

Abstract
Introduction
Radioiodinated meta-iodobenzylguanidine (MIBG), a norepinephrine transporter (NET) substrate, has been extensively used as an imaging agent to study the pathophysiology of the heart and for the diagnosis and treatment of neuroendocrine tumors. The goal of this study was to develop an 18F-labeled analogue of MIBG that like MIBG itself could be synthesized in a single radiochemical step. Towards this end, we designed 4-fluoropropoxy-3-iodobenzylguanidine (FPOIBG).
Methods
Standards of FPOIBG and 4-fluoropropoxy-3-bromobenzylguanidine (FPOBBG) as well as their tosylate precursors for labeling with 18F, and a tin precursor for the preparation of radioiodinated FPOIBG were synthesized. Radiolabeled derivatives were synthesized by nucleophilic substitution and electrophilic iododestannylation from the corresponding precursors. Labeled compounds were evaluated for NET transporter recognition in in vitro assays using three NET-expressing cell lines and in biodistribution experiments in normal mice, with all studies performed in a paired-label format. Competitive inhibition of [125I]MIBG uptake by unlabeled benzylguanidine compounds was performed in UVW-NAT cell line to determine IC50 values.
Results
[18F]FPOIBG was synthesized from the corresponding tosylate precursor in 5.2 ± 0.5% (n = 6) overall radiochemical yields starting with aqueous fluoride in about 105 min. In a paired-label in vitro assay, the uptake of [18F]FPOIBG at 2 h was 10.2 ± 1.5%, 39.6 ± 13.4%, and 13.3 ± 2.5%, in NET-expressing SK-N-SH, UVW-NAT, and SK-N-BE(2c) cells, respectively, while these values for [125I]MIBG were 57.3 ± 8.1%, 82.7 ± 8.9%, and 66.3 ± 3.6%. The specificity of uptake of both tracers was demonstrated by blocking with desipramine. The 125I-labeled congener of FPOIBG gave similar results. On the other hand, [18F]FPOBBG, a compound recently reported in the literature, demonstrated much higher uptake, albeit less than that of co-incubated [125I]MIBG. IC50 values for FPOIBG were higher than that obtained for MIBG and FPOBBG. Unlike the case with [18F]FPOBBG, the heart uptake [18F]FPOIBG in normal mice was significantly lower than that of MIBG.
Conclusion
Although [18F]FPOIBG does not appear to warrant further consideration as an 18F-labeled MIBG analogue, analogues wherein the iodine in it is replaced with a chlorine, fluorine or hydrogen might be worth pursuing.
Advances in knowledge and implications for patient care
An 18F-labeled analogue of the well-known radiopharmaceutical MIBG could have significant impact, potentially improving imaging of NET related disease in cardiology and in the imaging of neuroendocrine tumors. Although 18F-labeled analogues of MIBG have been reported including LMI1195, we undertook this work hypothesizing that based on its greater structural similarity to MIBG, FPOIBG might be a better analogue than LMI1195.
Keywords: MIBG, fluorine-18, norepinephrine transporter, PET imaging, neuroendocrine tumors
1. Introduction
Radioiodinated meta-iodobenzylguanidine (MIBG) [1], originally developed as an adrenomedullary imaging agent [2], has been widely used for imaging the pathophysiology of the heart [3] and in the diagnosis and therapy of neuroendocrine tumors [4]. For diagnostic applications, planar scintigraphy and single photon emission computed tomography (SPECT) are the modalities that are often used in tandem with [131I]MIBG and [123I]MIBG, respectively. Positron emission tomography (PET) has several inherent advantages over planar imaging and SPECT. Thus, an MIBG analogue labeled with a positron-emitter could have important applications in both cardiology and oncology. Given that 124I is a positron-emitting radioisotope of iodine, the logical option is [124I]MIBG, which was first synthesized more than two decades ago; improvements in its synthesis reported later [5, 6]. Although a number of studies evaluating the potential usefulness of [124I]MIBG have appeared [7–11], as noted by others [12, 13], 124I is not an ideal radionuclide for PET. MIBG analogues labeled with other positron emitters such as 76Br and 11C also have been reported [14, 15]. Bromine-76 also has a relatively long half-life and is not readily available; on the other hand, the half-life of 11C is so short that radiochemical synthesis of 11C-labeled compounds can often be challenging.
