

Abstract
RASopathies are a class of genetic syndromes caused by germline mutations in genes encoding Ras/MAPK pathway components. Cardio-facio-cutaneous (CFC) syndrome is a RASopathy characterized by distinctive craniofacial features, skin and hair abnormalities, and congenital heart defects caused by activating mutations of BRAF, MEK1, MEK2, and KRAS. We define the phenotype of seven patients with de novo deletions of chromosome 19p13.3 including MEK2; they present with a distinct phenotype but have overlapping features with CFC syndrome. Phenotypic features of all seven patients include tall forehead, thick nasal tip, underdeveloped cheekbones, long midface, sinuous upper vermilion border, tall chin, angular jaw, and facial asymmetry. Patients also have developmental delay, hypotonia, heart abnormalities, failure to thrive, obstructive sleep apnea, GE reflux and integument abnormalities. Analysis of EGF stimulated fibroblasts revealed that P-MEK1/2 was ~50% less abundant in cells carrying the MEK2 deletion compared to the control. Significant differences in total MEK2 and Sprouty1 abundance were also observed. Our cohort of seven individuals with MEK2 deletions has overlapping features associated with RASopathies. This is the first report suggesting that, in addition to activating mutations, MEK2 haploinsufficiency can lead to dysregulation of the MAPK pathway.
Keywords: MAP2K2, MEK2, deletion 19p13.3, RASopathy, Ras/MAPK pathway, MEK2, CFC, Cardio-facio-cutaneous syndrome
INTRODUCTION
The Ras/mitogen-activated protein kinase (Ras/MAPK) pathway is a critical signaling pathway with an important role in cellular proliferation, differentiation, growth, senescence and death – all of which are critical to normal development (1,2). This pathway regulates signals transduced from the cell surface to the nucleus and codes for proteins that serve as molecular on/off switches which activate or inhibit downstream effectors.
Germline mutations of genes encoding components of this pathway have been described in Noonan (NS; OMIM 163950), cardio-facio-cutaneous (CFC; OMIM 115150), Legius (OMIM 611431), and Costello (CS; OMIM 218040) syndromes, capillary malformation-arteriovenous malformation (OMIM 608354) and neurofibromatosis type 1 (NF1; OMIM 162200). These conditions are collectively referred to as RASopathies and, as a group, are characterized by cardiac defects, distinct craniofacial features, skin abnormalities, variable degrees of cognitive deficits, growth retardation, and a predisposition to benign and malignant neoplasia (1). The majority of the mutations identified in the RASopathies are mutations which increase Ras/MAPK pathway signaling, many of which are missense mutations (1). However, whole gene deletions and duplications have also been reported in patients with NF1 and Noonan syndrome, respectively (3–5).
There is both locus and allelic heterogeneity within the syndromes that comprise the RASopathies, however, heterozygous missense mutations in MAP2K2 (hereafter referred to as MEK2), which codes for the protein kinase MEK2, have been associated only with CFC syndrome to date (6). CFC is a multiple congenital anomaly disorder characterized by distinct facial features, ectodermal manifestations, cardiac defects, gastrointestinal abnormalities and mild to severe intellectual disability (7–9). The craniofacial features resemble those observed in Noonan syndrome, however, in general, individuals with CFC syndrome tend to present with more severe medical complications and developmental disability (10). To date all of the identified MEK2 mutations that cause CFC syndrome are heterozygous gain-of-function mutations that activate the downstream pathway (6,12). There have been no reports of CFC associated with deletions or haploinsufficiency of MEK2 (13–15). Two patients with microdeletions of 19p13.3 which include MEK2 have been reported; (16,17) their phenotype is similar to that observed in the patients that are the subject of this report.
We report seven patients with submicroscopic deletions in 19p13.3 in whom the smallest region of overlap contains only one known gene, MEK2. These seven patients have clinical features suggestive of an abnormality in the Ras/MAPK pathway yet are not diagnostic for classical CFC or any of the other previously described RASopathies. Through functional assays of the MAPK pathway, we show that the deletion and the resultant haploinsufficiency of MEK2 causes dysregulation of the Ras/MAPK pathway, and we propose that haploinsufficiency of MEK2 may be a mechanism for a novel RASopathy phenotype.
METHODS
Patients
We obtained clinical data and results of laboratory testing on seven patients with microdeletions that included MEK2. The microdeletion breakpoints have been converted to human genome build 19 using the UCSD Genome Browser on-line tool. Clinical photographs of all seven patients in our cohort were reviewed by M.J.N, U.M. and K.A.R. (clinical geneticists). Informed consents to publish the photographs of all the patients were obtained from the patients’ guardians as were the consents for publication of the clinical data and sample procurement.
Primary human fibroblasts and cell culture conditions
Primary human fibroblasts were derived from a skin biopsy obtained from patient 1. The control primary human fibroblast cell line is a commercially available line derived from a skin biopsy of a healthy 3 year old male with a normal karyotype (Coriell Institute, GM00498). Primary cells were cultivated under standard conditions in either complete media [High Glucose Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (FBS; JRScientific, 43652), 0.1 mM MEM Non-Essential Amino Acid Solution (Gibco, 11140-050), 110 mg/L sodium pyruvate, 292 mg/L L-glutamine, and penicillin-streptomycin] or serum starvation media [complete media with FBS decreased to 0.1%]. Cells were cultured at 37°C in 5% CO2. All experiments were performed in triplicate between cell passages 6 and 17.
Ras/MAPK pathway stimulation experiments
Primary human fibroblasts were seeded in complete media at a density of 80,000 cells/35 mm dish. Cell culture dishes were incubated at 37°C in 5% CO2. At subconfluency, cells were serum starved and incubated overnight (16 hr). Cells remaining in standard 10% FBS served as a control. Post serum starvation, cells were treated with or without serum starvation media supplemented with 2 ng/ml Epidermal Growth Factor (EGF) (Sigma, E9644). Cells were harvested at the indicated time points after EGF exposure.
