

Abstract
Background
Pediatric embryonal brain tumors (PEBTs), which encompass medulloblastoma (MB), primitive neuroectodermal tumor (PNET) and atypical teratoid/rhabdoid tumor (AT/RT), are the second most prevalent pediatric brain tumor type. AT/RT is highly malignant and is often misdiagnosed as MB or PNET. The distinction of AT/RT from PNET/MB is of clinical significance because the survival rate of patients with AT/RT is substantially lower. The diagnosis of AT/RT relies primarily on morphologic assessment and immunohistochemical (IHC) staining for a few known markers such as the lack of INI1 protein expression. However, in our clinical practice we have observed several AT/RT-like tumors, that fulfilled histopathological and all other biomarker criteria for a diagnosis of AT/RT, yet retained INI1 immunoreactivity. Recent studies have also reported preserved INI1 immunoreactivity among certain diagnosed AT/RTs. It is therefore necessary to re-evaluate INI1(+), AT/RT-like cases.
Method
Sanger sequencing, array CGH and mRNA microarray analyses were performed on PEBT samples to investigate their genomic landscapes.
Results
Patients with AT/RT and those with INI(+) AT/RT-like tumors showed a similar survival rate, and global array CGH analysis and INI1 gene sequencing showed no differential chromosomal aberration markers between INI1(−) AT/RT and INI(+) AT/RT-like cases. We did not misdiagnose MBs or PNETs as AT/RT-like tumors because transcriptome profiling revealed that not only did AT/RT and INI(+) AT/RT-like cases express distinct mRNA and microRNA profiles, their gene expression patterns were different from those of MBs and PNETs. The most similar transcriptome profile to that of AT/RTs was the profile of embryonic stem cells. However; the transcriptome profile of INI1(+) AT/RT-like tumors was more similar to that of somatic neural stem cells, while the profile of MBs was closer to that of fetal brain tissue. Novel biomarkers were identified that can be used to distinguish INI1(−) AT/RTs, INI1(+) AT/RT-like cases and MBs.
Conclusion
Our studies revealed a novel INI1(+) ATRT-like subtype among Taiwanese pediatric patients. New diagnostic biomarkers, as well as new therapeutic tactics, can be developed according to the transcriptome data that were unveiled in this work.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-015-0103-3) contains supplementary material, which is available to authorized users.
Keywords: Atypical teratoid/rhabdoid tumor, INI1, Pediatric embryonal brain tumor, Transcriptome, Stem cell
Background
Pediatric brain tumors are second only to neoplasms of the lymphoid-hematopoietic system in childhood in terms of the number of cases and mortality [1, 2]. Pediatric embryonal brain tumors (PEBTs) are the second most prevalent type of pediatric brain tumors and include medulloblastoma (MB), CNS primitive neuroectodermal tumor (CNS-PNET) and atypical teratoid/rhabdoid tumor (AT/RT). AT/RTs of the central nervous system (CNS) were first described by Rorke et al. in 1987. This tumor type was later recognized as a rare, highly malignant entity of CNS embryonal tumors that predominantly occurs in infants and young children with a peak incidence between birth and 3 years of age [3–6]. The distinction of AT/RT from CNS-PNET and MB is of clinical significance because the reported 2-year survival rate of patients with AT/RTs is substantially lower than that of patients with CNS-PNETs or MBs: in Taiwan, the reported 2-year survival rate of patients with AT/RTs is 18 %, while that of patients with a standard-risk MB is 83.9 % [7].
Clinically, cerebellar AT/RT is often misdiagnosed as a PNET or a MB. The diagnosis of AT/RT is based on the presence of rhabdoid tumor cells, which are medium-sized, round-to-oval cells with distinct borders, a large amount of cytoplasm and eccentrically located nuclei. Other morphological features include the presence of small cells with the morphology of PNET cells as well as epithelial and mesenchymal components [5]. AT/RT cells are immunoreactive for a wide range of epithelial, mesenchymal, glial and neural markers, including epithelial membrane antigen (EMA), vimentin (VIM), smooth muscle actin (SMA), and glial fibrillary acidic protein (GFAP) [5, 7, 8]. MB cells are negative by staining for EMA and SMA but are positive for synaptophysin (SYN) [5, 7, 8]. The most widely known tumor suppressor and diagnostic biomarker for AT/RT is INI1 (also known as SMARCB1, hSNF5, BAF47). INI1 belongs to a core member of the ATP-dependent SWI/SNF chromatin-remodeling complex, which is a master regulator of gene expression that is involved in cancer. Genetic studies have shown that deletion or mutation of the INI1 gene, which is located on 22q11.2, occurs in approximately 75 % [3, 9] to 98 % [10] of AT/RTs. Immunohistochemical (IHC) staining for INI1 is considered a sensitive and highly specific approach for the diagnosis of AT/RT and in the differentiation of this tumor type from PNET and MB [11]. The lack of INI1 protein immunoreactivity has been shown in 100 % [4, 9, 12] to 84 % [13] of AT/RT cases. In contrast, retained INI1 expression (INI1 positive) was noted in all cases of PNETs/MBs [4, 9, 11–13].
With respect to the histopathologic diagnosis of PEBT, in our clinical practice, INI1 as well as EMA, VIM, SMA, GFAP and SYN are included in a panel of IHC markers to distinguish AT/RT from other PEBTs, especially MB. Unexpectedly, we observed several AT/RT-like cases, which fulfilled all other biomarker and histopathologic criteria for a diagnosis of AT/RT, yet still demonstrated INI1 immunoreactivity. It is therefore necessary to understand these atypical cases and to classify them more accurately. For this purpose, we performed a thorough pathological review of our PEBT cases and conducted a systemic genomic analysis. Transcriptomic analysis was also performed using fresh tissues of histopathologically confirmed cases.
Materials and methods
Study materials and clinical data
Study cases were retrieved from the surgical pathology files of the Department of Pathology and Laboratory Medicine, Taipei Veterans General Hospital (VGH-TPE), Taiwan. The Parent/legal guardian of patients in this study provided informed consent, and all procedures were approved by the Institutional Review Board of VGH-TPE (VGHIRB No.:2011-11-007GA & 2011-11-008GA). Fresh tumor tissues that were removed during surgery were snap-frozen and stored in liquid nitrogen until DNA and RNA extraction. The overall survival time was calculated as the time from surgery until death or the time from surgery until the last follow-up appointment for the patients who survived. A Mann–Whitney test was used to compare age differences among the different groups of patients. The differences in survival times were assessed with the log-rank test.