Fluorine-18 has a half-life of 110 min, which is compatible with the biological half-life of MIBG, and the need for an MIBG analogue labeled with 18F has been advocated. Our laboratory was the first to develop 18F-labeled MIBG analogues 3-[18F]fluorobenzylguanidine ([18F]MFBG) and 4-[18F]fluorobenzylguanidine ([18F]PFBG) [16]. MFBG was shown to be a better analogue of MIBG than PFBG; however, its radiochemical synthesis was more difficult. Both analogues were inferior to MIBG with respect to uptake in NET-expressing SK-N-SH human neuroblastoma cells in vitro and myocardial uptake in normal mice in vivo; however, interest in [18F]MFBG for imaging has recently been revived [17, 18]. Hypothesizing that the lackluster performance of [18F]MFBG and [18F]PFBG might be due to the lack an iodine substituent on its benzene ring, which may be essential for bioactivity, we synthesized 4-[18F]fluoro-3-iodobenzylguanidine ([18F]FIBG) that is essentially MIBG with an added fluorine [19]. Indeed, FIBG was demonstrated to be a superior MIBG analogue [19, 20]; however, its structure was not amenable for single step 18F-labeling via the classical SNAr substitution. As a result, its synthesis involved four radiochemical steps with a low overall radiochemical yield, making further evaluation of [18F]FIBG difficult. Parenthetically, novel methods reported for 18F-labeling of substrates that are not activated for SNAr substitutions can be adapted for [18F]FIBG synthesis and efforts are under way in this direction in our laboratory. These methods include use of diaryl iodonium salts [21, 22] and iodonium ylide [23] precursors, and those mediated by nickel, palladium, and copper [24–27].
Other 18F-labeled MIBG analogues that lack an iodo substituent have been reported [28]. Radiofluorination by SN2 substitution on a sp3 carbon can be performed often in a single step, which motivated the development of 18F-labeled MIBG analogues containing a fluoroalkyl side chain [29]. However, these analogues, which lack an iodine in their structure, performed poorly compared to MIBG in both in vitro and in vivo assays. Hypothesizing that the presence of an iodine substituent is a sine qua non for MIBG-like behavior, we undertook the work reported herein. We opted for a fluoropropoxy group rather than the fluoroalkyl groups used by Lee et al. [29] because multiple 18F-labeled compounds bearing [18F]fluoroalkoxy arenes have been reported, which can often be generated by fluoroalkylation of corresponding phenols. More relevant to MIBG, the 4-hydroxy derivative (HIBG) has been shown to be a good analogue of MIBG [30]. We have developed a method for the synthesis of 4-[18F]fluoropropoxy-3-iodobenyzylguanidine ([18F]FPOIBG) and evaluated its potential as an MIBG analogue in vitro using NET-expressing cancer cells and in vivo in normal mice. Since starting this work, an analogous compound containing bromine instead of iodine has been reported [31].
2. Materials and Methods
2.1. General
All chemicals, unless specified otherwise, were obtained from Sigma-Aldrich or Alpha Aesar and used as such. Sodium [125I]iodide (81 TBq/mmol) and sodium [131I]iodide (44 TBq/mmol) as a solution in 0.1N NaOH were procured from Perkin Elmer Life and Analytical Sciences (Boston, MA). A method reported in the literature [32] was used for the synthesis of 4-hydroxy-3-iodo-toluene (1a) from para-cresol. MIBG radiolabeled with 125I was prepared at a carrier-free level from a tin precursor as reported [6]. Fluorine-18 was obtained either by in house cyclotron irradiation of [18O]H2O as described before [33] or from PET-NET solutions (Durham, NC). For labeling reactions, 18F activity trapped in a QMA cartridge was eluted with a mixture of Kryptofix (28 mg) and potassium carbonate (2.4 mg) in 0.75 mL of 95% acetonitrile, and dried by azeotroping with acetonitrile three times. Aluminum-backed sheets (Silica gel 60 F254) used for analytical TLC and silica gel 60 used for normal-phase column chromatography were obtained from EM Science (Gibbstown, NJ). In some cases, chromatography was also performed using the Biotage Isolera chromatography system (Charlotte, NC) with their prepacked columns. High-performance liquid chromatography (HPLC) was performed using the following systems: 1) for analytical and semi-preparative HPLC of unlabeled compounds - Waters Model Delta 600 semi-preparative system with a Model 600 controller and a Model 2487 dual wavelength absorbance detector; data were acquired using Millenium software; and 2) for radiochemical synthesis - Beckman Gold HPLC system equipped with a Model 126 programmable solvent module, a Model 166 NM variable wavelength detector, a Model 170 radioisotope detector and a Beckman System Gold remote interface module SS420X; data were acquired using the 32 Karat® software. Recently, the gamma detector in this system was replaced with a LabLogic ScanRam—RadioTLC scanner/HPLC detector combination (Brandon, Fl) and later radio HPLC analyses were performed with that detector. Reversed-phase HPLC was performed using a Waters 4.6 × 250-mm XTerra RP18 (5 μm) column and a 19 × 150-mm XTerra RP18 (7 μm) column for analytical and semi-preparative runs, respectively. Proton NMR spectra of samples were obtained on a 400 MHz spectrometer (Varian/Agilent; Inova) and chemical shifts are reported in δ units using the residual solvent peaks as a reference. Mass spectra were recorded using an Agilent LC/MSD Trap for electrospray ionization (ESI) LC/MS or an Agilent LCMS-TOF with DART, a high resolution mass spectrometer used for ESI, DART and LC-MS.