Western blotting
Total cellular protein was prepared using 1X Cell Lysis Buffer (Cell Signaling, 9803) with 1 mM phenylmethylsulfonyl fluoride (PMSF). Protein concentrations were determined using the DC Protein Assay Kit (Biorad). Samples were diluted with NuPAGE LDS sample buffer (Invitrogen) with 5% beta-mercaptoethanol (BME). Two micrograms of denatured protein was separated on 4–12% Bis Tris gels (Invitrogen, NP0329BOX) by gel electrophoresis using the XCell SureLock Mini-Cell (Invitrogen, EI0002). Proteins were transferred to PVDF membrane using the X Cell II blot module (Invitrogen, EI0002). The following primary antibodies were used: P-MEK1/2 (Cell Signaling, 2338), P-ERK1/2 (Cell Signaling, 9101), Total MEK1 (Cell Signaling, 2352), Total MEK2 (Cell Signaling, 9147), Total ERK (Cell Signaling, 9102), Sprouty 1 (Zymed, 40–1800), Anti-GAPDH (Ambion, AM4300). Proteins were detected by chemiluminescence using the Visualizer Western Blot Detection Kit (Millipore, 64–202 or 64–201). Western blot exposures were quantified using Image J software (18). Three separate experiments were quantified and used to calculate mean relative intensity and standard error. Statistical significance of the mean relative band intensities of the control compared to patient 1 was determined by performing a Student’s T-test.
RESULTS
Patient 1
This boy was referred at 14 months for assessment of macrocephaly, developmental delay and dysmorphic features (Fig. 1A and Table 1). Karyotype was 46,XY at a 550 band level. At age 11 years, he was profoundly delayed with single words only, and had a history of G-tube feeding for failure to thrive, sleep apnea and right bundle branch block. BAC-based chromosome microarray analysis (Kleberg Cytogenetics Laboratory CMA V 5.0) detected a deletion of 19p13.3 (1 clone: RP11-454N6) which includes MEK2. Whole genome oligonucleotide array CGH showed a 1.6 Mb deletion spanning chr19:3,308,930–4,894,921 (hg19; Fig. 2). Parental studies were normal.
Figure 1. Craniofacial features of patients.
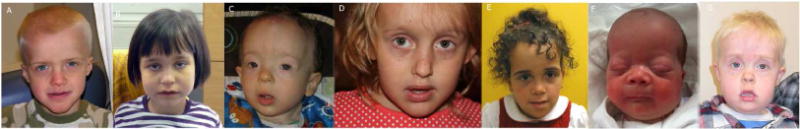
Panels A–G, patients 1–7. Note the tall forehead, underdeveloped cheekbones, thick nasal tip, sinuous upper vermilion border and thick lower vermilion, and prominent, and tall chin with angular jaw; facial asymmetry is also seen.
Table 1.
Clinical features of nine patients with MAP2K2/MEK2 deletions.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 (16) |
Patient 9 (17) |
|
|---|---|---|---|---|---|---|---|---|---|
| Sex | M | F | M | F | F | M | M | F | F |
| Age | 10y | 5y 9mo | 3y | 4y | 6y | 2mo | 5y | 4y | 15y |
| Deletion (hg19) Chr19: |
3,308,930–4,894,921 | 3,832,931–5,654,449 | 3,957,908–4,349,804 | 4,089,863–4,145,062 | 3,068,450–4,102,449 | 3,533,803–4,585,171 | 3,379,721–4,182,307 | 2,904,809–4,837,388 | 4,090,320–4,124,126 |
| Deletion Size [Mb] | 1.6 | 1.82 | 0.392 | 0.06 | 1.03 | 1.05 | 0.803 | 1.93 | 0.174 |
| Growth | |||||||||
| BW | 3.6 kg | 2.460 kg | 2.47kg | 3.84 kg | 3.66 kg | 1.11kg (27/40) | 2.90k (41/40) | 90% | na |
| Failure to thrive | + | + | + | + | + | + | − | + | − |
| HC | +2.5 SD | −3.5 SD | −2.5 SD | +3.5 SD | +5.5 SD | +1 SD | +3.5 SD | +3 SD | +4.55 SD |
| Intellectual disability | Severe delay no speech | Severe delay no speech | mild delay | mild delay | severe delay | na* | mild to moderate delay | severe delay | mild delay |
| Reason for referral | DD | DD | DF | DD+DF | R/O RAS | MCA | DD+DF | DD | OW |
| Facial Features | |||||||||
| Prominent forehead | + | + | + | + | + | + | + | + | + |
| Horizontal/downslanting PF | + | + | + | + | + | + | + | + | + |
| Facial asymmetry | + | + | + | + | + | + | − | na** | na** |
| Long midface | + | + | + | + | + | + | + | + | + |
| Tall chin/angular jaw | + | + | + | + | + | + | + | + | + |
| Sinuous upper vermilion border | + | + | + | + | + | + | + | + | + |
| Thin upper vermilion border | + | − | + | − | + | + | − | + | + |
| Sparse eyebrows | + | − | − | + | + | + | − | + | + |
| Underdeveloped cheek bones | + | − | + | − | + | + | − | + | + |
| Cardiovascular system: | |||||||||
| Bicuspid aortic valve | + | − | − | − | − | − | − | − | − |
| Tetralogy of Fallot | − | − | − | − | − | − | − | + | − |
| ASD | + | − | − | + | + | − | − | − | − |
| Heart block | + | − | − | − | − | − | − | − | − |
| Gastrointestinal system: | |||||||||
| GI reflux | + | + | + | + | + | + | + | + | − |
| G-tube feeding | + | + | − | − | + | + | − | − | − |
| Umbilical hernia | − | − | + | + | − | − | − | − | − |
| Hepatomegaly | − | + | − | + | − | − | − | − | − |
| Splenomegaly | − | − | − | + | − | − | − | − | − |
| Central nervous system: | |||||||||
| Dysgenesis/agenesis of CC | − | + | − | na | + | − | − | − | na |
| Cerebellar hypoplasia | + | − | − | − | − | − | − | − | na |
| Hypotonia | − | + | + | + | − | − | +(mild) | + | |
| Hypomyelination | − | + | − | − | − | − | − | − | na |
| Obstructive sleep apnea | + | + | + | − | + | + | + | − | − |
| Integument | |||||||||
| Keratosis pilaris | + | + | − | + | − | − | − | na | na |
| Eczema | severe | − | − | − | + | − | + | na | na |
| Capillary hemanigomata | − | +scalp trunk buttocks | +forehead | − | − | +on right testis | +glabellar | − | − |
| Dysplastic nails | + | − | − | + | − | − | + | na | na |
| Widely-spaced teeth | + | − | − | + | + | na* | + | + | − |
| Sparse hair | + | − | + | − | − | + | − | + | − |
| Cutis aplasia | − | + | − | − | − | − | − | + | − |
| Other | irregular pigmented patch L leg | multiple pigmented nevi | pre-auricular skin tag | thin skin tooth enamel hypoplasia | |||||
| Other | low WBC NYD | athetosis | low platelets NYD | BPD | significant anxiety, HG, OMs | dilated aorta | BPD |
NYD – not yet diagnosed; na – information not available
patient was too young to assess
angle of photograph prevents assessment of facial symmetry; CC – corpus callosum, PF – palpebral fissures; BPD – bronchopulmonary dysplasia of prematurity; HG – hypoglycemia; OMs – recurrent otitis media; RAS – RASopathy; R/O – rule out; DD – developmental delay; MCA – multiple congenital anomalies; DF – dysmorphic features; OW – overweight
Figure 2. Schematic of the deletions in nine patients showing the relative positions of MAP2K2/MEK2 and DAPK3.
