

Abstract
In contrast to observations with carbohydrates, experiments with 4-alkoxy-substituted acetals indicate that an alkoxy group can accelerate acetal hydrolysis by up to 20-fold compared to substrates without an alkoxy group. The acceleration of ionization in more flexible acetals can be up to 200-fold when compensated for inductive effects.
Keywords: Electrostatic Effects, Kinetics, Carbocations, Carbohydrates
Studies of the relative reactivities of glycosyl donors, often determined by measuring the kinetics of acetal hydrolysis,[1-2] provide critical information used for designing iterative oligosaccharide synthesis.[3] These experiments reveal that the relative rates of substitution reactions of carbohydrate-derived acetals[3-6] depend upon the stereochemical configurations of substituents. For example, the methyl acetal of galactose (2) hydrolyzed five times faster than the glucose-derived acetal 1, which differs only in the stereochemical configuration at C-4 (Figure 1).[1,7-10] This difference in rate has been attributed to favorable electronic interactions between the electron-rich oxygen atom at C-4 and the developing positive charge at C-1 in the transition state for hydrolysis.[7,11-12] This stabilizing electrostatic effect, however, does not compensate for the electron-withdrawing influence of the oxygen atom at C-4: inductive destabilization by the hydroxyl group slows hydrolysis by 4-30 times compared to reactions where the alkoxy group was replaced with a substituent that is not electron-withdrawing (for example, 3 in Figure 1).[7,13-14]
Figure 1.
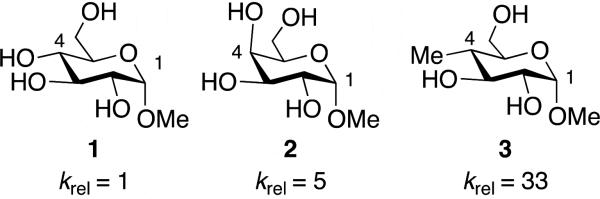
Relative rates of hydrolysis of α-methyl pyranosides (2.0 M HCl, 74 °C).[7]
In this Communication, we provide evidence that the electrostatic stabilization conferred by an alkoxy group four carbons away from an acetal group, the arrangement found in pyranosides 1 and 2, can overcome inductive effects and accelerate acetal hydrolysis if the system is more conformationally flexible than carbohydrates are. Flexibility likely allows the alkoxy group to approach the acetal functional group during substitution to provide up to a 20-fold increase in the rate of acetal hydrolysis compared to substrates that do not have an alkoxy group. When rates are compensated for inductive destabilization of the transition state for hydrolysis, through-space electrostatic stabilization of developing charge can accelerate hydrolysis by up to 200-fold.
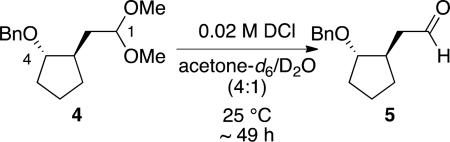
To evaluate the difference in reactivity exerted by a 4-alkoxy group based upon its position in space, a series of 4-alkoxy-substituted acetals resembling 4 ([Eq. (1)]) were synthesized and their rates of acetal hydrolysis were measured. These compounds were designed so the ability of the alkoxy group to approach the acetal carbon atom could be varied systematically. The two key functional groups were arrayed around a ring, just as for the carbohydrate systems (Figure 1), so entropic effects[15] would be similar for all substrates. Protection of the alkoxy group as the benzyl ether was chosen because the fate of the benzyl group could reveal the structures of reactive intermediates (vide infra). The hydrolysis reactions follow the conditions depicted in [Eq. (1)].[16]
 |
(1) |
The relative rates of acid-catalyzed hydrolysis are summarized in Chart 1, listed in order of increasing rate. Relative rates were normalized to the rate of hydrolysis of alkyl acetal 8. This simple acetal should be sterically equivalent to the other ß-substituted acetals because the two-carbon side-chains of acetals 4, 6, and 10-12 should adopt conformations where the acetal groups are oriented away from the alkoxy groups.[17-18] Comparison of the rates of ionization of α-branched acetal 9 and ß-branched acetal 8 indicates that steric effects play only a minor role in the hydrolysis of acetals in this series.[19]
Chart 1.

