Keuls test when the interaction was significant (p < 0.05).
Results
Body weight and growth
Consistent with our previous studies [18,20], baseline body weight of mice was not different between WT (31.3 ± 0.8 g) and cKO (31.2 ± 0.9 g) (p > 0.05). Body weight of control mice did not change from day 0 at any time point (p > 0.05; Fig. 1A). However, there was a significant 8% decrease in the body weight of WT-Suspended mice and 5% decrease in the weight of cKO-Suspended mice at day 7 (p < 0.05), although there was no difference between genotypes (p > 0.05). HLS mice regained a similar amount of body weight, ending the experiment at a similar −5% and −3% of day 0 for WT-Suspended and cKO-Suspended, respectively (p < 0.05). These findings are similar to our previous studies [20] and not unexpected following HLS of mature rodents [28]. We measured bone length as an index of growth [29]. The length of femur (Fig. 1B) or tibia (Fig. 1C) was not different between groups.
Fig. 1.
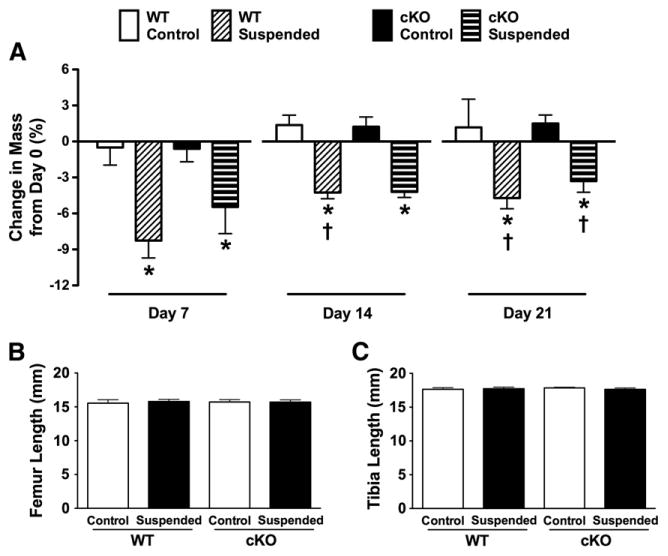
(A) Change in body weight from baseline (day 0) in wild-type (WT) and Cx43 conditional knockout (cKO) mice following three weeks of normal loading conditions (Control) or mechanical unloading via hindlimb suspension (Suspended). There were no significant differences between any of the groups with respect to the length of the (B) femur or (C) tibia. Comparisons via two-way ANOVA. Mean ± SE. For body weight, * indicates a significant difference (p < 0.05) between Control and Suspended within a genotype at a given time point, while † indicates a significant difference (p < 0.05) between that experimental group and the equivalent group at day 7. n = 5/group.
Connexin 43 deficient mice have numerous empty and TUNEL-positive cortical lacunae
There were more empty lacunae in femur cortical bone in cKO-Control (63% empty lacunae) vs. WT-Control mice (9% empty lacunae) (p < 0.05; Fig. 2A). HLS resulted in a greater number of empty lacunae: WT-Suspended mice had twice as many empty lacunae as WT-Control (p < 0.05). There was no difference between cKO-Suspended and cKO-Control (p > 0.05). Representative H&E-stained sections of femur cortical bone highlight the numerous empty lacunae in cKO vs. WT (Fig. 2B). There were no empty lacunae in trabecular bone of cKO mice at the proximal tibia or distal femur (Supplementary Fig. 1), a finding consistent with studies of lumbar vertebrae from Cx43 deficient mice [30,31].
Fig. 2.