2.2. Tert-butyl (3-(2-iodo-4-methylphenoxy)propoxy)dimethylsilane (2a)
A mixture of 1a (1.40 g; 6 mmol), potassium iodide (0.2 g; 1.20 mmol), anhydrous potassium carbonate (2.49 g; 18 mmol), and (3-bromopropoxy)(tert-butyl)dimethylsilane (2.28 g; 9 mmol) in 30 mL acetonitrile was refluxed for 2–3 h. The reaction mixture was cooled to 20°C and the insoluble components were filtered and washed with acetonitrile. Acetonitrile was evaporated from the filtrate and the crude product was subjected to chromatography with 5% ethyl acetate in hexanes to yield 2.40 g (5.9 mmol; 98.4%) of a colorless oil: 1H-NMR-400 MHz (CDCl3) δ 0.02 (s, 6H), 0.87 (s, 9H), 1.99 (m, 2H), 2.23 (s, 3H), 3.85 (t, 2H), 4.04 (t, 2H), 6.68 (d, 1H), 7.04 (dm, 1H), 7.57 (dm, 1H). LRMS (LCMS-ESI) m/z: 407.1 (M+H)+; HRMS (DART) calcd for C16H28IO2Si (M+H)+ 407.0903, found 407.0902 ± 0.0002 (n = 4).
2.3. Tert-butyl(3-(2-bromo-4-methylphenoxy)propoxy)dimethylsilane (2b)
This reaction was done following a procedure similar to that used for 2a starting with 0.56 g (2.99 mmol) of 2-bromo-4-methyl phenol. Chromatography was performed using a gradient of 0–5% ethyl acetate in hexanes to render 565 mg (1.57 mmol, 53%) of a clear oil: 1H-NMR-400 MHz (CDCl3) δ 0.00 (s, 6H), 0.84 (s, 9H), 1.96 (m, 2H), 2.22 (s, 3H), 3.80 (t, 2H), 4.04 (t, 2H), 6.75 (d, 1H), 6.98 (d, 1H), 7.30 (d, 1H). LRMS (LCMS-ESI and DART) m/z: 359 and 361 (M+H)+, 381 and 383 (M+Na)+, 397 and 399 (M+K)+; HRMS (DART) calcd for C16H2879BrO2Si (M+H)+ 359.1042, found 359.1035 ± 0.0001 (n = 4).
2.4. (3-(4-(Bromomethyl)-2-iodophenoxy)propoxy)(tert-butyl)dimethylsilane (3a)
A mixture of 2a (2.89 g; 7.10 mmol), NBS (1.45 g; 8.17 mmol), benzoyl peroxide (188 mg; 0.78 mmol) in 70 mL of dry dichloromethane was heated at reflux for about 2 h while irradiating with an incandescent lamp. Dichloromethane was evaporated and the resultant brown oil was loaded onto a column of silica gel that was eluted with 5% ethyl acetate in hexanes. Crude 3a (1.40 g), obtained by evaporating dichloromethane from the eluate, was used as such for the next step. 1H-NMR-400 MHz (CDCl3) δ 0.02 (s, 6H), 0.85 (s, 9H), 2.00 (m, 2H), 3.83 (t, 2H), 4.08 (t, 2H), 6.73 (d, 1H), 7.28 (dd, 1H), 7.77 (dd, 1H).
2.5. (3-(4-(Bromomethyl)-2-bromophenoxy)propoxy)(tert-butyl)dimethylsilane (3b)
This compound was prepared in essentially similar manner to 3a starting with 359 mg (1.00 mmol) of 2b. The crude mixture was passed over a bed of silica and eluted with 2–5% ethyl acetate in hexanes to obtain 340 mg (0.77 mmol, 77%). This was used as such for the next step.
2.6. (3-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-iodophenoxy)propoxy)(tert- butyl)dimethylsilane (4a)
A solution of potassium tert-butoxide in THF (4.5 mL, 4.5 mmol) was transferred under argon atmosphere to a dried flask and the THF was evaporated with a gentle stream of argon. BisBoc-guanidine (1.23 g, 4.75 mmol) was added to the above followed by 4 mL of dry DMF. About 5 min later, a solution of crude 3a (see 2.4.) in 6 mL DMF was added to the above solution and the mixture stirred at 20°C under argon for about 90 min. The reaction mixture was partitioned between ethyl acetate and water, and the crude product subjected to chromatography using 5% ethyl acetate in hexanes as the mobile phase to get 460 mg (0.68 mmol; 24%) of a white solid: 1H-NMR-400 MHz (CDCl3) δ0.00 (s, 6H), 0.84 (s, 9H), 1.37 (s, 9H), 1.48 (s, 9H), 1.99 (m, 2H), 3.84 (t, 2H), 4.45 (t, 2H), 5.02 (s, 2H), 6.71 (d, 1H), 7.22 (camouflaged by CHCl3 peak, 1H), 7.72 (d, 1H), 9.30 (br d, 2H). LRMS (LCMS-ESI) m/z: 664.2 (M+H)+, 564.2 (M-Boc)+, 464.2 (M-2Boc)+; HRMS (DART) calcd for C22H39IN3O4Si (M-Boc)+ 564.1755, found 564.1751 ± 0.0002 (n = 4).