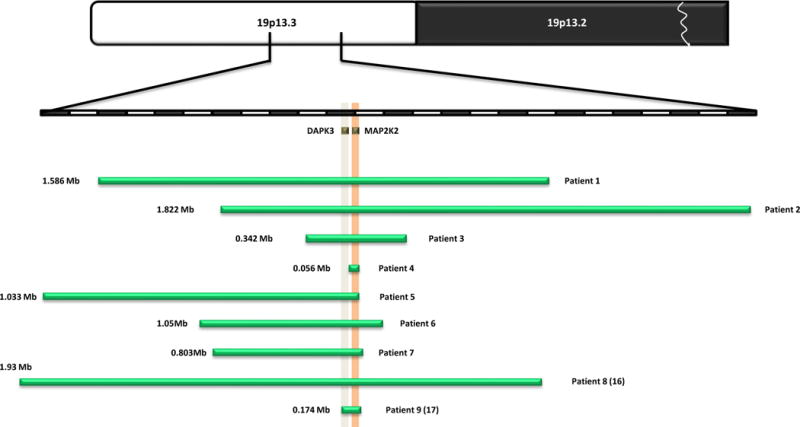
The size, extent and genomic content of deletions of chromosome 19p13.3 are shown. The upper seven green bars depict the deletions in our cohort of patients; the bottom two bars represent the patients reported by Al-Kateb et al (16), and De Smith et al. (17). MEK2 and DAPK3 are shown in orange and grey respectively. The upper chromosomal diagram is not to scale.
Patient 2
This girl was referred at 4 years, 2 months of age. At that time she presented with severe developmental delay, movement disorders, G-tube feeding for failure to thrive, frequent obstructive sleep apnea, scalp defect, a history of capillary hemangioma requiring surgical treatment, and dysmorphic features (Fig. 1B, Table 1). Karyotype was 46,XX at a 550 band level. Molecular karyotyping by 6.0 SNP-array showed a deletion of 1.82Mb in chromosome region 19p13.3: chr19:3,832,931–5,654,449 (hg19; Fig. 2). The deletion was confirmed by FISH analysis with BAC clone RP11–500M22 specific for 19p13.3. Parental studies were normal.
Patient 3
This boy was first evaluated at one day of age because of dysmorphic features (Fig. 1C) and thromobocytopenia. He developed capillary hemangiomata and craniosynosotosis required surgical correction. He had ongoing problems with apnea (Table 1). Karyotype was 46,XY at a 550 band level. Oligoarray comparative genomic hybridization showed a duplication and a deletion in chromosome 19p13.3. The duplication was from bp 41,898–3,894,256; and the deletion from bp 3,957,908–4,349,804 (hg19; Fig. 2). Parental studies were normal.
Patient 4
This female patient was referred at 4 years 7 months with minimal developmental delay and dysmorphic features (Fig. 1D) and a history of failure to thrive (Table 1). Karyotype was 46,XX at a 550 band level. Microarray analysis showed an ~ 60 kb deletion at chr19:4,089,863–4,145,062 (hg19; v.6.4 array, 44,000 oligos; Fig. 2). This deletion encompasses at least exons 2–7 of MEK2, however, the gene could be entirely deleted since the boundaries are not precise in this array version. Parental studies were normal.
Patient 5
This 6 year old girl with significant developmental delay (Fig. 1E) has a history of failure to thrive in infancy, obstructive and central apnea requiring tracheostomy and of gastroesophageal reflux requiring Nissen fundoplication (Table 1). Dysmorphic features are shown in Fig. 1E. Karyotype was 46,XX at a 550 band level. Microarray testing utilizing a BAC array with 4,200 clones (CytoChip v.2; BlueGnome Ltd, Cambridge, UK) showed deletion of one clone on the short arm of chromosome 19 within band p13.3 (Fig. 2). The deletion was confirmed by FISH with the clone RP11-60H2. A SNP microarray with 1 million markers (1M-Quad; Illumina Inc, San Diego, USA) confirmed the deletion and refined the distal and proximal breakpoints to genomic positions 3,068,450 and 4,102,449 (hg19). The size was 1.03 Mb. Parental studies were normal.
Patient 6
This twin boy was born at 27 weeks of gestation, following the death of the co-twin with trisomy 13 at 23 weeks gestation. Dysmorphic features were noted at birth (Fig. 1F). He had failure to thrive, apnea and gastroesophageal reflux (Table 1). He died at age 3 months from respiratory failure. Karyotype performed after amniocentesis showed 46,XY at 550 band level. Microarray showed a 1.05 Mb deletion on chromosome 19p13.3 (3,533,803–4,585,171) (hg19; Fig. 2). Parental studies were normal.
Patient 7
This 4 year and 11 month old boy (Fig. 1G) was first evaluated at 24 months of age for developmental delay, hypotonia and facial dysmorphism. At that visit, he was also noted to be macrocephalic. He had a history of gastroesophageal reflux (requiring medical, but not surgical treatment), constipation, eczema and tonsillar hypertrophy associated with obstructive sleep apnea. Microarray testing (Boston University Center for Human Genetics) utilizing an Affymetrix 6.0 SNP array with 900,000 SNPs and 940,000 CNV probes showed an 803 kb deletion of the short arm of chromosome 19p13.3 (3,428,721–4,231,307)(hg19). The deletion was confirmed by FISH with the clone RP11-671I19. Parental FISH studies were normal.