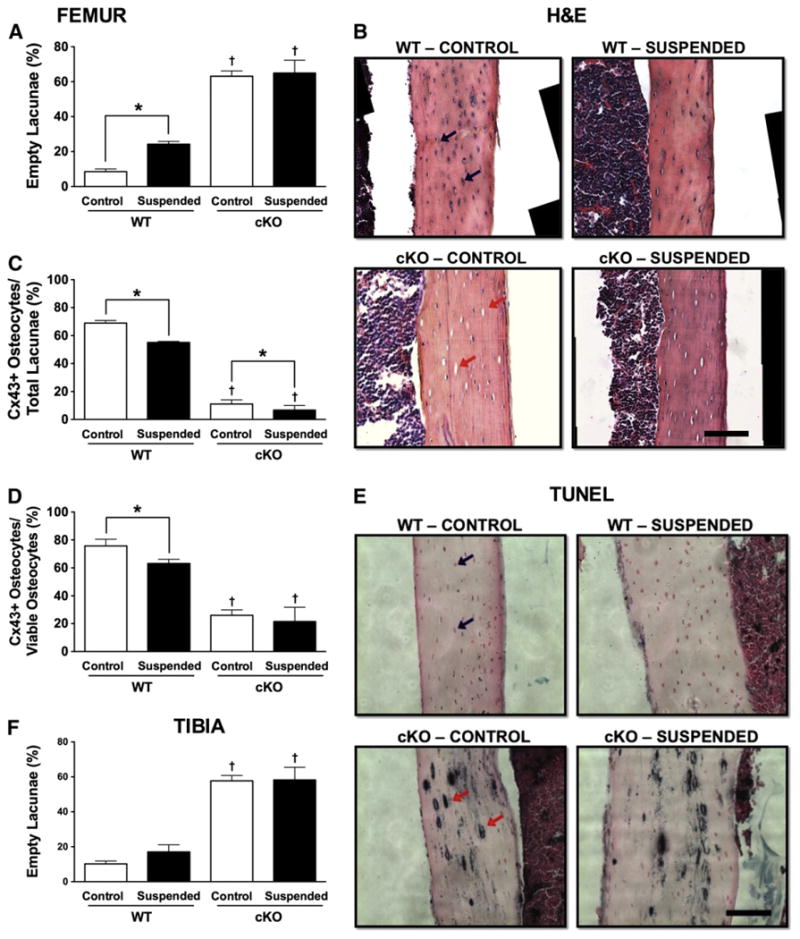
(A) Quantification of the relative number of empty lacunae within femur cortical bone obtained from wild-type (WT) and Cx43 conditional knockout (cKO) mice following three weeks of normal loading conditions (Control) or mechanical unloading via hindlimb suspension (Suspended). (B) Representative H&E section of femur cortical bone. Cx43+ osteocytes were quantified relative to both (C) total lacunae and (D) occupied lacunae. (E) TUNEL staining of femur cortical bone. (F) Empty lacunae quantified in tibia cortical bone. In both images, the black bar represents 100 μm. Comparisons via two-way ANOVA. Mean ± SE. * indicates a significant difference (p < 0.05) between Control and Suspended within a genotype, while † indicates a significant difference (p < 0.05) between WT and cKO within a loading condition. n = 3–5/group.
Relative to the total number of lacunae present, cortical bone of cKO-Control mice had 84% fewer Cx43+ osteocytes vs. WT-Control (p < 0.05; Fig. 2C). In order to account for the presence of empty cortical lacunae in cKO mice, we also determined the percentage of Cx43 + cells relative to occupied lacunae (Fig. 2D). As a percentage of occupied lacunae, there was a 66% reduction in Cx43+ cells in cKO-Control mice relative to WT-Control (p < 0.05).
Relative to the total number of lacunae, WT-Suspended mice had 20% fewer Cx43+ osteocytes vs. WT-Control, while cKO-Suspended had 40% fewer compared to cKO-Control (p < 0.05 for both; Fig. 2C). Accounting for only occupied lacunae, there was a similar 17% reduction in Cx43+ cells for WT-Suspended vs. WT-Control (p < 0.05; Fig. 2D); however, there was no difference between cKO-Suspended and cKO-Control (p > 0.05).
TUNEL + cells found throughout femur cortical bone sections suggest an apoptotic process accounting for the empty lacunae (Fig. 2E). Unfortunately, there were not enough sections available to accurately quantify TUNEL + cells. TUNEL staining was not different between WT and cKO for trabecular bone in the tibia or femur (Supplementary Fig. 2).