Histopathologic diagnosis of AT/RT and MB
A diagnosis of AT/RT was based on the morphologic features of the tumor and the results of IHC, as described in our previous reports [7, 14]. With regard to the morphologic features, rhabdoid cells, which were either large, pale, bland cells or the classical type similar to those observed in malignant rhabdoid tumors of the kidney, were essential for diagnosis [8, 15]. Other features, such as cells that are similar to primitive neuroectodermal cells, an epithelial component and a mesenchymal component, could also be observed. An IHC diagnostic panel included the following markers: epithelial membrane antigen [EMA; monoclonal, dilution 1:40, Dako, Glostrup, Denmark, Histostain SP Broad Spectrum (HRP), Zymed Lab., Carlsbad, USA (Histostain); antigen retrieval using a microwave, three cycles for 5 min each (M)], vimentin (VIM; monoclonal, 1:600, Dako, Histostain, M), smooth muscle actin (SMA, HHF-35; monoclonal, 1:75, Dako, Carpinteria, CA, USA, Histostain, M), and glial fibrillary acidic protein (GFAP; monoclonal, 1:300, Dako, Histostain, M) [5, 8, 14]. The rhabdoid cells were immunoreactive for two or more of the above-listed antibodies. IHC for INI1 (anti-BAF47; monoclonal, 1:40, BD Transduction Laboratories, San Diego, CA, USA, Histostain, M) and SMARCA4 (anti-BRG1; monoclonal, 1:100, Abcam, Cambridge, U.K., Histostain, M) was included in all cases in this study. With regard to a diagnosis of MB, besides the presence of the known morphology of this tumor type, the features of AT/RT as listed above were absent. Immunostaining for synaptophysin (SYN; monoclonal, 1:50, Novocastra, Newcastle upon Tyne, U.K., Histostain, M) was included to confirm the diagnosis and to distinguish it from AT/RT; IHC for SYN was also performed in all AT/RT cases. Positive and negative controls were included with each batch of sections to confirm the consistency of the analysis in all of the stains that were performed in this study. As for the INI1 staining, positive control consisted of endothelial cells within the tumor. Negative controls consisted of staining without applying the primary antibody and staining of a known INI1 negative AT/RT.
Direct sequencing, reverse transcription-PCR (RT-PCR) and Quantitative real-time reverse transcription-PCR (qRT-PCR)
Genomic DNAs and total RNA were isolated from fresh-frozen tumor samples by the DNeasy Blood& Tissue Kit and RNAeasy (Qiagen) according to the manufacturer’s instruction (Qiagen, GmbH, Germany), respectively. Genomic DNAs were used to peform PCR using specific INI1 gene primers and then sequenced by direct sequencing. For RT-PCR and qRT-PCR, 1 μg of total RNA was used to perform reverse transcription (RT) using the RevertAid™ Reverse transcriptase kit (Cat. K1622; Fermentas, Glen Burnie, Maryland, USA) as directed by the manufacturer. For INI1 RT-PCR, a paired-primer encompassing exon 5 and 6 was used. For INI1, the forward primer was 5′-AACAGGAACCGCATGGGCCG-3′, and the reverse primer was 5′-GCCCGTGTTCCGGATGGCAA-3′ (amplicon size: 579 bps). For GAPDH, the forward primer was 5′-CAAGGTCATCCATGACAACTTTG-3′, and the reverse primer was 5′-GTCCACCACCCTGTTGCTGTAG-3′ (amplicon size, 496 bps). Quantitative real-time PCR reactions were performed using Maxima™ SYBR Green qPCR Master Mix (Cat. K0222; Fermentas, Glen Burnie, Maryland, USA), and the specific products were detected and analyzed using the StepOne™ sequence detector (Applied Biosystems, USA). The expression level of each gene was normalized to GAPDH expression. For GAPDH, the forward primer was 5′-CCAGCCGAGCCACATCGCTC-3′ and the reverse primer was 5′-ATGAGCCCCAGCCTTCTCCAT-3′. For SOX4, the forward primer was 5′- TCGCTGTACAAGGCGCGGAC-3′ and the reverse primer was 5′-TTCTCCGCCAGGTGCTTGCC-3′. For ERBB2, the forward primer was 5′- AGTACCTGGGTCTGGACGTG-3′ and the reverse primer was 5′-CTGGGAACTCAAGCAGGAAG-3′. For OLIG2, the forward primer was 5′-CAGAAGCGCTGATGGTCATA-3′ and the reverse primer was 5′-TCGGCAGTTTTGGGTTATTC-3′.
Array CGH (aCGH) analysis
As described in our previous study [14], the samples were mixed with control DNA samples from healthy donors before they were subjected to the analysis. A Human Genome CGH Microarray Kit 244A (Agilent Technologies, USA) with 99,000 probes and an average probe spatial resolution of 15.0 kb was used. aCGH was performed according to the protocol suggested by Agilent. Data analysis was performed using CGH Analytics 3.4 (Agilent Technologies) using the default parameters. Briefly, chromosomal abbreviations were calculated using the ADM2 statistic algorithm with a moving average window of 1 Mb; additionally, the default thresholds of ADM2 recommended by Agilent were used to make an amplification or deletion call.
Gene expression microarray (GEM) and computational analyses
Array data on adult neural stem cells and embryonic stem cells were obtained in our previous study [16] and from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) dataset GSE9940. An mRNA expression array analysis was performed as previously described [16, 17]. Briefly, an Affymetrix™ HG-U133 Plus 2.0 whole genome array was used. RMA log expression units were calculated from the Affymetrix GeneChip array data with the ‘affy’ package of the Bioconductor (http://www.bioconductor.org/) suite software for the R statistical programming language (http://www.r-project.org/). The default RMA settings were used to background correct, normalize and summarize all expression values. Significant differences between sample groups were identified by the ‘limma’ package [16]. Briefly, a t-statistic was calculated as normal for each gene, and a p-value was then calculated with a modified permutation test [16]. To control for the multiple testing errors, a false discovery rate (FDR) algorithm was then applied to these p-values to calculate a set of q-values: thresholds of the expected proportion of false positives or false rejections of the null hypothesis. Heat maps were then created by dChip software (http://www.dchip.org/). Classical multidimensional scaling (MDS) was performed with the standard function of the R program to provide a visual impression of how the various sample groups are related. Gene annotation was performed by the ArrayFusion web tool (http://microarray.ym.edu.tw/tools/arrayfusion/) [18]. Principal component analysis (PCA) was performed with Partek Genomics Suite software (http://www.partek.com) to provide a visual impression of how the various sample groups are related. All array data have been submitted to the NCBI Gene Expression Omnibus (GEO) database, and the accession number is GSE65132 (Additional file 2).
MicroRNA microarray analysis
The Agilent Human miRNA Microarray Kit V2 (Agilent, Foster City, CA, USA) containing probes for 723 human microRNAs from the Sanger database v10.1 was used. GeneSpring GX 9 software (Agilent, USA) was used for value extraction. A 2-tailed Student’s t-test was then used for the calculation of the p value for each miRNA probe.