2.7. (3-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-bromophenoxy)propoxy)(tert- butyl)dimethylsilane (4b)
This compound was synthesized in similar manner to 4a starting with 340 mg (0.77 mmol) of 3b and chromatographed using 5% ethyl acetate in hexanes to yield 416 mg (0.68 mmol; 77%) of a white solid: 1H-NMR-400 MHz (CDCl3) δ0.00 (s, 6H), 0.84 (s, 9H), 1.37 (s, 9H), 1.47 (s, 9H), 1.99 (m, 2H), 3.83 (t, 2H), 4.07 (t, 2H), 5.05 (s, 2H), 6.80 (d, 1H), 7.18 (m, 1H), 7.48 (d, 1H), 9.30 (br d, 2H). LRMS (LCMS-ESI) m/z: 638.2, 640.2 (M+Na)+, 616.4, 618.4 (M+H)+, 560.2, 562.2 (M-tBu)+; (DART) 616.2 and 618.2 (M+H)+, 560.2 and 562.2. (M-tBu); HRMS (DART) calcd for C27H4779BrN3O6Si (M+H)+ 616.2418, found 616.2421± 0.0003 (n = 4).
2.8. 3-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-iodophenoxy)propanol (5a)
Tetrabutyl ammonium fluoride (1M, 1.51 mL; 1.51 mmol) was added to a solution of 4a (1.00 g; 1.51 mmol) in 50 mL of THF at 0–5°C and the mixture stirred at 20°C for 18 h. The volatiles were evaporated and the crude product was subjected to chromatography using 3:1 hexanes:ethyl acetate as the mobile phase to obtain 0.67g (1.22 mmol; 81%) of an oil: 1H-NMR-400 MHz (CDCl3) δ 1.37 (s, 9H), 1.46 (s, 9H), 2.05 (m, 2H), 2.31 (br s, 1H), 3.88 (t, 2H), 4.11 (t, 2H), 5.00 (s, 2H), 6.71 (d, 1H), 7.22 (overlapped with chloroform peak, 1H), 9.30 (br d, 2H). LRMS (LCMS-ESI) m/z: 550.2 (M+H)+; HRMS (DART) calcd for C21H33IN3O6 (M+H)+ 550.1414, found 550.1426 ± 0.0001 (n = 4).
2.9. 3-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-bromophenoxy)propanol (5b)
Compound 5b was synthesized from 4b (404 mg; 0.66 mmol) following the procedure used for the synthesis of 5a. Chromatography was performed with 30% ethyl acetate in hexanes to obtain 298 mg (0.59 mmol; 90%) of an oil: 1H-NMR-400 MHz (CDCl3) δ 1.39 (s, 9H), 1.48 (s, 9H), 2.08 (m, 2H), 3.90 (br m, 2H), 4.16 (t, 2H), 5.06 (s, 2H), 6.82 (d, 1H), 7.22 (m; overlapped with CHCl3 peak, 1H), 7.51 (s, 1H), 9.35 (br d, 2H). LRMS (LCMS-ESI) m/z: 446, 448 (M-tBu)+, 502.1, 504.1 (M+H)+; (DART) 502.2 and 504.2 (M+H)+, 486.1 and 448.1 (M- tBu)+, 390.0 and 392.0 (M-2 tBu)+, 243 and 245, 185 and 187. (M+H)+ HRMS (DART) calcd for C21H3379BrN3O6 502.1553, found 502.1558 ± 0.0005 (n = 4).
2.10. 3-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-iodophenoxy)propyl 4-methylbenzenesulfonate (6a)
Methyl triflate (23 μL; 0.20 mmol) was added to a solution of tosyl imidazole (49 mg; 0.22 mmol) in 1.6 mL of THF at 0°C under argon atmosphere. Thirty minutes later, a solution of 5a (97.5 mg; 0.18 mmol) and 1-methyl imidazole (14.6 mg; 0.18 mmol) in 1 mL THF was added. The reaction mixture was allowed to gradually warm to 20°C and left stirring overnight. The volatiles were evaporated, and the crude mixture was subjected to preparative TLC using 7:3 hexanes:ethyl acetate as the mobile phase to obtain 106 mg (0.15 mmol; 84%) of an oil: 1H-NMR-400 MHz (CDCl3) δ1.41 (s, 9H), 1.48 (s, 9H), 2.13 (m, 2H), 2.33 ( s, 3H), 3.93 (t, 2H), 4.30 (t, 2H), 5.03 (s, 2H), 6.58 (d, 1H), 7.16 (d, 2H), 7.22 (d; overlapped with CHCl3 peak, 1H), 7.71 (d, 2H), 7.72 (s, 1H), 9.30 (br d, 2H). LRMS (LCMS-ESI) m/z: 704.3 (M+H)+, 726.3 (M+Na)+, 742.3 (M+K)+; HRMS (DART) calcd for C28H39IN3O8S (M+H)+ 704.1503, found 704.1504 ± 0.0004 (n = 8).