There was a similar pattern of empty lacunae in tibia cortical bone when comparing cKO-Control to WT-Control (Fig. 2F), although there was no significant effect of unloading on the percentage of empty lacunae in WT mice at this site (p > 0.05). TUNEL-positive staining was also abundant in tibia cortical bone from cKO mice (data not shown).
Cx43 deficient mice have attenuated whole bone sclerostin following unloading
cKO-Control mice displayed a trend for Sost mRNA levels that were nearly three times lower than WT-Control mice, although the difference was not significant (p = 0.36, Fig. 3A). HLS resulted in Sost mRNA levels in WT mice that were 433% greater than those in WT-Control (p < 0.05). There was no difference between cKO-Suspended and cKO-Control mice (p > 0.05).
Fig. 3.
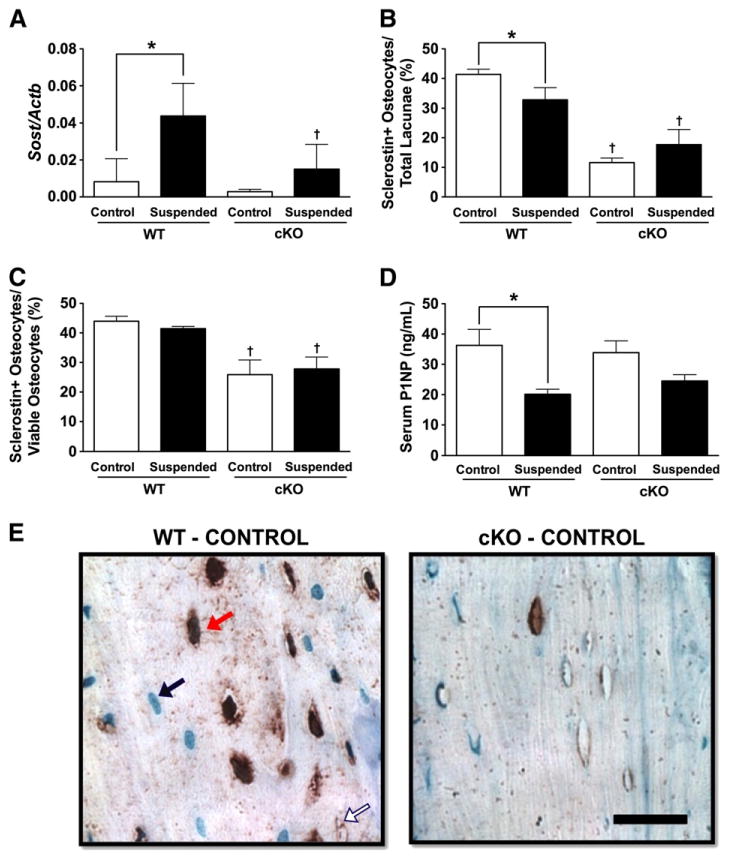
(A) mRNA levels of the gene encoding sclerostin (Sost) were determined from whole tibias and femurs obtained from wild-type (WT) and Cx43 conditional knockout (cKO) mice following three weeks of normal loading conditions (Control) or mechanical unloading via hindlimb suspension (Suspended). Quantification of the number of sclerostin + osteocytes was performed in femur cortical bone relative to (B) total lacunae and (C) occupied lacunae. (D) Serum levels of P1NP. (E) A representative section of femur cortical bone. The black bar represents 25 μm. Comparisons via two-way ANOVA. Mean ± SE. * indicates a significant difference (p < 0.05) between Control and Suspended within a genotype, while † indicates a significant difference (p < 0.05) between WT and cKO within a loading condition. n = 5/group, measured in triplicate, for RT-PCR. n = 3–5/group for serum and osteocyte quantification.
Sclerostin + osteocytes were quantified at the femur midshaft. Relative to the total number of lacunae, cKO-Control mice had 72% fewer sclerostin + osteocytes than WT-Control (p < 0.05; Fig. 3B). Surprisingly, HLS reduced the number of sclerostin + osteocytes by 21% in WT mice (p < 0.05), while there was no difference between cKO-Suspended and cKO-Control (p > 0.05). Excluding empty lacunae, cKO-Control mice had 41% fewer sclerostin + osteocytes compared to WT-Control (p < 0.05; Fig. 3C). There was no difference between Suspended and Control for either genotype (p > 0.05). A representative image of sclerostin+ cells is shown in Fig. 3E.