Relative rates of hydrolysis of acetals by DCl in acetone-d6/D2O (4:1), listed in order of increasing rate.
The acetal models illustrated in Chart 1 share trends in reactivity with their carbohydrate relatives. The presence of an alkoxy group in acetals cis- and trans-7 decreased the rate compared to alkyl acetal 9 because an alkoxy group would inductively destabilize positively charged intermediates.[13,20] The relative reactivity of the acetals cis- and trans-7 is also reminiscent of the carbohydrate systems: the slower-reacting acetal cis-7 positions the alkoxy group at C-4 further from the acetal carbon atom (Figure 2), much as the slower-reacting glucose isomer does (Figure 1).[21] The small difference between the rates of the stereoisomers likely reflects the fact that the alkoxy group in trans-7 is far enough away to exert little stabilization of the transition state for hydrolysis.[22-23]
Figure 2.
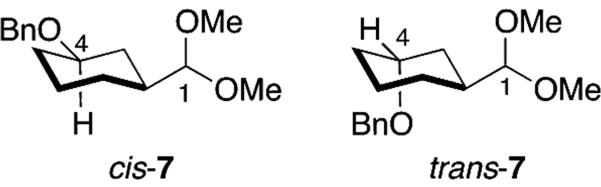
Chair conformations of acetals cis- and trans-7 illustrating the distances between the alkoxy groups and the acetal carbon atoms.
In contrast to the results exemplified in Figure 1,[24] an alkoxy group at C-4 of an acetal can accelerate the hydrolysis of an acetal. Whereas the cyclopentane-derived acetal 4 is less reactive than the alkyl acetal 8, the larger cycloalkyl-containing acetals 10-12 reacted up to 20 times faster (Chart 1). This trend is consistent with the idea that the larger, more flexible ring[25] allowed the alkoxy group to approach the acetal carbon more closely in the transition state for ionization[1] without incurring additional strain (13, Figure 3). As observed for galactosides compared to glucosides (Figure 1),[7] the closer the alkoxy group can approach the acetal carbon atom,[11-12] the faster hydrolysis occurred.
Figure 3.
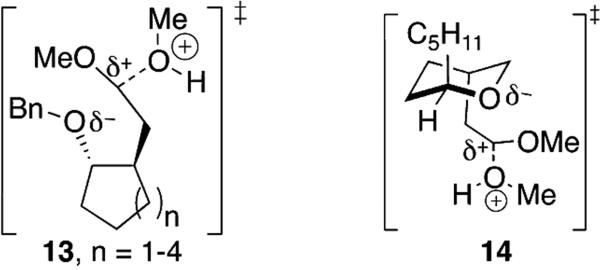
Structural requirements for acceleration of acetal hydrolysis by an alkoxy group.