2.11. 3-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-bromophenoxy)propyl 4-methylbenzenesulfonate (6b)
This compound was synthesized in similar manner to 6a starting with 262 mg (0.52 mmol) of 5b. Chromatography was done using 7:3 hexanes:ethyl acetate to obtain 320 mg (0.49 mmol; 93%) of a white solid: 1H-NMR-400 MHz (CDCl3) δ 1.40 (s, 9H), 1.48 (s, 9H), 2.16 (m, 2H), 2.35 ( s, 3H), 3.93 (t, 2H), 4.28 (t, 2H), 5.05 (s, 2H), 6.67 (d, 1H), 7.18 (d, 3H), 7.47 (d, 1H), 7.72 (d, 2H), 9.33 (br d, 2H). LRMS (LCMS-ESI) m/z: 600.0 and 602.0 (M-tBu)+, 656.1, 658.1 (M+H)+, 678.1, 680.1 (M+Na)+, 694.0, 696.0 (M+K)+; (DART) 656.2 and 658.2 (M+H)+, 600.1 and 602.1 (M- tBu)+, 544.0 and 546.0 (M-2 tBu)+. HRMS (DART) calcd for C28H3979BrN3O8S (M+H)+ 656.1641, found 656.1633 ± 0.0006 (n = 4).
2.12. 3-(2-Iodo-4-methylphenoxy)propan-1-ol (7)
A mixture of 1a (3.00 g, 12.82 mmol), 3-bromo-propan-1-ol (2.67 g, 19.23 mmol), and potassium carbonate (5.31 g, 38.5 mmol) in acetonitrile (50 mL) was heated at reflux for 4 h. Acetonitrile was evaporated and the residual material was partitioned between water and ethyl acetate. The pooled ethyl acetate extract was dried with MgSO4, the drying agent was filtered off, and ethyl acetate was evaporated from the filtrate. Silica gel chromatography using 9:1 hexanes:ethyl acetate afforded 3.5 g (11.98 mmol, 93%) of a solid: 1H-NMR-400 MHz (CDCl3) δ 2.07 (m, 2H), 2.18 (br s, 1H), 2.23 (s, 3H), 3.90 (br m, 2H), 4.11 (t, 2H), 6.69 (d, 1H), 7/05 (dd. 1H), 7.56 (d, 1H). LRMS (DART) m/z: 275.0 (M-OH)+, 293.0 (M+H)+; HRMS (DART) calcd for C10H14IO2 (M+H)+ 293.0038, found 293.0035± 0.0009 (n = 4).
2.13. 1-(3-Fluoropropoxy)-2-iodo-4-methylbenzene (8)
A mixture of 7 (3.5 g; 11.98 mmol), triethylamine trihydrofluoride (2.90 g; 17.97 mmol), and XtalFluor-E®/(diethylamino)difluorosulfonium tetrafluoroborate [34] (4.12 g, 17.97 mmol) in 50 mL of acetonitrile was stirred under argon at 20°C overnight. Acetonitrile was evaporated, and the residual material was partitioned between ethyl acetate and water. Ethyl acetate was evaporated from the pooled organic layers and the crude mixture subjected to silica gel chromatography to obtain 2.50 g (8.50 nmol, 71%) of a clear oil: 1H NMR-400 MHz (CDCl3) δ 2.23 (s, 3H), 2.11–2.27 (m, 2H), 4.08 ( t, 2H), 4.65 (t, 1H), 4.77 (t, 1H), 6.69 (d, 1H), 7.06 (dd, 1H), 7.57 (d, 1H). LRMS (GCMS) m/z: 107, 234, 294.0 (M+). GC trace indicated this compound to be more than 99% pure.