Results
Clinical features of the included primary pediatric embryonal brain tumors
The diagnosis of AT/RTs and other PEBTs, especially MBs, was based on the morphologic and IHC features described in our previous reports (Additional file 1-A) [7, 14]. Fresh tissues from 45 patients with PEBT (9 INI1- AT/RT, 5 INI1+ AT/RT-like, and 31 INI1+ MB) were used in the genomics studies (Table 1). With regard to IHC assays, EMA, VIM, SMA, GFAP and INI1 were used as diagnostic markers, and all AT/RT or AT/RT-like cases demonstrated positivity for at least two of these markers (Table 2). The locations within the CNS of the AT/RT and INI1+ AT/RT-like tumors included the cerebellum and the lateral ventricle. Pediatric cerebellar MB cases were also collected as control samples, and all of those tumors were INI1+ (Table 1). Examples of loss (INI1-) and preservation (INI1+) of INI1 expression according to IHC in AT/RTs and AT/RT-like cases are shown in Fig. 1a and are as described in our previous work [14]. The IHC results were clear because the tumor cells were either diffusely positive or diffusely negative (Fig. 1a). Mutations in the SMARCA4 subunit are considered alternative mutations that may be present in AT/RT-like tumors [19, 20]; however, by IHC, we found no SMARCA4 loss in our AT/RTs and AT/RT-like tumors (Table 1 and Additional file 1-B).
Table 1.
Clinical details and INI1 IHC data of PEBT cases enrolled in aCGH and transcriptome studies
| Case | Age | Sex | Site | Histo. | INI1 | SMARCA4 | aCGH | mRNA | microRNA |
|---|---|---|---|---|---|---|---|---|---|
| No. | (yr) | Dx | IHC | IHC | array | array | |||
| L01 | 8.3 | m | cblm | AT/RT-like | + | + | + | + | + |
| L02 | 4.2 | m | cblm | AT/RT-like | + | + | + | ND | ND |
| L06 | 3.5 | f | cblm | AT/RT-like | + | + | + | ND | ND |
| L07 | 9.8 | m | cblm | AT/RT-like | + | + | + | ND | ND |
| L08 | 9.4 | f | cblm | AT/RT-like | + | + | + | + | + |
| A03 | 2.3 | f | cblm | AT/RT | - | + | + | ND | ND |
| A04 | 4.5 | f | lat. vent. | AT/RT | - | + | + | + | + |
| A05 | 0.1 | m | cblm | AT/RT | - | + | + | ND | ND |
| A09 | 5.2 | f | cblm | AT/RT | - | + | + | + | + |
| A10 | 1.6 | m | cblm | AT/RT | - | + | ND | + | + |
| A11 | 1.4 | f | cblm | AT/RT | - | + | ND | + | ND |
| A12 | 0.6 | f | cblm | AT/RT | - | + | ND | + | ND |
| A13 | 5 | m | cblm | AT/RT | - | + | ND | + | ND |
| A14 | 1.6 | m | cblm | AT/RT | - | + | ND | + | ND |
| M01 | 8.7 | m | cblm | MB, cl | + | + | + | ND | ND |
| M02 | 9.2 | f | cblm | MB, an | + | + | + | ND | ND |
| M03 | 4 | m | cblm | MB, cl | + | + | + | ND | ND |
| M04 | 6.3 | m | cblm | MB, ds | + | + | + | ND | ND |
| M05 | 3.5 | f | cblm | MB, ds | + | + | + | ND | ND |
| M06 | 9.4 | m | cblm | MB, cl | + | + | + | ND | ND |
| M07 | 7.6 | m | cblm | MB, an | + | + | + | ND | ND |
| M08 | 14.3 | f | cblm | MB, cl | + | + | + | ND | ND |
| M09 | 13.6 | m | cblm | MB, cl | + | + | + | + | ND |
| M10 | 12.2 | f | cblm | MB, cl | + | + | + | ND | ND |
| M11 | 4.1 | f | cblm | MB, cl | + | + | + | + | ND |
| M12 | 4.3 | m | cblm | MB, cl | + | + | ND | + | ND |
| M13 | 3.2 | m | cblm | MB, cl | + | + | ND | + | + |
| M14 | 3 | m | cblm | MB, cl | + | + | ND | + | + |
| M15 | 1.5 | f | cblm | MB, an | + | + | ND | + | + |
| M16 | 9.1 | f | cblm | MB, an | + | + | ND | + | + |
| M17 | 13 | f | cblm | MB (*) | + | + | ND | + | + |
| M18 | 18.2 | m | cblm | MB, cl | + | + | ND | + | + |
| M19 | 5.8 | f | cblm | MB, cl | + | + | ND | + | + |
| M20 | 5.1 | m | cblm | MB, cl | + | + | ND | + | + |
| M21 | 3 | m | cblm | MB, cl | + | + | ND | + | + |
| M22 | 8.5 | f | cblm | MB, an | + | + | ND | + | + |
| M23 | 2.2 | m | cblm | MB, ds | + | + | ND | + | + |
| M24 | 5.8 | f | cblm | MB (*) | + | + | ND | + | ND |
| M25 | 7.6 | f | cblm | MB, cl | + | + | ND | + | + |
| M26 | 12.2 | m | cblm | MB, an | + | + | ND | + | ND |
| M27 | 6.5 | f | cblm | MB, an | + | + | ND | + | ND |
| M28 | 2.1 | m | cblm | MB, ds | + | + | ND | + | ND |
| M29 | 6.3 | m | cblm | MB, cl | + | + | ND | + | ND |
| M30 | 10 | m | cblm | MB, an | + | + | ND | + | ND |
Site: cblm: cerebellum, lat.vent: lateral ventricle
Histo. Dx. (Histopathological diagnosis): cl: classic, ds: demoplastic, an: anaplastic
INI1 IHC: −: loss of INI1 expression (INI1-), +: retained INI1 expression (INI1+);
SMARCA4 IHC: −: loss of SMARCA4 expression, +: retained SMARCA4 expression;
aCGH & GEM: +:performed, ND: not determined
(*)M17 with focal anaplasia; M24 with myogenic and melanocytic differentiation
Table 2.
IHC features of AT/RT and AT/RT-like cases enrolled
| Case no. | INI1 | EMA | VIM | SMA | GFAP |
|---|---|---|---|---|---|
| L01 | + | - | + | + | + |
| L02 | + | - | + | + | - |
| L06 | + | + | + | + | + |
| L07 | + | + | - | + | + |
| L08 | + | + | + | + | - |
| A03 | - | + | + | + | + |
| A04 | - | + | + | + | + |
| A05 | - | + | + | - | + |
| A09 | - | + | + | + | - |
| A10 | - | + | + | + | + |
| A11 | - | + | + | + | + |
| A12 | - | + | + | + | + |
| A13 | - | + | + | + | + |
| A14 | - | + | + | + | + |
Fig. 1.