Serum P1NP, a marker of bone formation, was not different between cKO- and WT-Control (p > 0.05; Fig. 3D). P1NP levels in WT-Suspended mice were 45% lower than those in WT-Control (p < 0.05). P1NP levels did not decrease significantly in cKO-Suspended (p > 0.05).
Cx43 deficiency attenuates the unloading-induced increase in osteoclast number
There was no difference between WT- and cKO-Control animals with respect to Oc.N (Fig. 4A) or Oc.S (Fig. 4B) in tibia trabecular bone (p > 0.05 for both). Unloading resulted in increased Oc.N (+134%) and Oc.S (+122%) in WT mice (p < 0.05 for both). There was no difference between cKO-Control and cKO-Suspended (p > 0.05). Trabecular osteoclasts can be seen in Fig. 4H. There was a trend for greater Oc.N along the endocortical surface of femurs for cKO-Control vs. WT-Control (+117%, p = 0.08; Fig. 4C). Oc.S, however, was 85% greater for cKO-Control vs. WT-Control (p < 0.05; Fig. 4D). Similar to trabecular bone, unloading resulted in a significant increase in both femur endocortical Oc.N (+259%) and Oc.S (+213%) in WT mice (p < 0.05 for both), with no differences in cKO (p > 0.05). Cortical osteoclasts can be seen in Fig. 4I.
Fig. 4.

(A) Osteoclast number (Oc.N/BS) and (B) osteoclast surface (Oc.S/BS) were determined in proximal tibia trabecular bone from wild-type (WT) and Cx43 conditional knockout (cKO) mice following three weeks of normal loading (Control) or mechanical unloading via hindlimb suspension (Suspended). Osteoclast parameters were also quantified along the endocortical surface of femur, including (C) Oc.N/BS and (D) Oc.S/BS. mRNA levels of (E) Tnsfs11, the gene encoding RANKL, (F) Tnsfs11b, the gene encoding OPG, and (G) serum levels of CTX. Representative images show (H) trabecular and (I) cortical osteoclasts. The black bars represent 50 μm. Comparisons via two-way ANOVA. Mean ± SE. * indicates a significant difference (p < 0.05) between Control and Suspended within a genotype, while † indicates a significant difference (p < 0.05) between WT and cKO within a loading condition. n = 5/group, measured in triplicate, for RT-PCR. n = 3–5/group for serum and osteoclast quantification.
mRNA levels of Tnsfs11 (encodes RANKL) were not different between WT- and cKO-Control (p > 0.05; Fig. 4E). Tnsfs11 levels were four-fold greater for WT-Suspended vs. WT-Control (p < 0.05), whereas Tnsfs11 levels in cKO-Suspended mice were just two-fold greater than cKO-Control (p < 0.05); however, there was no significant difference in Tnsfs11 mRNA levels between WT- and cKO-Suspended. There was no difference between any groups with respect to Tnsfs11b (encodes OPG) (p > 0.05; Fig. 4F) or serum CTX (p > 0.05; Fig. 4G).
Discussion
In the present study, we found that cortical bone in the tibia and femur of Cx43 deficient mice contained numerous empty lacunae and abundant TUNEL staining, suggestive of an apoptotic process. These findings are similar to a previous study by Bivi et al., which documented increased osteocyte apoptosis in cortical bone from Cx43 deficient mice [30]. Unloading of WT mice led to increased Sost mRNA expression and decreased serum P1NP. Conversely, Sost expression and P1NP levels were not different between control and unloaded Cx43 deficient mice, nor was cortical bone formation rate, as demonstrated in our previous study [20]. There was a large increase in cortical osteoclast indices in unloaded WT mice, while Cx43 deficient mice saw no increase. These results suggest that a baseline decrease in cortical osteocyte viability induced by Cx43 deficiency may lead to increased bone formation and reduced bone resorption, relative to WT mice, during mechanical unloading. Attenuated trabecular osteoclast indices, despite no change in osteocyte viability in this compartment, suggest that an alternative mechanism may also be important in trabecular bone.