2.14. 4-(Bromomethyl)-1-(3-fluoropropoxy)-2-iodobenzene (9)
A mixture of 8 (118 mg; 0.40 mmol), NBS (81 mg; 0.45 mmol), benzoyl peroxide (4.86 mg; 0.02 mmol) in 4 mL of dichloromethane was heated at 60°C while irradiating with an incandescent lamp for about 3 h. An additional amount (42 mg; 0.24 mmol) of NBS was added and the heating and irradiation continued for another 75 min. Volatiles were evaporated and the residue taken in ethyl acetate. The ethyl acetate solution was washed with sodium metabisulphite solution, dried and evaporated. Preparative TLC of the crude material using 5% ethyl acetate in hexanes as mobile phase gave 68 mg (0.18 mmol; 45%) of a brown solid: 1H-NMR-400 MHz (CDCl3) δ 2.14–2.26 (m, 2H), 4.11 (t, 2H), 4.40 (s, 2H), 4.65 (t, 1H), 4.77 (t, 2H), 6.74 (d, 1H), 7.31 (dd, 1H), 7.79 (dd, 1H). This material was taken without further purification to the next step.
2.15. 1-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-iodophenoxy)-3-fluoropropane (10)
Potassium tert-butoxide in THF (1M, 275 μL; 0.28 mmol) was added to bis-Boc-guanidine (71 mg; 0.27 mmol). A hazy solution of 9 (68 mg; 0.18 mmol) in 3 mL of dry THF was added to the above solution and the resultant mixture stirred at 20°C under argon for 26 h. THF was evaporated and the resultant material was partitioned between ethyl acetate and water. The crude material thus obtained was taken in chloroform and applied to two preparative TLC plates and the plates were developed with 99.75:0.25 chloroform:methanol to yield 9.8 mg (0.02 mmol, 10%) of a thick oil: 1H-NMR-400 MHz (CDCl3) δ 1.40 (s, 9H), 1.49 (s, 9H), 2.13 (m, 2H), 2.16 (t, 1H), 2.23 (t, 1H), 4.10 (t, 2H), 4.65 (t, 1H), 4.77 (t, 1H), 5.03 (s, 2H), 6.72 (d, 1H), 7.26 (dd, 1H), 7.75 (d,1H), 9.30 (br d, 2H). LRMS (LCMS-ESI) m/z: 552.2 (M+H)+; HRMS (DART) calcd for C21H32FIN3O5 (M+H)+ 552.1371, found 552.1371 ± 0.0002 (n = 4).
2.16. 4-(3-Fluoropropoxy)-3-iodobenzylguanidine (FPOIBG, 11)
A 95:2.5:2.5 (v/v/v) mixture of trifluoroacetic acid:water:tri-isopropylsilane (1 mL) was added to 10 (52.8 mg; 0.09 mmol) and the homogeneous mixture left at 20°C for 1 h. All volatiles were evaporated and the residue subjected to preparative HPLC. For this, the semi-preparative column was eluted at a flow rate of 7 mL/min using a gradient mobile phase consisting of 0.1% TFA in both water (A) and acetonitrile (B); the proportion of B was linearly increased from 5% to 100% in 30 min. The fractions containing the major peak that eluted with a tR = 16 min were isolated, and the solvents from the pooled fractions evaporated to obtain 32.9 mg (0.07 mmol; 74%) of an oil: 1H-NMR-400 MHz (CD3CN) δ 2.10 – 2.24 (m, 2H), 4.15 (t, 2H), 4.24 (d, 2H), 4.64 (t, 1H), 4.76 (t, 1H), 6.94 (d, 1H), 6.94 (br s; overlapped with another set of peaks, 4H), 7.30 (dd, 1H), 7.75 (d,1H), 8.03 (br s, 1H). LRMS (LCMS-ESI) m/z: 352.1 (M+H)+; HRMS (DART) calcd for C11H16FIN3O (M+H)+ 352.0322, found 352.0314 ± 0.0004 (n = 4).
2.17. 1-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-bromophenoxy)-3-fluoropropane (12)
THF from a 1 M solution of TBAF in THF (50.4 μL; 0.05 mmol) was evaporated with an argon stream and the residue dried with a high vacuum pump. Compound 6b (33 mg; 0.05 mmol) was added to the above followed by 0.2 mL of dry acetonitrile. The mixture was stirred at 20°C for 3 h and at 50°C for another 3 h. An additional amount TBAF (0.05 mmol) in acetonitrile was added and heated at 50°C for 1 h. Acetonitrile was evaporated, and the residue partitioned between ethyl acetate and hexanes. The crude material was subjected to preparative TLC using 8:2 hexanes:ethyl acetate as the eluent to obtain 12 mg (0.024 mmol, 48%) of a thick oil: 1H-NMR-400 MHz (CDCl3) δ 1.39 (s, 9H), 1.49 (s, 9H), 2.14–2.29 (m, 2H), 4.12 (t, 2H), 4.63 (t, 1H), 4.75 (t, 1H), 5.04 (s, 2H), 6.80 (d, 1H), 7.21 (dd, 1H), 7.52 (d,1H), 9.35 (br d, 2H). LRMS (LCMS-ESI) m/z: 447.8 and 449.8 (M-tBu)+, 504.0 and 505.0 (M+H)+ 520.0 and 522.0 (M+Na)+; HRMS (DART) calcd for C21H3279BrFN3O5 (M+H)+ 504.1509, found 504.1517 ± 0.0002 (n = 4).