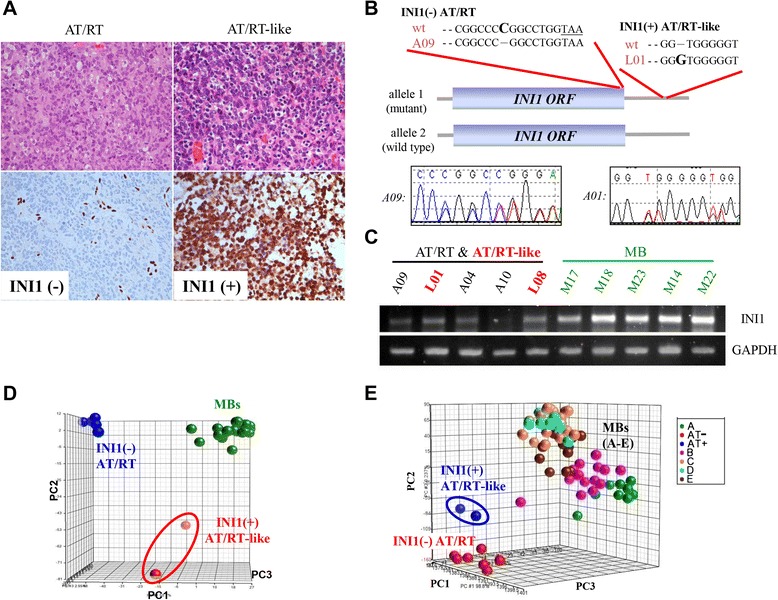
Identification of INI1(+) AT/RT-like cases among Taiwanese pediatric embryonal brain tumor cases. (a) Hematoxylin and eosin stain and INI1 stain of pediatric AT/RT and AT/RT-like cases, one with negative INI1 immunoreactivity (left) and one with positive INI1 immunoreactivity (right; anti-INI1, 400x). (b) Schematic representation of the results of INI1 gene sequencing. Patient A09 has only one mutated allele in the AT/RT: a single C deletion (in bold) was detected just before the INI1 stop codon (TAA; underlined). This resulted in a frame-shift mutation. In patient L01 with an INI1(+) AT/RT-like tumor, a single G insertion (in bold) was detected in the 3′UTR region of one INI1 allele. No protein abrogation was expected in this case. ORF indicates the open reading frame. (c) An examination of INI1 mRNA expression in tumor tissues by RT-PCR. (d) A PCA plot was drawn according to mRNAs that are differentially expressed between MB, AT/RT, and AT/RT-like cases (q < 10−5). INI1(+) AT/RT-like tumors have distinct mRNA expression profiles similar to those of AT/RTs and MBs. (e) Gene expression analysis of Taiwanese INI1(+) AT/RT-like cases and another published Caucasian MB data set. A PCA plot drawn according to the whole transcriptome again showed that INI1(+) AT/RT-like tumors were different from MBs and INI1(−) AT/RTs (A: Wnt subgroup; B: SHH subgroup; C: subgroup which expressed neuronal differentiation characteristic; D: subgroup which expressed neuronal and photoreceptor characteristic; E: subgroup which expressed photoreceptor characteristic and increased protein biosynthesis/cell cycle)
Similar genomic DNA aberrations in INI1 (−) AT/RTs and INI1(+) AT/RT-like cases
AT/RT cases A03-05 and A09-10, which were all INI1(−), were used in our previous study [14]. By direct sequencing, we determined that the INI1 gene region in these 5 cases was intact, while the expression of the INI1 protein in these cases was paradoxically lost. A novel yet unidentified posttranscriptional regulatory mechanism that occurs as part of INI1 protein synthesis likely exists in AT/RT tumor cells [14]. We conducted a similar analysis of the genomic DNA from cases in our current series, which included 5 AT/RT-like tumors (L01-L02 & L06-L08, Table 1) and an additional 4 new AT/RT cases (A11-A14, Table 1). PCR-amplified genomic DNA samples that were isolated from fresh-frozen tumors were used for direct INI1 gene sequencing, and mutations were found in only two cases. L01 (INI1+) showed a G insertion in exon 9 in one allele. Because this insertion was located in the 3′-UTR region of the INI1 mRNA, no abrogation of the protein would be expected (Fig. 1b, inserted G in bold). With regard to the AT/RTs, only one case (A09), which was documented in our previous report [14], acquired a C deletion in exon 9 in one allele (Fig. 1b). Such a deletion would lead to a frame-shift mutation and would generate a new INI1 protein with an extra 100-aa tail (Fig. 1b, deleted C in bold and TAA stop codon underlined). Thus, no differences in the INI1 gene region were detected between the INI1(+) AT/RT-like and INI1(−) AT/RT populations. RT-PCR experiments confirmed the expression of INI1 transcripts in both INI1(+) AT/RT-like cases (L01 and L08, Fig. 1c) and INI1(−) AT/RTs [14] (A04 and A09, Fig. 1c).
Deletion of the INI1 gene, which is located at 22q11.2, has been proposed to be a unique feature of AT/RT tumors [3, 9]. However, our previous array CGH analysis on Taiwanese INI1(−) AT/RT cases showed no deletion around the INI1 gene (Table 3) [14]. We extended the aCGH analysis to INI1(+) AT/RT-like cases, and still no 22q11.2 deletion could be found (Table 3). The INI1(+) and INI1(−) cases shared common chromosomal aberrations, including gains in 1q21.31 (150836484 ~ 150848568), 11p15.5 ~ p15.4 (274838 ~ 2917590), 11q11 (55129329 ~ 55195049), 14q32.33 (105602756 ~ 105630148), and 22q13.31 (44813553 ~ 44849176) (Table 3). The areas that were most commonly deleted were 7q36.1 (151592619 ~ 151887233), 8p11.23 (39356595 ~ 39482044), 12p13.31 (7696275 ~ 8068377), 15q11.2 (18810004 ~ 19910926) and 21p11.1 (10117898 ~ 10144936) (Table 3). Chromosomal aberrations in MB, including the well-known iso-17q mutation (17p11.2 ~ q25.3, 20851757 ~ 78653717 [14], were not detected in any of INI1(+) AT/RT-like or INI(−) AT/RT cases, which indicates that no patients with MB were misdiagnosed or mistaken to have INI1(+) AT/RT-like tumors.
Table 3.