Osteocyte-derived sclerostin is the primary mediator of unloading-induced bone loss [21]. Consequently, a paucity of osteocytes would be expected to desensitize bone to the catabolic effects of unloading. Tatsumi et al. found that acute osteocyte ablation was able to prevent a rise in sclerostin expression during HLS [32]. Similarly, we did not observe an increase in Sost expression with unloading in Cx43 deficient mice. We also noted fewer sclerostin-positive cortical osteocytes in unloaded cKO mice compared to WT. These findings are consistent with our previous report of preserved endocortical and periosteal bone formation rate in cKO mice subjected to HLS [20] and suggest that the de-sensitization of cortical bone to unloading is at least partly the result of a fewer viable osteocytes leading to lower whole bone sclerostin.
Mechanical unloading has been documented to result in increased osteocyte apoptosis [33]. Indeed, we noted an increased proportion of empty lacunae in cortical bone of WT-Suspended vs. WT-Control mice. In addition, we found that WT mice subjected to unloading had fewer sclerostin-positive cortical osteocytes compared to WT control mice; however, sclerostin-positive osteocytes as a fraction of occupied lacunae remained the same. This finding suggests that unloading dramatically increases individual osteocyte expression of Sost, rather than increasing the total number of sclerostin-positive cells. Consistent with this finding, Robling et al. found increased Sost levels in whole tibias following HLS, with no change in sclerostin-positive osteocytes relative to occupied lacunae [34].
There was no difference in empty lacunae or TUNEL staining within the trabecular bone of cKO mice. Similarly, Plotkin et al. found no change in osteocyte apoptosis in trabecular bone of the lumbar vertebrate from Cx43 deficient mice [31]. Despite this, we have previously documented attenuated trabecular bone loss following unloading of Cx43 deficient mice [20]. These findings suggest the involvement of another mechanism, independent of osteocyte apoptosis, in the attenuated response of these mice to unloading. An intriguing possibility is that sclerostin from more abundant cortical osteocytes could be influencing trabecular bone, overriding the effects of trabecular osteocytes. However, while sclerostin secreted by cortical osteocytes is able to traverse the canalicular network to act on effector cells on cortical bone surfaces [35], it is unclear if it could influence more distant trabecular bone cells.
The specific mechanism by which Cx43 deficiency affects sclerostin and possibly RANKL levels in a manner that protects against unloading induced changes in cortical and trabecular bones is unknown. However, emerging evidence suggest a link between Cx43 and the sclerostin/Wnt/β-catenin pathway. For example, binding of β-catenin by Cx43 has been documented in non-bone tissues [4,7,36]. Recently, Bivi et al. found that β-catenin protein expression is increased in Cx43 deficient bone and there is increased expression of several β-catenin target genes in Cx43 knockdown MLOY-4 osteocytic cells [37]. Thus, during Cx43 deficiency, more β-catenin would be available for subsequent transcriptional activation of osteogenic genes [38]. Regardless, the current evidence suggests that osteocyte apoptosis is likely not the sole factor mediating the protective effect of Cx43 deficiency.
We found increased endocortical osteoclast activity in cKO-Control mice, consistent with the cortical thinning and expansion of the marrow cavity documented previously by our laboratory [18,20] and others [24,30]. We did not find any difference in trabecular osteoclast indices between WT- and cKO-Control, which is consistent with the lack of baseline differences in trabecular bone microstructural phenotype in this model of Cx43 deficiency. Following unloading of WT mice, we noted significantly greater osteoclast surface at both cortical and trabecular bone compartments, with the response being attenuated in cKO mice. These findings indicate that Cx43 deficiency reduces the induction of osteoclast activity during unloading. Similarly, Grimston et al. detected attenuated osteoclast indices following hindlimb immobilization of mice with an α1(I)-collagen-driven deficiency of Cx43 [19].