2.18. 4-(3-Fluoropropoxy)-3-bromobenzylguanidine (FPOBBG; 13)
A 95:2.5:2.5 (v/v/v) mixture (100 μL) of trifluoroacetic acid, triisopropylsilane, and water was added to 4.3 mg (8.5 μmol) of 12 and the mixture left at 20°C for 30 min. Volatiles were evaporated and the residue triturated with acetonitrile and after evaporating the acetonitrile the residue was pumped dry to get ~3.5 mg (quantitative) of a thick oil: 1H-NMR-400 MHz (CD3CN) δ 2.10 – 2.30 (m, 2H), 4.17 (t, 2H), 4.26 (d, 2H), 4.62 (t, 1H), 4.73 (t, 1H), 6.87 (br s, 4H), 7.04 (d, 1H), 7.27 (dd, 1H), 7.53 (d,1H), 8.02 (br s, 1H). LRMS (LCMS-ESI) m/z: 244.9 and 246.9, 260.9 and 262.9, 304.0 and 306.0 (M+H)+, 320.0 and 322.0 (M+Na)+; (DART) 304.04 and 306.04 (M+H)+, HRMS (DART) calcd for C11H1679BrFN3O (M+H)+ 304.0461, found 304.0451 ± 0.0003 (n = 4). Synthesis of this compound was repeated starting with 55 mg (0.09 mmol) of 6b. Compound 12 obtained from this batch was deprotected without chromatographic purification. The reaction mixture after deprotection of 12 was subjected to preparative HPLC using conditions used for the purification of 11 to obtain 5.8 mg (0.014 mmol; 17% for two steps) of 13. This was used for the determination of IC50 by competitive inhibition assay.
2.19. 1-(4-(1,2-Bis(tert-butoxycarbonyl)guanidino)methyl-2-(trimethylstannyl)phenoxy)-3-fluoropropane (14)
A mixture of 10 (61 mg; 0.11 mmol), hexamethyl ditin (400 mg; 1.22 mmol) and bis(triphenylphosphine) palladium(II) dichloride (12 mg; 0.02 mmol) in 1 mL dioxane was heated at 90°C for 2 h. The flask was cooled to 20°C and the mixture filtered over a bed of Celite. The Celite bed was washed with ethyl acetate and solvents were evaporated from the combined filtrate. The residual oil was applied to a bed of silica containing a fluorescent indicator that was packed in a quartz frit funnel, and the excess tin reagent and other nonpolar compounds were eluted with hexanes. The desired product and polar byproducts were eluted with ethyl acetate. Ethyl acetate was evaporated and the residual material was taken in 0.5 mL of chloroform and applied to a preparative TLC plate, which was developed with 10% ethyl acetate in hexanes. The product band was isolated and extracted with ethyl acetate to obtain 46.4 mg of an oil. This was further purified by PTLC using 5% ethyl acetate in hexanes as solvent to get 32.4 mg (0.06 mmol, 50%) a thick oil: 1H-NMR-400 MHz (CDCl3) δ 0.26 (s, 9H [119Sn-H d]), 1.42 (s, 9H), 1.50 (s, 9H), 2.14 (dm, 2H), 4.06 (t, 2H), 4.57 (t, 1H), 4.69 (t, 1H), 5.09 (s, 2H), 6.74 (d, 1H), 7.27 (d, 1H), 7.39 (d, 1H), 9.36 (br d, 2H). LRMS (DART) cluster peaks at 534.14 (M-tBu)+, 590.21 (M+H)+. HRMS (DART) calcd for C24H41FN3O5120Sn (M+H)+ 590.2052, found 590.2059 ± 0.0005 (n = 4).
2.20. 4-(3-[18F]Fluoropropoxy)-3-iodobenzylguanidine ([18F]FPOIBG; [18F]11)
A solution of 6a (7 mg) in 0.5 mL dry acetonitrile was added to the dried 18F activity (~1.85 GBq), and the mixture heated at 50°C for 30 min. The mixture was partitioned between ether and water, and the ethereal solution was concentrated, the residual radioactivity re-dissolved in acetonitrile and injected on to the Xterra RP18 5μm 4.6 × 250 mm analytical HPLC column that was eluted at 1 mL/min with a gradient consisting of 0.1% TFA in both water (A) and acetonitrile (B); the proportion of B was linearly increased from 30% to 100% over a period of 25 min. Under these conditions, 10 eluted with a tR of 17–18 min. HPLC fractions containing [18F]10 were pooled, extracted with ether, and the ethereal solution dried by passage through a Na2SO4 cartridge (BondElut, Agilent Technologies). Ether was evaporated, and the residual radioactivity was treated with 0.5 mL TFA at 20°C for 30 min. TFA was evaporated, and the resultant [18F]11 was reconstituted in PBS. For quality control, an aliquot of this was injected onto the HPLC column; compound 11 eluted with a tR of 8–9 min under these conditions.