The slow hydrolysis of tetrahydropyran-derived acetal 6 reinforces the importance of flexibility on the rates of hydrolysis of a 4-alkoxy-substituted acetal. If there were any interaction of the oxygen atom of the ring of acetal with the protonated acetal group during hydrolysis (14, Figure 3), the ring would need to adopt an orientation resembling a strained bicyclic structure[26] with substituents in disfavored axial orientations.[27] These energetic penalties and the inductive destabilization of the alkoxy group[13,20] must outweigh any benefit the alkoxy group can confer on hydrolysis. The hydrolysis of this compound provides a point of comparison for the hydrolysis of the other alkoxy-substituted acetals. Using the rate of hydrolysis of the constrained acetal 6 to control for inductive effects, it can be estimated that electrostatic effects imparted by an alkoxy group can accelerate hydrolysis by up to 200-fold.[28]
The relatively slow ionization of cyclopentane-derived acetal 4 suggests the origin of the accelerating influence of an alkoxy group on the hydrolysis in acetals. Two types of interactions of an alkoxy group during hydrolysis can be envisioned (illustrated in Scheme 1 for acetal 4). At one limit, a new covalent bond would form between the alkoxy group and the acetal carbon in the transition state for hydrolysis (pathway a), as occurs in neighboring-group participation during solvolytic displacement reactions.[29] Alternatively, the alkoxy group might only approach the acetal carbon atom in the transition state (pathway b) without forming a new covalent bond (as suggested for the influence of acyl groups on the hydrolysis of 2-acyloxy-substituted acetals[24]). If formation of an intermediate oxonium ion such as 16 were required (Scheme 1, pathway a), ionization of acetal 4 would form a strained trans-bicyclo[3.3.0]octane ring system.[26,30] By contrast, ionization of cyclohexane-derived acetal 10 would form a much less strained oxonium ion (by about 10 kcal/mol[26,30]). Consequently, the cyclohexane-derived acetal 10 should hydrolyze much faster than the cyclopentane-derived acetal 4. The difference in rate is only 20-fold, however, which is a small rate difference considering the large difference in strain.
Scheme 1.
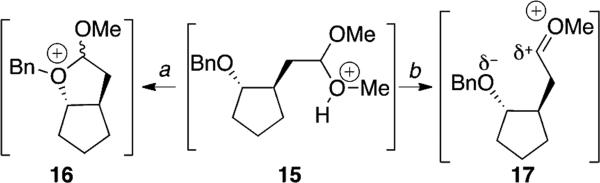
Interaction of an alkoxy group during hydrolysis: [a] formation of a new covalent bond in the transition state, and [b] electrostatic stabilization in the transition state.
Electrostatic stabilization of the oxocarbenium ion 17 that forms upon hydrolysis is consistent with the relative rates (Scheme 1, pathway b).[11-12] Electrostatic stabilization would require the alkoxy group to be close to the acetal carbon,[11-12] but the carbon–oxygen bond distances would be much longer[31] than they would be for an oxonium ion.[32] As a result, the relative rates of hydrolysis should not track the ring strain closely because a new ring would not be fully formed.
Other observations also suggest that oxonium ion intermediates are not involved in acetal hydrolysis. In none of the hydrolyses were products observed that were consistent with loss of a benzyl group, as might be expected if an oxonium ion had formed.[33] By contrast, solvolysis of 4-alkoxy-substituted tosylate[34] 18 reveals that the model ring systems can form oxonium ions if doing so were required for solvolysis (Scheme 2). The presence of significant quantities of cyclized, debenzylated product 21 along with benzyl esters 22 and 23 implicates the formation of oxonium ion 19 during solvolysis.[34]
Scheme 2.

Solvolysis of tosylate 18 resulted in benzyl substitution products.
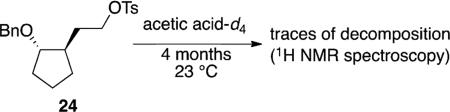
The tosylate model system also showed how large the difference in rates can be between similar substrates if oxonium ions were reactive intermediates. In contrast to the relatively rapid solvolysis of cyclohexane-derived tosylate 18, the cyclopentane-derived tosylate 24 was resistant to solvolysis: after four months at 23 °C, a sample of tosylate 24 in acetic acid-d4 showed only traces of decomposition [Eq. (2)]. The low reactivity of this tosylate can be attributed to the difficulty of engaging in neighboring-group participation during solvolysis[34] through a highly strained trans-bicyclo[3.3.0]octane-like oxonium ion resembling 16 (Scheme 1).[26] Because the acetal substrates do not show as large a difference in solvolysis rate as the tosylates do provides additional evidence against the formation of an oxonium ion during acetal hydrolysis.
 |
(2) |
Experiments with alkoxy-substituted acetals suggest that an alkoxy group can accelerate the ionization of acetals. The accelerating influence of the alkoxy group, which can be attributed to electrostatic stabilization of the developing oxocarbenium ion intermediate, can outweigh the decelerating inductive effect by about 200-fold.