Summary of the clinical findings can be found in Table 1. Complete clinical data for the patients are described in the supplementary material.
Clinical data
Three patients were female and four were male; the ages ranged from 3 months to 10 years at the time of last assessment. They all present with similar craniofacial features comprising a prominent and tall forehead, long midface with underdeveloped cheekbones, facial asymmetry, and prominent and angular jaw with tall chin (Fig. 1). The patients have horizontal or down-slanting palpebral fissures and a thickened nasal tip. The mouth has a sinuous upper vermilion border and prominent (thick) lower vermilion. Somatic and physical findings observed in all the patients included gastrointestinal dysmotility manifesting as swallowing difficulties, feeding problems, gastroesophageal reflux, and failure to thrive (Table 1).
Four patients had documented macrocephaly (patients 1, 4, 5 and 7), while two others were microcephalic (patients 2, 3); macrocephaly was not correlated to size of the deletion. Congenital heart defects such as bicuspid aortic valve and ASD were seen in three patients and cardiac rhythm abnormalities in one patient (Table 1). Six had obstructive sleep apnea and three of those required nighttime CPAP or ventillatory support (Table 2). Abnormalities of bone marrow function were observed in two patients: patient 1 had lymphopenia, and patient 3 self-resolving thrombocytopenia of prenatal onset, both conditions were of unknown etiology. Cutaneous manifestations observed in the patients with MEK2 deletion were present in a similar frequency to those observed in individuals with CFC syndrome;18 particularly, multiple nevi, hemangiomata and keratosis pilaris were observed in our cohort. Hyperkeratosis pilaris was seen in three patients and three patients had severe eczema, two others had multiple and large capillary hemangiomata over the head and body, one had a large nevus flammeus (Table 1). Abnormally brittle and hyperconvex nails were seen in three patients, and one patient had congenital cutis aplasia. All patients in out cohort had gastoresophageal reflux, in four percutaneous tube feeding was required (Table 1).
Table 2.
| start | stop | Symbol | O | Cyto | Description |
|---|---|---|---|---|---|
| 3359616 | 3463603 | NFIC | + | 19p13.3 | nuclear factor I/C (CCAAT-binding transcription factor) |
| 3474405 | 3480540 | C19orf77 | − | 19p13.3 | chromosome 19 open reading frame 77 |
| 3490819 | 3500938 | DOHH | − | 19p13.3 | deoxyhypusine hydroxylase/monooxygenase |
| 3506295 | 3536755 | FZR1 | + | 19p13.3 | fizzy/cell division cycle 20 related 1 (Drosophila) |
| 3538263 | 3557571 | C19orf28 | − | 19p13.3 | chromosome 19 open reading frame 28 |
| 3539155 | 3544028 | C19orf71 | + | 19p13.3 | chromosome 19 open reading frame 71 |
| 3572943 | 3579081 | HMG20B | + | 19p13.3 | high mobility group 20B |
| 3585569 | 3593539 | GIPC3 | + | 19p13.3 | GIPC PDZ domain containing family, member 3 |
| 3594504 | 3606831 | TBXA2R | − | 19p13.3 | thromboxane A2 receptor |
| 3607245 | 3613928 | C19orf29-AS1 | + | 19p13.3 | C19orf29 antisense RNA 1 (non-protein coding) |
| 3610626 | 3626813 | C19orf29 | − | 19p13.3 | chromosome 19 open reading frame 29 |
| 3630179 | 3700477 | PIP5K1C | − | 19p13.3 | phosphatidylinositol-4-phosphate 5-kinase, type I, gamma |
| 3728374 | 3750682 | TJP3 | + | 19p13.3 | tight junction protein 3 (zona occludens 3) |
| 3750771 | 3761673 | APBA3 | − | 19p13.3 | amyloid beta (A4) precursor protein-binding, family A, member 3 |
| 3762665 | 3767563 | MRPL54 | + | 19p13.3 | mitochondrial ribosomal protein L54 |
| 3769087 | 3772219 | RAX2 | − | 19p13.3 | retina and anterior neural fold homeobox 2 |
| 3777967 | 3801810 | MATK | − | 19p13.3 | megakaryocyte-associated tyrosine kinase |
| 3804022 | 3869027 | ZFR2 | − | 19p13.3 | zinc finger RNA binding protein 2 |
| 3870792 | 3872205 | LOC100131661 | + | 19p13.3 | ferritin, light polypeptide pseudogene 5 |
| 3880618 | 3928080 | ATCAY | + | 19p13.3 | ataxia, cerebellar, Cayman type |
| 3933101 | 3942414 | ITGB1BP3 | + | 19p13.3 | integrin beta 1 binding protein 3 |
| 3958452 | 3969826 | DAPK3 | − | 19p13.3 | death-associated protein kinase 3 |
| 3961412 | 3961510 | MIR637 | − | 19p13.3 | microRNA 637 |
| 3976054 | 3985461 | EEF2 | − | 19pter-q12 | eukaryotic translation elongation factor 2 |
| 3982505 | 3982570 | SNORD37 | − | 19p13.3 | small nucleolar RNA, C/D box 37 |
| 4007749 | 4038067 | PIAS4 | + | 19p13.3 | protein inhibitor of activated STAT, 4 |
| 4041105 | 4043154 | LOC100652920 | − | putative uncharacterized protein UNQ9165/PRO28630-like | |
| 4045216 | 4066816 | ZBTB7A | − | 19p13.3 | zinc finger and BTB domain containing 7A |
| 4090319 | 4124126 | MAP2K2 | − | 19p13.3 | mitogen-activated protein kinase kinase 2 |
| 4153629 | 4173048 | CREB3L3 | + | 19p13.3 | cAMP responsive element binding protein 3-like 3 |
| 4174106 | 4182596 | SIRT6 | − | 19p13.3 | SIR2-like protein 6 |
| 4183351 | 4224811 | ANKRD24 | + | 19p13.3 | ankyrin repeat domain 24 |