Array CGH analysis showed no differential chromosomal aberration between INI1(−) AT/RTs & INI1(+) AT/RT-like cases
| Array CGH results | |||
|---|---|---|---|
| Case No. | INI1 | Gain | loss |
| L01* | + | 1q21.3, 11q11, 14q32.33, | 8p11.23 |
| L02 | + | 1q21.3, 14q32.33, 22q11.23 | 7q36.1, 12p13.31, 21p11.1 |
| L06 | + | 1q21.3, 11q11, 22q11.23, 22q13.31 | 7q36.1, 12p13.31, 15q11.2, 21p11.1 |
| L07 | + | 11p15.5 ~ p15.4, 14q32.33, 22q11.23, 22q13.31 | 7q36.1, 8p11.23, 12p13.31, 21p11.1 |
| L08 | + | 11p15.5 ~ p15.4, 11q11, 14q32.33, 22q11.23, 22q13.31 | 15q11.2, 21p11.1 |
| A03 | - | 1q21.3, 11p15.5 ~ p15.4, 11q11, 14q32.33, 22q11.23 | 8p11.23, 15q11.2 |
| A04 | - | 11p15.5 ~ p15.4, 14q32.33, 22q13.31 | 7q36.1, 8p11.23, 21p11.1 |
| A05 | - | 15q11.2, 21p11.1 | |
| A09** | - | 14q32.33, 22q11.23 | 7q36.1, 12p13.31 |
* Mutation on 1 allele; no mutation on the protein level
** Mutation on 1 allele; results in a new protein with additional 79 a.a
Distinct gene expression patterns among different subgroups of PEBTs
To further verify that INI1(+) AT/RT-like cases were not misdiagnosed MBs, as well as to provide new insights into the molecular differences among MB, AT/RT, and INI1(+) AT/RT-like cases, we examined the transcriptome patterns of these 3 closely related embryonal brain tumors by microarray analysis. A principle component analysis (PCA) plot based on genes that differentiate MB and AT/RTs and AT/RT-like cases (with a positive false discovery rate (pFDR) threshold of q < 10−5, 496 probe sets) showed that the AT/RT and MB cases we diagnosed were distinct from each other at the mRNA level (Fig. 1d, Additional file 2). Furthermore, the genetic profiles of AT/RTs and INI1(+) AT/RT-like tumors were distinct (Fig. 1d).
With another independent MB data set published by Kool et al. [21] (GEO database accession No. GSE10327) as an independent cohort, we still observed distinct transcriptome patterns among MBs, AT/RTs and INI1(+) AT/RT-like tumors (Fig. 1e). We also compared the transcriptomes of AT/RTs and INI1(+) AT/RT-like tumors to those of PNETs. An MDS plot showed that the transcriptomes of either AT/RTs or INI1(+) AT/RT-like tumors were significantly different from those of PNETs (Additional file 1-C), which indicates that the INI1(+) AT/RT-like cases in our series were not misdiagnosed PNETs.
Stem cell traits of different groups of embryonal brain tumors reflect their distinct clinical prognoses
To evaluate the survival outcomes of patients with AT/RTs and INI1(+) AT/RT-like tumors, we expanded the case numbers in the Kaplan-Meier (KM) estimator analysis. KM analysis and a log-rank test revealed that patients with INI1(−) AT/RTs and INI1(+) AT/RT-like tumors had a similar overall survival rate (19 AT/RTs and 16 AT/RT-like cases; Fig. 2a). In contrast, all 35 patients with AT/RTs or AT/RT-like tumors had a worse overall survival than patients with MBs (P < 0.0001; Fig. 2b). It is clear that cancer cells possess traits that are reminiscent of those ascribed to normal stem cells and that the degree of stem cell gene expression correlates with patient prognosis. Histologically poorly differentiated tumors or late-stage tumors show preferential overexpression of genes that are normally enriched in ES cells (ESCs) or in somatic stem cells, which may partly explain why these tumors are more malignant [22, 23]. We hypothesized that the distinct clinical survival trends of different PEBTs might be reflected by their gene expression traits. The array data of pluripotent ESCs, multipotent somatic neural stem cells (NSCs) and terminally differentiated fetal brain (FB) cells were included in a comparative transcriptome analysis. As expected, MBs were more similar to FB tissue (Fig. 2c). The transcriptome profiles of INI1(−) AT/RTs and INI1(+) ATRT-like tumors were most similar to those of ESCs and NSCs, respectively (Fig. 2c). Such impressions were quantified by the measurement of the average linkage distances between different embryonal brain tumors with respect to ESCs, to NSCs, or to FB cells (Fig. 2d).
Fig. 2.
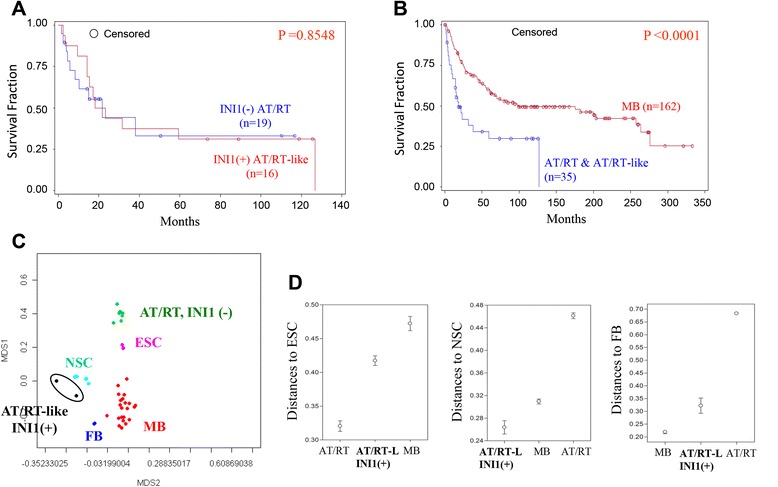
Gene signatures reflect the clinical prognostic status of different subtypes of embryonal brain tumors. (a) Overall survival rates of the INI1(−) AT/RTs (n = 19) and INI1(+) AT/RT-like (n = 16) cases included in the IHC analysis. (b) Survival curves of 199 cases of primary pediatric CNS embryonal tumors (162 MBs, 19 AT/RTs, and 16 INI1(+) AT/RT-like cases) that were included in the IHC validation study. All patients with AT/RTs or AT/RT-like tumors had a worse overall survival than patients with MBs (P < 0.0001). (c) An MDS plot based on genes that are differentially expressed in the 3 subtypes of embryonal brain tumors (q < 0.001) show the relationships among AT/RTs, MBs, INI1(+) AT/RT-like tumors and different stem or progenitor cells. ESC: embryonic stem cell; NSC: adult neural stem cell; FB: fetal brain tissues. (d) Analysis of the transcriptome distance between AT/RTs, MBs, INI1(+) AT/RT-like tumors and different stem or progenitor cells. AT/RT-L: AT/RT-like
Novel biomarkers for MBs, INI1(−) AT/RTs, and INI1(+) AT/RT-like tumors
To identify novel biomarkers and the pathogenetic mechanisms of these 3 subtypes of embryonal brain tumors, genes that are differentially expressed between each tumor type were also identified. A gene expression heat map for these genes indicated their unique expression patterns in these tumor subtypes (Fig. 3a). The FGFR2 growth factor receptor, the S100A4 stemness gene and the ERBB2 (HER2/neu) oncogene were dominant in the INI1(−) AT/RT cases (Fig. 3a, underlined). CXXC5 and SOX4 were enriched in MBs, while OLIG2, SOX6, SOX8, and SOX10 were expressed in 2 INI1(+) AT/RT-like samples (Fig. 3a). We also profiled the microRNA patterns in INI1(+) AT/RT-like samples, INI1(−) AT/RTs and MBs. Five microRNAs (miR-128, −138, −219-5p, −219-2-3p and −346) were found to be abundantly expressed in INI1(+) AT/RT-like cases (Fig. 3b).
Fig. 3.