Supplementary Material
Footnotes
This research was supported by the National Institutes of Health, National Institute of General Medical Sciences (GM-61066). We thank Ms. Lisa Barker for her intellectual contributions and Dr. Chin Lin for assistance with NMR spectroscopy and mass spectrometric data. D.A.L.O. was supported by a Margaret Strauss Kramer Fellowship from the NYU Department of Chemistry.
Supporting information for this article is given via a link at the end of the document.
References
- 1.Capon B. Chem. Rev. 1969;69:407–498. [Google Scholar]
- 2.Sinnott ML. Carbohydrate Chemistry and Biochemistry. The Royal Society of Chemistry; Cambridge, UK: 2007. pp. 66–139. [Google Scholar]
- 3.Hsu C-H, Hung S-C, Wu C-Y, Wong C-H. Angew. Chem. Int. Ed. 2011;50:11872–11923. doi: 10.1002/anie.201100125. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123:12076–12129. [Google Scholar]
- 4.Mikkola S, Oivanen M. ARKIVOC. 2009;iii:39–53. [Google Scholar]
- 5.Douglas NL, Ley SV, Lücking U, Warriner SL. J. Chem. Soc., Perkin Trans. 1998;1:51–65. [Google Scholar]
- 6.Zhang Z, Ollmann IR, Ye X-S, Wischnat R, Baasov T, Wong C-H. J. Am. Chem. Soc. 1999;121:734–753. [Google Scholar]
- 7.Jensen HH, Bols M. Org. Lett. 2003;5:3419–3421. doi: 10.1021/ol030081e. [DOI] [PubMed] [Google Scholar]
- 8.The small difference in relative rates of reactions of the α- and β-anomers observed in reference 7 is consistent with earlier observations (see ref 1).
- 9.Activation of thioglycosides is faster for sugars bearing axial groups at C-4 (ref 3).
- 10.Septanosides with axial hydroxyl groups at C-5 hydrolyze about eight times faster than ones bearing equatorial hydroxyl groups: Markad SD, Miller SM, Morton M, Peczuh MW. Tetrahedron Lett. 2010;51:1209–1212.
- 11.Woods RJ, Andrews CW, Bowen JP. J. Am. Chem. Soc. 1992;114:859–864. [Google Scholar]
- 12.Miljković M, Yeagley D, Deslongchamps P, Dory YL. J. Org. Chem. 1997;62:7597–7604. [Google Scholar]
- 13.Hydrolysis of dinitrophenyl galactosides occured five-fold faster than for glucosides: Namchuk MN, McCarter JD, Becalski A, Andrews T, Withers SG. J. Am. Chem. Soc. 2000;122:1270–1277.
- 14.Inductive effects decrease the rate of acetolysis of trans-2-acetoxycyclohexyl tosylate compared to cyclohexyl tosylate even though neighboring-group participation occurs in the former, not the latter: Winstein S, Grunwald E, Buckles RE, Hanson C. J. Am. Chem. Soc. 1948;70:816–821.
- 15.Page MI. Chem. Soc. Rev. 1973;2:295–323. [Google Scholar]
- 16.The rates of hydrolysis of acetals were measured by 1H NMR spectroscopy, and the rates conformed to first-order kinetics as determined by plots of ln[acetal] versus time over one to six half-lives, which gave lines with R2 ≥ 0.96. Details are provided as Supporting Information.