| 4229540 | 4237525 | EBI3 | + | 19p13.3 | Epstein-Barr virus induced 3 |
| 4247111 | 4269085 | CCDC94 | + | 19p13.3 | coiled-coil domain containing 94 |
| 4278598 | 4290720 | SHD | + | 19p13.3 | Src homology 2 domain containing transforming protein D |
| 4292224 | 4302428 | TMIGD2 | − | 19p13.3 | transmembrane and immunoglobulin domain containing 2 |
| 4304591 | 4323843 | FSD1 | + | 19p13.3 | fibronectin type III and SPRY domain containing 1 |
| 4324040 | 4338847 | STAP2 | − | 19p13.3 | signal transducing adaptor family member 2 |
| 4343524 | 4360083 | MPND | + | 19p13.3 | MPN domain containing |
| 4360364 | 4400565 | SH3GL1 | − | 19p13.3 | SH3-domain GRB2-like 1 |
| 4402660 | 4443394 | CHAF1A | + | 19p13.3 | chromatin assembly factor 1, subunit A (p150) |
| 4445003 | 4457791 | UBXN6 | − | 19p13 | UBX domain protein 6 |
| 4445975 | 4446045 | MIR4746 | + | microRNA 4746 | |
| 4472255 | 4502223 | HDGF2 | + | 19p13.3 | hepatoma-derived growth factor-related protein 2 |
| 4502192 | 4517716 | PLIN4 | − | 19p13.3 | perilipin 4 |
| 4522543 | 4535208 | PLIN5 | − | 19p13.3 | perilipin 5 |
| 4537227 | 4540036 | LRG1 | − | 19p13.3 | leucine-rich alpha-2-glycoprotein 1 |
| 4542600 | 4559771 | SEMA6B | − | 19p13.3 | sema domain, transmembrane domain (TM), and cytoplasmic domain, (semaphorin) 6B |
| 4621556 | 4622369 | LOC100287614 | + | 19p13.3 | ribosomal protein S10 pseudogene |
| 4639527 | 4655580 | TNFAIP8L1 | + | 19p13.3 | tumor necrosis factor, alpha-induced protein 8-like 1 |
| 4657557 | 4670415 | C19orf10 | − | 19p13.3 | chromosome 19 open reading frame 10 |
| 4675243 | 4723855 | DPP9 | − | 19p13.3 | dipeptidyl-peptidase 9 |
| 4679294 | 4685960 | LOC100131094 | + | 19p13.3 | hypothetical LOC100131094 |
| 4724082 | 4724153 | TRNAG3 | + | 19p13.3 | transfer RNA glycine 3 (anticodon UCC) |
| 4724647 | 4724719 | TRNAV32 | − | transfer RNA valine 32 (anticodon CAC) | |
| 4769117 | 4772568 | MIR7-3HG | + | 19p13.3 | MIR7-3 host gene (non-protein coding) |
| 4770682 | 4770791 | MIR7-3 | + | 19p13.3 | microRNA 7-3 |
| 4791728 | 4795571 | FEM1A | + | 19p13.3 | fem-1 homolog a (C. elegans) |
| 4800220 | 4801278 | LOC100507004 | − | hypothetical LOC100507004 | |
| 4815938 | 4831737 | TICAM1 | − | 19p13.3 | toll-like receptor adaptor molecule 1 |
| 4838346 | 4867780 | PLIN3 | − | 19p13.3 | perilipin 3 |
| 4890449 | 4902879 | ARRDC5 | − | 19p13.3 | arrestin domain containing 5 |
All patients had a degree of intellectual impairment, however, it was quite variable spanning from minimal delays in gross motor and fine motor milestone acquisition (patient 4) to severe intellectual disability with minimal speech (patient 1).
The following central nervous system abnormalities were noted: dysgensis of corpus callosum in two patients, and cerebellar hypoplasia in one; one patient had hypomyelination, this latter patient also had a movement disorder of unknown etiology.
Functional characterization and studies of fibroblasts with MEK2 deletion
In order to examine the effect of MEK2 deletion in our patients, functional assays were performed using primary fibroblast cell lines derived from patient 1 and a healthy control. Equal levels of total MEK1 protein were observed in both cell lines. However, the level of total MEK2 was significantly reduced in patient 1 compared to the control (Fig. 3A). When fibroblasts were treated with EGF, a factor known to stimulate the MAPK pathway, MEK1/2 phosphorylation peaked at 5 minutes in both cell lines and decreased over time (Fig. 3A). However, P-MEK1/2 was ~50% less abundant in the haploinsufficient MEK2 cell line compared to the control (P=0.0006 and 0.01 for the 5 and 15 min time points respectively) (Fig. 3A, B). Differences in the ERK1/2 phosphorylation pattern were also observed. In the control cell line, P-ERK1/2 was most abundant at 5 and 15 min post EGF stimulation and then gradually decreased over time. In cells derived from patient 1, P-ERK1/2 peaked 15 min post stimulation and decreased over time. Although the maximum level of ERK1/2 phosphorylation observed in patient 1 and the control did not show a significant difference (P=0.95), phosphorylation appeared to decreased towards basal levels faster in the fibroblasts of patient 1 (Fig. 3A, 3C). The observed differences in the ERK phosphorylation were not due to variances in the amount of ERK protein available for phosphorylation as the levels of total ERK were similar in patient 1 and the control (Fig. 3A). To study Ras/MAPK regulation, we also examined the level of Sprouty 1, a member of the Sprouty protein family. Sprouty proteins are known modulators of MAPK pathway signaling in a variety of cell types (20,21). Protein levels of Sprouty 1 were significantly increased in patient 1 compared to the control with p-values ranging from 0.004 to 0.02 for the 0, 5, 30, and 60 min time points (Fig. 3A & D). Taken together, these data demonstrate that regulation of the Ras/MAPK pathway is altered in cells carrying a MEK2 deletion.
Figure 3. Regulation of the Ras/MAPK Pathway is altered when MEK2 is deleted.