In our previous study, we did not detect a difference in cortical bone loss between WT and Cx43 deficient mice using in vivo pre- and post-unloading MicroCT [20]. Despite this, we did find preservation of histomorphometric cortical bone formation rate following unloading of cKO mice, consistent with the attenuated Sost expression and higher serum P1NP levels we documented here. These findings suggested that a similar level of osteoclast-mediated bone resorption must dominate the response to unloading in these mice. Therefore, we were surprised to find attenuated cortical osteoclast parameters in cKO-Suspended mice in the present study. This may be due to the fact that osteoclast indices represent a single endpoint measurement, and do not reflect accumulated microstructural changes occurring over the course of the three-week unloading period. There may have been increased cKO osteoclast activity that subsequently declined following a period of initial acclimation to unloading. Indeed, previous studies have shown that osteoclast activity peaks early during HLS [39]. Future studies should sacrifice mice at multiple time points (e.g., days 3, 5, 7, 14) in order to determine the early effects of Cx43 deficiency on cortical microstructure and osteoclast indices during HLS. In addition, suppression of bone formation, rather than increased osteoclast activity, tends to dominate the response to unloading in younger mice [11]. It may be possible to detect the maintenance of cortical microstructure with unloading in younger mice if preservation of bone formation rate is the most important effect of Cx43 deficiency. Indeed, younger, 4-month-old Cx43 deficient mice have attenuated cortical bone loss as assessed by MicroCT following botox immobilization [19].
Following three weeks of unloading, Tnsfs11 (encodes RANKL) mRNA levels in WT mice were not significantly different from cKO. In addition, we detected no difference in the levels of Tnsfs11 (encodes OPG). This is surprising, considering our aforementioned finding of reduced osteoclast indices in cKO-Suspended versus WT-Suspended and that RANKL is a critical mediator of osteoclast differentiation and activation during unloading [32]. At first glance, these findings suggest that Cx43 deficiency affects osteoclast activity through a non-RANKL/OPG-dependent mechanism. For example, sclerostin has been shown to directly regulate osteoclast activity and bone resorption in vitro [22] and in vivo [12,40]. In addition, osteocyte apoptosis has been shown to precede bone loss during unloading [33] and the release of cell contents during this process is a critical step in osteoclast recruitment [30,41]. Given that osteocytes are significantly reduced in number at baseline in Cx43 deficient cortical bone, there are not likely to be as many osteocytes undergoing active apoptosis during unloading to recruit additional osteoclasts for bone resorption. Supporting this, the proportion of empty cortical lacunae increased significantly following unloading of WT mice, with no change for Cx43 deficient mice. However, it is also important to note that, despite the lack of a significant difference in expression of Tnsfs11 at day 21, there was a trend for lower levels in cKO-Suspended. As was the case for CTX, it may be that Tnsfs11 levels have begun to return to baseline following three weeks of unloading. Thus, measurement of gene expression at earlier time points may allow us to observe significant attenuation of Tnsfs11 expression in cKO mice. This would not be unexpected, given our finding of reduced baseline osteocyte viability and that osteocytes are the primary source of RANKL mediating bone remodeling and resorption during unloading [42].
Conclusions
Previously, we documented attenuated trabecular bone loss and preservation of cortical bone formation in Cx43 deficient mice following mechanical unloading via hindlimb suspension. In the present study, we show that Cx43 deficient mice do not experience many of the deleterious cellular and molecular changes induced by mechanical unloading. At baseline, Cx43 deficient mice have abundant empty lacunae and increased TUNEL staining throughout the cortical bone, suggesting increased osteocyte apoptosis. When subjected to unloading, these mice have reduced whole bone Sost expression and attenuation of osteoclast indices. Additional studies are needed to define the role of Cx43 deficiency in the trabecular compartment, given that trabecular bone loss and osteoclast indices are reduced with unloading, yet there is no difference in trabecular osteocyte viability. Regardless of the specific mechanism, these data demonstrate that Cx43 deficiency desensitizes bone to the effects of unloading by modulation of both arms of bone remodeling.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This work was supported by grants R01 AG013087 from the National Institute on Aging and MA02802 from the National Space Biomedical Research Institute. Thanks to Dr. Arthur Berg, Assistant Professor of Biostatistics and Bioinformatics, for guidance on statistical analyses.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bone.2013.07.022.
Conflict of interest: The authors have no financial interests or conflicts of interest to disclose.