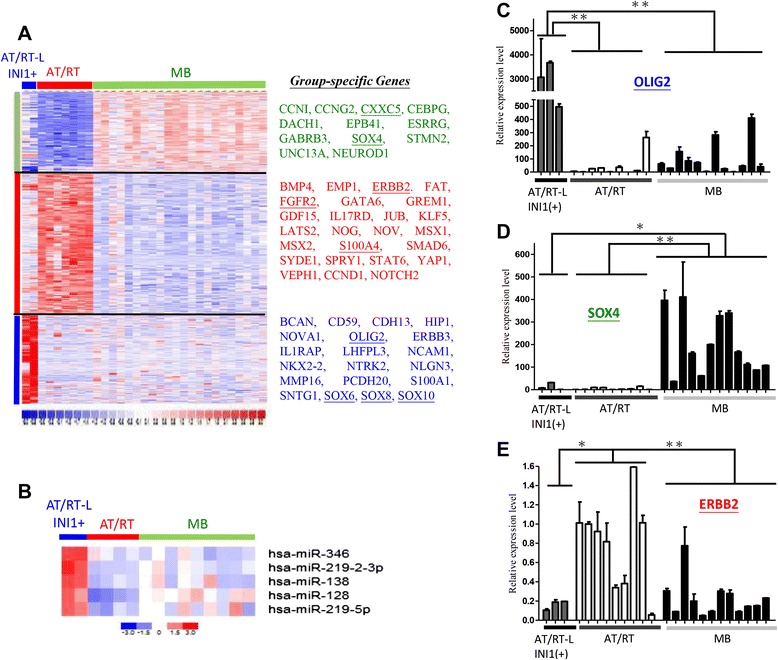
Genes that distinguish AT/RTs, MBs, and INI1(+) AT/RT-like tumors. (a-b) Heat maps show group-specific genes (a) and microRNAs (b). AT/RT-L: AT/RT-like. Red: up-regulation, Blue: down-regulation. (c-e) Real-time PCR validation of OLIG2 (c), SOX4 (D) and ERBB2 (e) array data on new batches of patient RNAs. * P < 0.05; ** P < 0.01
Array data on OLIG2, SOX4 and ERBB2 were validated by quantitative PCR (qPCR) (Fig. 3c-e, Additional file 2). OLIG2 was shown to be significantly overexpressed in INI1(+) AT/RT-like tumors (Fig. 3c), while SOX4 was overexpressed in MBs (Fig. 3e). Such a difference in the expression levels of these genes can be used to distinguish MBs from AT/RTs and, more critically, can be used to distinguish AT/RTs from INI1(+) AT/RT-like tumors.
Discussion
CNS AT/RTs are a specific entity of pediatric embryonic tumors. The ability to distinguish AT/RT from MB is of clinical significance because the reported survival rate of patients with AT/RTs is significantly lower than that of patients with a standard-risk MB [3]. The dismal outcome of patients with AT/RT is due to their poor response to conventional adjuvant therapy protocols, but recent studies have shown that improvements in patient survival can be achieved with intensive aggressive therapy or radiotherapy as an initial treatment [6, 24, 25]. Here, we report the identification of a new subtype of AT/RT-like tumors among cases of primary pediatric embryonal brain tumors in Taiwan and our data support the reliability of our original pathological diagnoses.
The INI1-positive AT/RT-like subtype may not be restricted to cases of PEBT in this Taiwanese population. A recent case report from Italy illustrates an INI1(+) AT/RT case in a 9-month-old boy whose tumor showed retained INI1/SMARCB1 expression by IHC and lacked genetic alterations in the INI1 gene [19]. Instead, the tumor had a nonsense mutation and loss of protein expression of another member of the SWI/SNF chromatin-remodeling complex, the ATPase subunit SMARCA4 (BRG1) due to a homozygous SMARCA4 mutation [c.2032C > T (p.Q678X)] [19]. Preserved INI1 protein expression and a SMARCA4 mutation were also observed in familial rhabdoid tumor predisposition syndrome (RTPS), which has been linked to heterozygous SMARCB1 germline mutations. Two sisters with RTPS whose tumors lacked mutations in INI1/SMARCB1 were diagnosed in Germany [20]; instead of a mutation in INI1/SMARCB1, mutations were again found in SMARCA4 in both patients. This result implied that SMARCA4 is a second member of the SWI/SNF complex involved in cancer predisposition and that SMARCA4 may play an essential role in the pathogenesis of new INI1/SMARCB1-positive AT/RT-like tumors. Whether SMARCA4 or other proteins within the SWI/SNF chromatin-remodeling complex are also involved in the pathogenesis of these Taiwanese INI1(+) AT/RT-like cases is currently under investigation.
Based on array CGH analysis, the abnormalities in the genomic DNA of the AT/RTs and the AT/RT-like cases we studied were indistinguishable yet distinct from those of published MB cases [14]. The INI1 region is intact in both INI1(+) AT/RT-like and INI1(−) AT/RT cases (Fig. 1b-c). Genetic studies of western cohorts have shown that deletion or mutation of the INI1 gene occurs in approximately 75 % [3, 9] to 98 % [10] of the AT/RTs studied. Nevertheless, in our previous study, we found no deletion around the INI1 gene in Taiwanese patients [14]. Subgroups of patients with AT/RTs have tumors that express INI1 mRNA, even though the tumors in our series were negative for INI1 protein by IHC [14]. Our study reveals that a novel yet unidentified posttranscriptional regulatory mechanism(s) that occurs after INI1 protein synthesis exists in AT/RT tumor cells. The application of INI1 genomic DNA as an essential diagnostic tool is therefore not invariably suitable, at least in Taiwanese cases.
Tumor development, progression, and prognosis remain positioned at the front line of medical research. Clinically, cancer cells with a poor differentiated pathological grade usually have a worse therapeutic response [22]. It has been demonstrated that cMyc, but not other tested oncogenes, is sufficient to reactivate the ESC-like program in normal and cancer cells [26]. The convergence of dedifferentiation and cancer malignancy also comes from the discovery that the epithelial-mesenchymal transition (EMT), which is a critical mechanism that mediates embryogenesis and cancer metastasis, can induce the formation of cancer spheres from transformed breast cancer or colon cancer epithelial cells. This concept is also derived from the discovery that tumor stem cells that have undergone EMT are more motile and show greater metastatic ability [27–29]. Among the three subtypes of PEBTs that we analyzed, we also discovered distinct stem/progenitor cell signatures in these tumors as well as expression levels of stemness genes that correlated well with patient prognosis (Fig. 2). Genes that are responsible for tumor stemness and resistance to therapy remain to be elucidated and require further investigation.