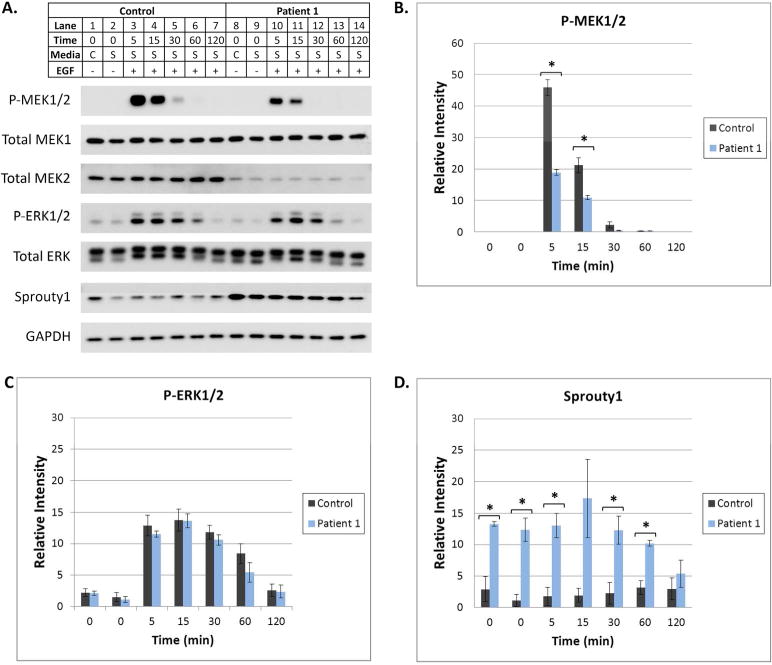
(A) At subconfluency, fibroblasts were placed in complete (C, 10% FBS) or serum starvation (S, 0.1% FBS) media for 16 hours. Serum starved cells were then treated with EGF for 5–120 min. Cells were harvested and protein lysates were subjected to Western blot analysis utilizing antibodies specific for various MAPK pathway components. A representative Western blot is shown. GAPDH was utilized as a loading control. Differences in the levels of P-MEK1/2, P-ERK1/2, Total MEK2, and Sprouty1 were observed as described in the Results. (B, C, and D) Image J software was used to quantify P-MEK1/2, P-ERK1/2, and Sprouty 1 bands on Western blots from 3 separate experiments. Error bars represent standard error of the mean relative intensity calculated for 3 experiments. Asterisks indicate p-values <0.05 for the control compared to patient 1. Statistically significant differences in P-MEK1/2 and Sprouty1 were observed.
DISCUSSION
We report seven patients with deletions of 19p13.3 whose smallest region of overlap (chr19:4,086,696–4,100,886; hg19) contains only one known gene, MEK2 (Fig. 2). These patients present with a recognizable pattern of malformations, craniofacial features, failure to thrive, gastrointestinal dysfunction, cutaneous abnormalities, obstructive sleep apnea, and intellectual disabilities (Fig. 1, Table 1). The seven patients reported herein share facial and somatic features with two other patients previously reported in the literature with microdeletions that included MEK2 (16,17) presenting further evidence of the existence of a recognizable phenotype (Table 1).
Activating gain-of-function missense mutations in MEK2 are responsible for CFC syndrome. To date there have been no known reports of classical CFC syndrome associated with deletions of MEK2. Studies performed in mice suggest that MEK1 is required for normal embryo development and that MEK2 is expendable (22,23). To determine the functional consequences of MEK2 deletion in our patients, we performed assays using primary fibroblasts isolated from patient 1 and a healthy control. These assays used serum starvation followed by EGF stimulation to uncover differences in Ras/MAPK pathway regulation, a method previously used to detect the pathway changes observed in other RASopathies (24,25). In our experiments, we detected significant differences in the protein levels and phosphorylation patterns of multiple Ras/MAPK pathway components, including MEK2, P-MEK1/2, and Sprouty1, demonstrating that MAPK pathway regulation is altered in this patient with a deletion of MEK2.
In addition to MEK2, the deletion found in patient 1 contains 62 other genes (supplemental Table 1). Six of these genes are known to be associated with a disease phenotype, GIPC3, TBXA2R, PIP5K1C, RAX2, ATCAY, and SH3GL1 (supplemental Table 2). Studies performed in mice suggest that the GIPC3 protein, which localizes to spiral ganglion and inner ear sensory hair cells, is essential for postnatal maturation of the hair bundle (27). Frameshift and missense mutations in GIPC3 cause an autosomal recessive form of nonsyndromic sensorineural deafness in humans (27,28). The TBXA2R gene encodes the thromboxane A2 receptor, a G-protein coupled receptor involved in hemostasis. Heterozygous missense mutations in TBXA2R result in loss-of-function and are associated with an increased susceptibility to platelet-type bleeding disorder 13 which exhibits an autosomal dominant mode of inheritance (29,30). However, Mumford et al speculate that a second mutation in TBXA2R or another gene involved in hemostasis is required for clinically significant platelet dysfunction (30). The PIP5K1C gene encodes PIPKI-gamma, a protein that catalyzes the phosphorylation of phosphatidylinositol 4-phosphate thereby producing phosphatidylinositol 4,5-bisphosphate. Loss-of-function missense mutations in PIP5K1C are reported to significantly reduce the kinase activity of the enzyme and cause autosomal recessive lethal congenital contractural syndrome type 3 (31). RAX2 is a homeobox gene that is expressed in the retina. Heterozygous missense mutations and a 6 base pair insertion in this gene are associated with age-related macular degeneration and cone rod dystrophy. Functional studies suggest that these mutations result in either an increase or decrease in the ability of RAX2 to transactivate Ret-1, a cis-acting DNA element located within the Rhodopsin promoter (32). The ATCAY gene encodes caytaxin, a neuron-restricted protein. Missense and splice site mutations in this gene can cause autosomal recessive Cayman cerebellar ataxia, a disorder in which affected individuals present with clinical features including hypotonia, intention tremor, dysarthria, nystagmus, truncal ataxia, and an ataxic gait (33). The SH3GL1 gene encodes a protein which contains a Src homology 3 (SH3) domain, a domain found in many proteins involved in signal transduction. In a study of a patient with acute myeloid leukemia, the SH3GL1 protein was found to be fused to MLL, a protein that commonly undergoes chromosomal translocation in leukemias (34). Although all six of these genes are deleted in patient 1, this child currently does not exhibit clinical features diagnostic for the diseases associated with these genes. Therefore, it may be unlikely that haploinsufficiency of these genes significantly contributes to the phenotype observed in patient 1.