References
- 1.Civitelli R, Beyer EC, Warlow PM, Robertson AJ, Geist ST, Steinberg TH. Connexin43 mediates direct intercellular communication in human osteoblastic cell networks. J Clin Invest. 1993;91:1888–96. doi: 10.1172/JCI116406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang JX, Siller-Jackson AJ, Burra S. Roles of gap junctions and hemichannels in bone cell functions and in signal transmission of mechanical stress. Front Biosci. 2007;12:1450–62. doi: 10.2741/2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bivi N, Lezcano V, Romanello M, Bellido T, Plotkin LI. Connexin43 interacts with betaarrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J Cell Biochem. 2011;112:2920–30. doi: 10.1002/jcb.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li MW, Mruk DD, Lee WM, Cheng CY. Connexin 43 and plakophilin-2 as a protein complex that regulates blood-testis barrier dynamics. Proc Natl Acad Sci U S A. 2009;106:10213–8. doi: 10.1073/pnas.0901700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loiselle AE, Jiang JX, Donahue HJ. Gap junction and hemichannel functions in osteocytes. Bone. 2013 Jun;54(2):205–12. doi: 10.1016/j.bone.2012.08.132. [DOI] [PubMed] [Google Scholar]
- 6.Plotkin LI. Connexin 43 and bone: not just a gap junction protein. Actual Osteol. 2011;7:79–90. [PMC free article] [PubMed] [Google Scholar]
- 7.Talhouk RS, Mroue R, Mokalled M, Abi-Mosleh L, Nehme R, Ismail A, et al. Heterocellular interaction enhances recruitment of alpha and beta-catenins and ZO-2 into functional gap-junction complexes and induces gap junction-dependant differentiation of mammary epithelial cells. Exp Cell Res. 2008;314:3275–91. doi: 10.1016/j.yexcr.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 8.Ton QV, Iovine MK. Determining how defects in connexin43 cause skeletal disease. Genesis. 2013;51:75–82. doi: 10.1002/dvg.22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loiselle AE, Paul EM, Lewis GS, Donahue HJ. Osteoblast and osteocyte-specific loss of connexin43 results in delayed bone formation and healing during murine fracture healing. J Orthop Res. 2013;31:147–54. doi: 10.1002/jor.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner CH, Warden SJ, Bellido T, Plotkin LI, Kumar N, Jasiuk I, Jasiuk N, et al. Mechanobiology of the skeleton. Sci Signal. 2009;2(pt3) doi: 10.1126/scisignal.268pt3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bikle DD, Halloran BP. The response of bone to unloading. J Bone Miner Metab. 1999;17:233–44. doi: 10.1007/s007740050090. [DOI] [PubMed] [Google Scholar]
- 12.patz J, Ellman R, Cloutier A, Louis L, van Vliet M, Suva L, et al. Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. J Bone Miner Res. 2013 Apr;28(4):865–74. doi: 10.1002/jbmr.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lloyd SA, Bandstra ER, Willey JS, Riffle SE, Tirado-Lee L, Nelson GA, et al. Effect of proton irradiation followed by hindlimb unloading on bone in mature mice: a model of long-duration spaceflight. Bone. 2012;51:756–64. doi: 10.1016/j.bone.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein TP. Weight, muscle and bone loss during space flight: another perspective. Eur J Appl Physiol. 2012 Nov 29; doi: 10.1007/s00421-012-2548-9. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 15.Maimoun L, Fattal C, Micallef JP, Peruchon E, Rabischong P. Bone loss in spinal cord-injured patients: from physiopathology to therapy. Spinal Cord. 2006;44:203–10. doi: 10.1038/sj.sc.3101832. [DOI] [PubMed] [Google Scholar]
- 16.Grimston SK, Brodt MD, Silva MJ, Civitelli R. Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the connexin43 gene (Gja1) J Bone Miner Res. 2008;23:879–86. doi: 10.1359/JBMR.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimston SK, Watkins M, Brodt M, Silva M, Civitelli R. Variable bone formation response to skeletal axial load in mice with a conditional deletion of the connexin43 (Cx43) gene (Gja1) 2011 Annual Meeting of the American Society of Bone and Mineral Research; San Diego, CA: 2011. p. S75. [Google Scholar]