- 17.Hoffmann RW, Kahrs BC, Reiβ P, Trieselmann T, Stiasny H-C, Massa W. Eur. J. Org. Chem. 2001:1857–1864. [Google Scholar]
- 18.Hoffmann RW, Kahrs BC, Schiffer J, Fleischhauer J. J. Chem. Soc., Perkin Trans. 1996;2:2407–2414. [Google Scholar]
- 19.Steric effects can influence the rates of acetal hydrolysis if stabilizing interactions in the intermediate oxocarbenium ions are affected: Fife TH, Hagopian L. J. Org. Chem. 1966;31:1772–1775.
- 20.Withers SG, MacLennan DJ, Street IP. Carbohydr. Res. 1986;154:127–144. [Google Scholar]
- 21.Jensen HH, Nordstrøm LU, Bols M. J. Am. Chem. Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 22.Calculations (HF 6-31G**) of the structure of a model acetal for the glucose and galactose α-methyl pyranosides show that the distances between the oxygen atom of a MeO group at C-4 and the acetal carbon atom are 3.64 Å and 2.94 Å, respectively (reference 12). Crystallographic studies in 3-alkylcyclohexanol derivatives reveal longer distances between the oxygen atom and the exocyclic carbon atoms: 4.85 Å for the diequatorial isomer Fleming I, Maiti P, Ramarao C. Org. Biomol. Chem. 2003;1:3989–4004. doi: 10.1039/b305880h. and 4.30 Å for the isomer where the alkoxy group is axial Bourque LE, Cleary PA, Woerpel KA. J. Am. Chem. Soc. 2007;129:12602–12603. doi: 10.1021/ja073758s.
- 23.Acetal cis-7 could adopt a sterically disfavored diaxial conformer positioning the alkoxy group closer to the acetal carbon atom, but the slower rate of acceleration compared to trans-7 suggests that that it does not.
- 24.Heuckendorff M, Pedersen CM, Bols M. Org. Lett. 2011;13:5956–5959. doi: 10.1021/ol202308y. [DOI] [PubMed] [Google Scholar]
- 25.Tran VT, Woerpel KA. J. Org. Chem. 2013;78:6609–6621. doi: 10.1021/jo400945j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allinger NL, Tribble MT, Miller MA, Wertz DH. J. Am. Chem. Soc. 1970;93:1637–1648. [Google Scholar]
- 27.Eliel EL, Hargrave KD, Pietrusiewicz KM, Manoharan M. J. Am. Chem. Soc. 1982;104:3635–3643. [Google Scholar]
- 28.By comparison, the involvement of an acyloxy group at the 2-position of a carbohydrate, which also does not cause considerable ring strain, has been suggested to accelerate hydrolysis by only up to 13-fold when controlled for inductive effects. This acceleration has been attributed to stabilization of the developing positive charge by the carbonyl oxygen atom, not formation of an onium ion (reference 24).
- 29.Capon B. Q. Rev. Chem. Soc. 1964;18:45–111. [Google Scholar]
- 30.Wiberg KB. Angew. Chem., Int. Ed. Engl. 1986;25:312–322. [Google Scholar]; Angew. Chem. 1986;98:312–322. [Google Scholar]
- 31.Chamberland S, Ziller JW, Woerpel KA. J. Am. Chem. Soc. 2005;127:5322–5323. doi: 10.1021/ja050830i. [DOI] [PubMed] [Google Scholar]
- 32.Gunbas G, Hafezi N, Sheppard WL, Olmstead MM, Stoyanova IV, Tham FS, Meyer MP, Mascal M. Nature Chem. 2012;4:1018–1023. doi: 10.1038/nchem.1502. [DOI] [PubMed] [Google Scholar]
- 33.Chao CS, Lin CY, Mulani S, Hung WC, Mong KK. Chem. Eur. J. 2011;17:12193–12202. doi: 10.1002/chem.201100732. [DOI] [PubMed] [Google Scholar]
- 34.Winstein S, Allred E, Heck R, Glick R. Tetrahedron. 1958;3:1–13. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.