In addition to the six genes with known gene-disease relationships, death associated protein kinase-3 (DAPK3, also called ZIPK) is another gene of potential interest within this deleted region. DAPK3 is a member of the death associated protein (DAP) family which also includes DAPK1, DAPK2, DRAK1, and DRAK2 (35). This protein family is involved in apoptotic and autophagic cell death, metastasis suppression, and tumor suppression (36). In a yeast two hybrid screen, one of the members of this protein family, DAPK1, was shown to directly interact with ERK, a major component of the Ras/MAPK pathway. When stimulated by EGF, ERK phosphorylates DAPK1 and enhances its kinase and apoptotic activity (35). DAPK1 directly interacts with and phosphorylates DAPK3. This phosphorylation event increases the death promoting activity of DAPK3 (36). The involvement of DAPK1 and DAPK3 in the MAPK pathway is particularly interesting because the DAPK3 gene is deleted in patients 1, 2, 3, 5 and 6 (Fig. 2). It is possible that deletion of DAPK3 may contribute to the phenotype observed in these patients. However, the phenotype observed in patients 1, 2, 3, 5 and 6 is very similar to that of patient 4, whose deletion contains only the MEK2 gene. Together, this evidence may suggest that haploinsufficiency of MEK2 is responsible for the majority of the phenotype observed in the patients reported in this study.
In summary, these seven patients with a microdeletion that includes MEK2 present with a recognizable phenotype that includes a characteristic craniofacial appearance and pattern of malformation and organ dysfunction. These somatic features are suggestive of dysregulation of the Ras/MAPK pathway and functional assays confirmed that Ras/MAPK pathway regulation is altered in one of these patients. Thus haploinsufficiency of MEK2 appears to be a new model of a RASopathy, where a deletion of one of the components of the pathway, MEK2, results in a RASopathy-like phenotype.
Supplementary Material
Acknowledgments
The authors are indebted to all families who contributed to this study. PASL is supported by the Harold Amos Faculty Development Program through the Robert Wood Johnson Foundation, NIDCR Supplement 3R37DE012711-13S1 and the CHLA-USC Child Health Research Career Development Program (NIH K12-HD05954).
Footnotes
Conflict of Interest. None of the authors have conflicts of interest relating to the contents of this manuscript.
References
- 1.Tidyman WE, Rauen KA. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–6. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4(5):373–84. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 3.Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, Mudd S, Penttinen M, Cordeiro I, Ponder M, Ponder BA, Krawczak M, Cooper DN. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998;102:591–7. doi: 10.1007/s004390050746. [DOI] [PubMed] [Google Scholar]
- 4.Shchelochkov OA, Patel A, Weissenberger GM, Chinault AC, Wiszniewska J, Fernandes PH, Eng C, Kukolich MK, Sutton VR. Duplication of chromosome band 12q24.11q24.23 results in apparent Noonan syndrome. Am J Med Genet. 2008;146A:1042–8. doi: 10.1002/ajmg.a.32215. [DOI] [PubMed] [Google Scholar]
- 5.Graham JM, Jr, Kramer N, Bejjani BA, Thiel CT, Carta C, Neri G, Tartaglia M, Zenker M. Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am J Med Genet. 2009;149A(10):2122–8. doi: 10.1002/ajmg.a.32992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds JF, Neri G, Herrmann JP, Blumberg B, Coldwell JG, Miles PV, Opitz JM. New multiple congenital anomalies/mental retardation syndrome with cardio-faciocutaneous involvement—The CFC syndrome. Am J Med Genet. 1986;25(3):413–27. doi: 10.1002/ajmg.1320250303. [DOI] [PubMed] [Google Scholar]
- 8.Allanson JE, Anneren G, Aoki Y, Armour CM, Bondenson M-L, Cave H, Gripp KW, Kerr B, Nystrom A-M, Sol-Church K, Verloes A, Zenker M. Cardio-Facio-Cutaneous syndrome: Does Genotype Predict Phenotype? Am J Med Genet C Semin Med Genet. 2011;157(2):129–35. doi: 10.1002/ajmg.c.30295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armour CM, Allanson JE. Further delineation of cardio-facio-cutaneous syndrome: Clinical features of 38 individuals with proven mutations. J Med Genet. 2008;45:249–54. doi: 10.1136/jmg.2007.054460. [DOI] [PubMed] [Google Scholar]
- 10.Yoon G, Rosenberg J, Blaser S, Rauen KA. Neurological complications of cardio-facio-cutaneous syndrome. Dev Med Child Neurol. 2007;49:894–9. doi: 10.1111/j.1469-8749.2007.00894.x. [DOI] [PubMed] [Google Scholar]
- 11.Rauen KA, Tidyman WE, Estep AL, Sampath S, Peltier HM, Bale SJ, Lacassie Y. Molecular and functional analysis of a novel MEK2 mutation in cardio-facio-cutaneous syndrome: transmission through four generations. Am J Med Genet. 2010;152A:807–14. doi: 10.1002/ajmg.a.33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dentici ML, Sarkozy A, Pantaleoni F, Carta C, Lepri F, Ferese R, Cordeddu V, Martinelli S, Briuglia S, Digilio MC, Zampino G, Tartaglia M, Dallapiccola B. Spectrum of MEK1 and MEK2 gene mutations in cardio-facio-cutaneous syndrome and genotype-phenotype correlation. Eur J Hum Genet. 2009;17:733–40. doi: 10.1038/ejhg.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz S, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 14.Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, Aoki Y, Matsubara Y, Arveiler B, Lacombe D, Pasmant E, Parfait B, Baumann C, Heron D, Sigaudy S, Toutain A, Rio M, Goldenberg A, Leheup B, Verloes A, Cave H. CFC and Noonan syndromes due to mutations in RAS/MAPK signaling pathway: Genotype/phenotype relationships and overlap with Costello syndrome. J Med Genet. 2007;44:763–71. doi: 10.1136/jmg.2007.050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulz AL, Albrecht B, Arici C, van der Burgt I, Buske A, Gillessen-Kaesbach G, Heller R, Horn D, Hubner CA, Korenke GC, Konig R, Kress W, Kruger G, Meinecke P, Mucke J, Plecko B, Rossier E, Schinzel A, Schulze A, Seemanova E, Seidel H, Spranger S, Tuysuz B, Uhrig S, Wieczorek D, Kutsche K, Zenker M. Mutation and phenotypic spectrum in patients with cardio-faciocutaneous and Costello syndrome. Clin Genet. 2008;73:62–70. doi: 10.1111/j.1399-0004.2007.00931.x. [DOI] [PubMed] [Google Scholar]