We uncovered unique gene expression patterns among AT/RTs, AT/RT-like tumors and MBs, and identified OLIG2 as a potential new biomarker for AT/RT-like cases (Fig. 3c). OLIG2 is expressed in neural progenitor cells [30], which corresponds with the finding that INI1(+) ATRT-like tumors shared a closest transcriptome profile to that of NSCs (Fig. 2c-d). OLIG2 also showed in subgroup of CNS-PNET [31], but according to our PCA plot (Additional file 1-C), they express distinct transcriptome. Therefore, INI1 plus OLIG2 and other existed biomarkers INI1 plus OLIG2 immunostains may help to accurately diagnose this new AT/RT-like subtype among cases of primary PEBTs. In addition, some genes were down-regulated in AT/RT-like tumors, such as SRD5A1 and CREB3L4. These genes have a chance to be biomarkers in AT/RT-like tumors, but it still need more samples to verify.
SOX4 is a potential new biomarker for MBs. Sox4 and Sox11, two HMG-box transcription factors, play central regulatory roles during neuronal maturation [32]. Other transcriptome analyses based on microarray or suppression subtraction hybridization also identified SOX4 as an abundant protein in human MBs [33, 34]. IHC studies have verified that the Sox4 and Sox11 proteins are strongly expressed in most classical MBs [34, 35]. Our array and RT-qPCR series contain classical MBs, desmoplastic MBs and anaplastic MBs, and all cases of MB expressed abundant SOX4 transcripts compared with cases of INI1(+) AT/RT-like cases or INI1(−) AT/RTs.
ERBB2 (HER2/neu) was found to be overexpressed in the AT/RT cases that we profiled. The ERBB2/HER2/neu oncogene, the human epidermal growth factor receptor 2, is a member of the epidermal growth factor receptor (EGFR/ErbB) family. The amplification or over-expression of this gene has been shown to play an important role in the pathogenesis and progression of certain aggressive types of breast cancer and advanced endometrioid carcinoma [23]. In recent years, ERBB2 has evolved to become an important prognostic biomarker and therapeutic target for breast cancer, and targeted therapies such as Herceptin™ (trastuzumab) and Omnitarg™ (pertuzumab) are now available clinically. The possibility that existing clinical drugs can be repurposed for the treatment of AT/RTs has yet to be evaluated.
In addition, we also find 5 miRNAs were up-regulated in AT/RT-like tumors. MiR-138 have been reported to serve as tumor suppressor in glioblastoma multiforme [36] or oncomiR in malignant glioma [37], respectively. MiR-128 and miR-219-5p are considered to be tumor suppressors, since which are found to inhibit proliferation, migration and progression in GBM [38] and gliomas [39]. Until now, miR-346 and miR-219-2-3p don’t publish in other brain tumor types. Therefore, miR-346 and miR-219-2-3p have a chance to be specific biomarkers for AT/RT-like tumors.
Conclusions
In conclusion, IHC and transcriptome studies of our primary embryonal brain tumor series identified a novel INI1 (+) AT/RT-like subtype among Taiwanese pediatric patients. Distinct gene expression patterns of closely related embryonal tumors were also provided. Careful diagnosis and clinical care of patients with different subtypes of embryonal brain tumors will benefit daily clinical practice. In addition, INI1(+) AT/RT-like cases are rare and hard to collect genetic data. Therefore, a collaborative international effort needs to happen immediately, increasing tumor numbers will help us to realize and supply specific biomarkers to distinguish between INI1(+) AT/RT-like tumors and other pediatric embryonal tumors.
Acknowledgments
The authors acknowledge the array services provided by the Microarray & Gene Expression Analysis Core Facility of the National Yang-Ming University Genome Research Center, which is supported by the National Research Program for Genomic Medicine, National Science Council (NSC).
This work was supported by the Ministry of Science and Technology [MOST; 103-2911-I-010-506, 102-2314-B-010-045, 101-2320-B-010-059-MY3 and 102-2314-B-038-060-MY3], ChengHsin General Hospital (103F003C01), Mackay Memorial Hospital (MMH-YM-10304), Veterans General Hospitals University System of Taiwan (VGHUST) Joint Research Program, Tsou’s Foundation [VGHUST103-G7-2-3], National Health Research Institutes [NHRI-EX103-10254SI], Taipei City Hospital [10401-62-014], National Yang-Ming University [Ministry of Education, Aim for the Top University Plan] Taipei Medical University Hospital Grant [No.104 TMU-TMUH-11] and Health and Welfare Surcharge of Tobacco Products [MOHW104-TDU-B-212-124-001]. This work was also supported in part by the UST-UCSD International Center for Excellence in Advanced Bioengineering sponsored by the Taiwan MOST I-RiCE Program [102-2911-I-009-101].
Additional files
(A) IHC features used for the diagnosis of AT/RT and MB in our clinical practice for pediatric embryonal brain tumors. (B) IHC results for SMARCA4 in AT/RT-like, AT/RT and MB samples. (C) Both INI1(+) AT/RT-like tumor and INI(−) AT/RT possess distinct mRNA profiles compared with those of PNETs. MDS plots were drawn based on genes that can differentiate AT/RT from AT/RT-like tumors (with a positive false discovery rate (pFDR) threshold of q < 0.001) show the relationships among AT/RTs, PNETs, and INI1(+) AT/RT-like tumors.
Expression data from AT/RTs, AT/RT-like tumors and medulloblastomas.
Footnotes
Donald Ming-Tak Ho and Chuan-Chi Shih contributed equally to this work.
Competing interests
The authors declare no competing financial or non-financial interests.
Authors’ contributions
MTH, CCS, HWW and TTW conceived the study and identified its value to pediatric tumor research. MTH, HWW and TTW designed the analysis approach. MLL, MEC, and TTW collected the tumor samples. MTH and SCL performed IHC staining. CYT, THH, CHT and TYC performed data analysis. MTH, HWW, and TTW provided biological guidance during the analysis process. The manuscript was written by HWW and submitted by THH, and all authors read and approved the final manuscript.
Contributor Information
Donald Ming-Tak Ho, Email: mtho@vghtpe.gov.tw.
Chuan-Chi Shih, Email: shihmd0316@yahoo.com.
Muh-Lii Liang, Email: mlliang@vghtpe.gov.tw.
Chan-Yen Tsai, Email: chanyentw@yahoo.com.tw.
Tsung-Han Hsieh, Email: thhsieh2003@gmail.com.
Chin-Han Tsai, Email: 4487@mmh.org.tw.
Shih-Chieh Lin, Email: diegolin@vghtpe.gov.tw.
Ting-Yu Chang, Email: taiwanbird@gmail.com.
Meng-En Chao, Email: chaomengen@gmail.com.
Hsei-Wei Wang, Email: hwwang@ym.edu.tw.
Tai-Tong Wong, Email: ttwong99@gmail.com.