- 18.Zhang Y, Paul EM, Sathyendra V, Davison A, Sharkey N, Bronson S, et al. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6:e23516. doi: 10.1371/journal.pone.0023516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res. 2011;26:2151–60. doi: 10.1002/jbmr.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd SA, Lewis GS, Zhang Y, Paul EM, Donahue HJ. Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J Bone Miner Res. 2012;27:2359–72. doi: 10.1002/jbmr.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24:1651–61. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 22.Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6:e25900. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bivi N, Aguirre JI, Vyas K, Allen M, Bellido T, Plotkin L. Increased osteocyte apoptosis and bone resorption, and decreased strength of cortical but not trabecular bone in mice lacking connexin43 in osteoblasts and osteocytes. J Bone Miner Res. 2009;24 [Google Scholar]
- 24.Watkins M, Grimston SK, Norris JY, Guillotin B, Shaw A, Beniash E, et al. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell. 2011;22:1240–51. doi: 10.1091/mbc.E10-07-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–4. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–12. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 27.Castro CH, Stains JP, Sheikh S, Szejnfeld VL, Willecke K, Theis M, et al. Development of mice with osteoblast-specific connexin43 gene deletion. Cell Commun Adhes. 2003;10:445–50. doi: 10.1080/cac.10.4-6.445.450. [DOI] [PubMed] [Google Scholar]
- 28.Morey-Holton ER, Globus RK. Hindlimb unloading rodent model: technical aspects. J Appl Physiol. 2002;92:1367–77. doi: 10.1152/japplphysiol.00969.2001. [DOI] [PubMed] [Google Scholar]
- 29.Halloran BP, Ferguson VL, Simske SJ, Burghardt A, Venton LL, Majumdar S. Changes in bone structure and mass with advancing age in the male C57BL/6J mouse. J Bone Miner Res. 2002;17:1044–50. doi: 10.1359/jbmr.2002.17.6.1044. [DOI] [PubMed] [Google Scholar]
- 30.Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res. 2012;27:374–89. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plotkin LI, Lezcano V, Thostenson J, Weinstein RS, Manolagas SC, Bellido T. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J Bone Miner Res. 2008;23:1712–21. doi: 10.1359/JBMR.080617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tatsumi S, Ishii K, Amizuka N, Li M, Kobayashi T, Kohno K, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–75. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 34.Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 35.Moester MJ, Papapoulos SE, Lowik CW, van Bezooijen RL. Sclerostin: current knowledge and future perspectives. Calcif Tissue Int. 2010;87:99–107. doi: 10.1007/s00223-010-9372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ai Z, Fischer A, Spray DC, Brown AM, Fishman GI. Wnt-1 regulation of connexin43 in cardiac myocytes. J Clin Invest. 2000;105:161–71. doi: 10.1172/JCI7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bivi N, Pacheco-Costa R, Brun LR, Murphy TR, Farlow NR, Robling AG, et al. Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J Orthop Res. 2013 Jul;31(7):1075–81. doi: 10.1002/jor.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu G, Vijayakumar S, Grumolato L, Arroyave R, Qiao H, Akiri G, et al. Canonical Wnts function as potent regulators of osteogenesis by human mesenchymal stem cells. J Cell Biol. 2009;185:67–75. doi: 10.1083/jcb.200810137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nabavi N, Khandani A, Camirand A, Harrison RE. Effects of microgravity on osteoclast bone resorption and osteoblast cytoskeletal organization and adhesion. Bone. 2011;49:965–74. doi: 10.1016/j.bone.2011.07.036. [DOI] [PubMed] [Google Scholar]
- 40.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 41.Kogianni G, Mann V, Noble BS. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J Bone Miner Res. 2008;23:915–27. doi: 10.1359/jbmr.080207. [DOI] [PubMed] [Google Scholar]
- 42.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hargens AR, Steskal J, Johansson C, Tipton CM. Tissue fluid shift, forelimb loading, and tail tension in tail suspended rats. Physiologist. 1984;27:S37–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.