- 16.Al-Kateb H, Hahn A, Gastier-Foster JM, Jeng L, McCandless SE, Curtis CA. Molecular characterization of a novel, de novo cryptic interstitial deletion on 19p13.3 in a child with a cutis aplasia and multiple congenital anomalies. Am J Med Genet. 2010;152A:3148–53. doi: 10.1002/ajmg.a.33738. [DOI] [PubMed] [Google Scholar]
- 17.De Smith AJ, van Haelst MM, Ellis RJ, Holder SE, Payne SJ, Hashim SK, Froguel P, Blakemore AIF. Chromosome 19p13.3 deletion in a patient with macrocephaly, obesity, mental retardation, and behavior problems. Am J Med Genet. 2011;155:1192–5. doi: 10.1002/ajmg.a.33986. [DOI] [PubMed] [Google Scholar]
- 18.Rasband WS. ImageJ. U.S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2012. http://imagej.nih.gov/ij/ [Google Scholar]
- 19.Seigel DM, McKenzie J, Frieden IJ, Rauen KA. Dermatological findings in 61 mutation-positive individuals with cardiofaciocutaneous syndrome. Br J Dermatol. 2011;164(3):521–9. doi: 10.1111/j.1365-2133.2010.10122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. 2008;11(1):53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 21.Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: modified modulators, matchmakers or missing links? Journal Endocrin. 2009;203:191–202. doi: 10.1677/JOE-09-0110. [DOI] [PubMed] [Google Scholar]
- 22.Belanger LF, Roy S, Tremblay M, Brott B, Steff AM, Mourad W, Hugo P, Erikson R, Charron J. Mek2 is dispensable for mouse growth and development. Mol Cell Biol. 2003;23(14):4778–87. doi: 10.1128/MCB.23.14.4778-4787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giroux S, Tremblay M, Bernard D, Cardin-Girard JF, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, Charron J. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr Biol. 1999;9(7):369–72. doi: 10.1016/s0960-9822(99)80164-x. [DOI] [PubMed] [Google Scholar]
- 24.Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39(1):70–4. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberger G, Meien S, Kutsche K. Oncogenic HRAS mutations cause prolonged PI3K signaling in response to epidermal growth factor in fibroblasts of patients with Costello syndrome. Hum Mutat. 2009;30(3):352–62. doi: 10.1002/humu.20855. [DOI] [PubMed] [Google Scholar]
- 26.Senawong T, Phuchareon J, Ohara O, McCormick F, Rauen KA, Tetsu O. Germline mutations of MEK in cardio-facio-cutaneous syndrome are sensitive to MEK and RAF inhibition: implications for therapeutic options. Hum Mol Genet. 2008;17(3):419–30. doi: 10.1093/hmg/ddm319. [DOI] [PubMed] [Google Scholar]
- 27.Charizopoulou N, Lelli A, Schraders M, Ray K, Hildebrand MS, Ramesh A, Srisailapathy CR, Oostrik J, Admiraal RJ, Neely HR, Latoche JR, Smith RJ, Northup JK, Kremer H, Holt JR, Noben-Trauth K. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat Commun. 2011;2:201. doi: 10.1038/ncomms1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rehman AU, Gul K, Morell RJ, Lee K, Ahmed ZM, Riazuddin S, Ali RA, Shahzad M, Jaleel AU, Andrade PB, Khan SN, Khan S, Brewer CC, Ahmad W, Leal SM, Riazuddin S, Friedman TB. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p. Hum Genet. 2011;130(6):759–65. doi: 10.1007/s00439-011-1018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirata T, Kakizuka A, Ushikubi F, Fuse I, Okuma M, Narumiya S. Arg60 to Leu mutation of the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J Clin Invest. 1994;94(4):1662–7. doi: 10.1172/JCI117510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mumford AD, Dawood BB, Daly ME, Murden SL, Williams MD, Protty MB, Spalton JC, Wheatley M, Mundell SJ, Watson SP. A novel thromboxane A2 receptor D304N variant that abrogates ligand binding in a patient with a bleeding diathesis. Blood. 2010;115(2):363–9. doi: 10.1182/blood-2009-08-236976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narkis G, Ofir R, Landau D, Manor E, Volokita M, Hershkowitz R, Elbedour K, Birk OS. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKI gamma of the phophatidylinsitol pathway. Am J Hum Genet. 2007;81(3):530–9. doi: 10.1086/520771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang QL, Chen S, Esumi N, Swain PK, Haines HS, Peng G, Melia BM, McIntosh I, Heckenlively JR, Jacobson SG, Stone EM, Swaroop A, Zack DJ. QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum Mol Genet. 2004;13(10):1025–40. doi: 10.1093/hmg/ddh117. [DOI] [PubMed] [Google Scholar]
- 33.Bomar JM, Benke PJ, Slattery EL, Puttagunta R, Taylor LP, Seong E, Nystuen A, Chen W, Albin RL, Patel PD, Kittles RA, Sheffield VC, Burmeister M. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35(3):264–9. doi: 10.1038/ng1255. [DOI] [PubMed] [Google Scholar]
- 34.So CW, Caldas C, Liu MM, Chen SJ, Huang QH, Gu LJ, Sham MH, Wiedemann LM, Chan LC. EEN encodes for a member of a new family of proteins containing an Src homology 3 domain and is the third gene located on chromosome 19p13 that fuses to MLL in human leukemia. Proc Natl Acad Sci USA. 1997;94(6):2563–8. doi: 10.1073/pnas.94.6.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Y, Hupp TR, Stevens C. Death-associated protein kinase (DAPK) and signal transduction: additional roles beyond cell death. FEBS J. 2010;277(1):48–57. doi: 10.1111/j.1742-4658.2009.07411.x. [DOI] [PubMed] [Google Scholar]
- 36.Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2(2):74–9. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.