References
- 1.Farwell J, Flannery JT. Cancer in relatives of children with central-nervous-system neoplasms. N Engl J Med. 1984;311(12):749–53. doi: 10.1056/NEJM198409203111201. [DOI] [PubMed] [Google Scholar]
- 2.Mori K, Kurisaka M. Brain tumors in childhood: statistical analysis of cases from the Brain Tumor Registry of Japan. Childs Nerv Syst. 1986;2(5):233–7. doi: 10.1007/BF00272492. [DOI] [PubMed] [Google Scholar]
- 3.Biegel JA. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus. 2006;20(1):E11. doi: 10.3171/foc.2006.20.1.12. [DOI] [PubMed] [Google Scholar]
- 4.Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol. 2004;28(5):644–50. doi: 10.1097/00000478-200405000-00013. [DOI] [PubMed] [Google Scholar]
- 5.Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56–65. doi: 10.3171/jns.1996.85.1.0056. [DOI] [PubMed] [Google Scholar]
- 6.Buscariollo DL, Park HS, Roberts KB, Yu JB. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer. 2012;118(17):4212–9. doi: 10.1002/cncr.27373. [DOI] [PubMed] [Google Scholar]
- 7.Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H. Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol. 2000;99(5):482–8. doi: 10.1007/s004010051149. [DOI] [PubMed] [Google Scholar]
- 8.Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22(9):1083–92. doi: 10.1097/00000478-199809000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF, et al. Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: Lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol. 2006;30(11):1462–8. doi: 10.1097/01.pas.0000213329.71745.ef. [DOI] [PubMed] [Google Scholar]
- 10.Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L, et al. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res. 2009;15(6):1923–30. doi: 10.1158/1078-0432.CCR-08-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol. 2007;14(5):335–9. doi: 10.1097/PAP.0b013e3180ca8b08. [DOI] [PubMed] [Google Scholar]
- 12.Sigauke E, Rakheja D, Maddox DL, Hladik CL, White CL, Timmons CF, et al. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol. 2006;19(5):717–25. doi: 10.1038/modpathol.3800581. [DOI] [PubMed] [Google Scholar]
- 13.Bourdeaut F, Freneaux P, Thuille B, Lellouch-Tubiana A, Nicolas A, Couturier J, et al. hSNF5/INI1-deficient tumours and rhabdoid tumours are convergent but not fully overlapping entities. J Pathol. 2007;211(3):323–30. doi: 10.1002/path.2103. [DOI] [PubMed] [Google Scholar]
- 14.Tsai CY, Wong TT, Lee YH, Chao ME, Lin SC, Liu DJ, et al. Intact INI1 gene region with paradoxical loss of protein expression in AT/RT: implications for a possible novel mechanism associated with absence of INI1 protein immunoreactivity. Am J Surg Pathol. 2012;36(1):128–33. doi: 10.1097/PAS.0b013e3182348cc4. [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharjee M, Hicks J, Langford L, Dauser R, Strother D, Chintagumpala M, et al. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. Ultrastruct Pathol. 1997;21(4):369–78. doi: 10.3109/01913129709021935. [DOI] [PubMed] [Google Scholar]
- 16.Huang TS, Hsieh JY, Wu YH, Jen CH, Tsuang YH, Chiou SH, et al. Functional network reconstruction reveals somatic stemness genetic maps and dedifferentiation-like transcriptome reprogramming induced by GATA2. Stem Cells. 2008;26(5):1186–201. doi: 10.1634/stemcells.2007-0821. [DOI] [PubMed] [Google Scholar]
- 17.Wu YH, Hu TF, Chen YC, Tsai YN, Tsai YH, Cheng CC, Wang HW: The manipulation of microRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood 2011. [DOI] [PubMed]
- 18.Yang TP, Chang TY, Lin CH, Hsu MT, Wang HW. ArrayFusion: a web application for multi-dimensional analysis of CGH, SNP and microarray data. Bioinformatics. 2006;22(21):2697–8. doi: 10.1093/bioinformatics/btl457. [DOI] [PubMed] [Google Scholar]
- 19.Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35(6):933–5. doi: 10.1097/PAS.0b013e3182196a39. [DOI] [PubMed] [Google Scholar]
- 20.Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86(2):279–84. doi: 10.1016/j.ajhg.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang SJ, Wang TY, Tsai CY, Hu TF, Chang MD, Wang HW. Increased epithelial stem cell traits in advanced endometrial endometrioid carcinoma. BMC Genomics. 2009;10:613. doi: 10.1186/1471-2164-10-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004;22(14):2877–84. doi: 10.1200/JCO.2004.07.073. [DOI] [PubMed] [Google Scholar]
- 25.Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23(7):1491–9. doi: 10.1200/JCO.2005.05.187. [DOI] [PubMed] [Google Scholar]
- 26.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2(4):333–44. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8):e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279–91. doi: 10.1053/j.gastro.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14(2):123–34. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Picard D, Miller S, Hawkins CE, Bouffet E, Rogers HA, Chan TS, et al. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol. 2012;13(8):838–48. doi: 10.1016/S1470-2045(12)70257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergsland M, Werme M, Malewicz M, Perlmann T, Muhr J. The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 2006;20(24):3475–86. doi: 10.1101/gad.403406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fevre-Montange M, Champier J, Szathmari A, Wierinckx A, Mottolese C, Guyotat J, et al. Microarray analysis reveals differential gene expression patterns in tumors of the pineal region. J Neuropathol Exp Neurol. 2006;65(7):675–84. doi: 10.1097/01.jnen.0000225907.90052.e3. [DOI] [PubMed] [Google Scholar]
- 34.de Bont JM, Kros JM, Passier MM, Reddingius RE, Sillevis Smitt PA, Luider TM, et al. Differential expression and prognostic significance of SOX genes in pediatric medulloblastoma and ependymoma identified by microarray analysis. Neuro Oncol. 2008;10(5):648–60. doi: 10.1215/15228517-2008-032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee CJ, Appleby VJ, Orme AT, Chan WI, Scotting PJ. Differential expression of SOX4 and SOX11 in medulloblastoma. J Neurooncol. 2002;57(3):201–14. doi: 10.1023/A:1015773818302. [DOI] [PubMed] [Google Scholar]
- 36.Qiu S, Huang D, Yin D, Li F, Li X, Kung HF, et al. Suppression of tumorigenicity by microRNA-138 through inhibition of EZH2-CDK4/6-pRb-E2F1 signal loop in glioblastoma multiforme. Biochim Biophys Acta. 2013;1832(10):1697–707. doi: 10.1016/j.bbadis.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 37.Chan XH, Nama S, Gopal F, Rizk P, Ramasamy S, Sundaram G, et al. Targeting glioma stem cells by functional inhibition of a prosurvival oncomiR-138 in malignant gliomas. Cell reports. 2012;2(3):591–602. doi: 10.1016/j.celrep.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 38.Rao SA, Arimappamagan A, Pandey P, Santosh V, Hegde AS, Chandramouli BA, et al. miR-219-5p inhibits receptor tyrosine kinase pathway by targeting EGFR in glioblastoma. PLoS One. 2013;8(5):e63164. doi: 10.1371/journal.pone.0063164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong Q, Cai N, Tao T, Zhang R, Yan W, Li R, et al. An axis involving SNAI1, microRNA-128 and SP1 modulates glioma progression. PLoS One. 2014;9(6):e98651. doi: 10.1371/journal.pone.0098651. [DOI] [PMC free article] [PubMed] [Google Scholar]