2.21. 4-(3-[18F]Fluoropropoxy)-3-bromobenzylguanidine ([18F]FPOBBG; [18F]16)
This compound was prepared starting from 6b essentially by the same method described above for [18F]11. Under the HPLC conditions described above (analytical column), [18F]12 eluted with a tR of 17–18 min. Deprotection of [18F]12 to yield [18F]13 and further work up was also performed as above. The retention time of [18F]13 under the same HPLC conditions (analytical column) as used above for [18F]11 was 8–9 min.
2.22. 4-(3-Fluoropropoxy)-3-[125I]iodobenzylguanidine ([125I]FPOIBG; [125I]11)
An aliquot of 14 (50 μg) in chloroform was transferred to a half-dram vial and the chloroform evaporated. A solution of [125I]iodide (1–3 μL; 18.5–55.5 MBq) was added to the above followed by 5 μL of a 1:3 (v/v) mixture of H2O2 (30% w/v):acetic acid. The vial containing the reaction mixture was sonicated for 20 s. A 95:2.5:2.5 (v/v/v) cocktail of TFA:water:tri-isopropylsilane (100 μL) was added and the vial sonicated for 1 min. The vial was left at 20°C for 10 min and sonicated again for 1 min. All the volatiles were evaporated with an argon stream, and the residual activity was taken in acetonitrile (25–50 μL) and injected onto an analytical reversed-phase HPLC column. The column was eluted with a gradient consisting of 0.1% TFA in both water (A) and acetonitrile (B) at a flow rate of 1 ml/min; the proportion of B was linearly increased from 30% to 70% over a period of 25 min. The HPLC fractions containing [131I]11 (tR = 8–9 min; tR of 10 under these conditions was 17–18 min) were collected and purged with argon for 10 min to evaporate most of acetonitrile, diluted with water and passed through a C18 solid-phase cartridge (Waters’ Sep-pak) that was activated with methanol and water. The cartridge was washed with water and then eluted with 0.2 mL portions of methanol. Methanol was evaporated from pooled fractions 3–6 that contained more than 95% of the radioactivity and the residual radioactivity was reconstituted in PBS.
2.23. Paired-label uptake by NET-expressing tumor cells: [18F]FPOIBG/[18F]FPOBBG versus [131I]MIBG and [125I]FPOIBG versus [131I]MIBG
NET-expressing cancer cells were used in these studies. SK-N-SH human neuroblastoma cells were obtained from ATCC (Rockville, MD). SK-N-BE(2c) human neuroblastoma cells and UVW human glioma cells transfected to express NET (UVW-NAT) were a generous gift from Dr. Rob Mairs, University of Glasgow. For SK-N-SH cells, the incubation medium was made by mixing 500 mL of RPMI 1640 (GIBCO), 55 mL of fetal calf serum, and 2.7 mL of penicillin-G/streptomycin (10,000 U of penicillin and 10,000 mg of streptomycin in 1ml of 0.85% saline). For SK-N-BE(2c) cells, the media consisted of DMEM (Cat No. 21969), 15% FCS, 1x L-glutamine, 1x penicillinG/streptomycin, 1x fungizone and 1x non-essential amino acids. For UVW-NAT cells, MEM (Cat No. 32360) containing 10% FCS, 1x L-glutamine, 1x penicillin-G/streptomycin, 1x fungizone and 1 g/L geneticin was used. Cells were plated at a density of 5 × 105 cells per well in 12-well plates and incubated overnight at 37°C in a humidified incubator containing 5% CO2. On the following day, the cell culture supernatants were removed and the cells were washed twice with PBS and supplemented with fresh medium (0.8 mL per well). The cells were incubated with the radiotracer pair at 37°C for 2 h (in some cases an additional time point of 4 h was used). In these experiments, about 3.7 kBq of radioiodinated compounds and 18.5 kBq of 18F-labeled compounds, each in 0.1 ml medium per well, were used. Non-specific uptake was determined by conducting a parallel experiment by co-incubating cells with 15 μL of 3.3 mM desipramine (DMI). In some cases, the effect of MIBG or FPOIBG (50 μM) on tracer uptake also was determined. At the end of the incubation, the cell culture supernatants were removed, and the cells were washed twice with cold PBS. Cells were lysed by incubation with 1.5 mL 1 N NaOH at 37°C for 10 min twice. Cell culture supernatants, lysed cells and input radioactivity standards were counted using a dual label program in an automated gamma counter. Cell-associated radioactivity was expressed as the percentage of input dose.
2.24. Competitive inhibition of uptake of [125I]MIBG by unlabeled MIBG, FPOBBG and FPOIBG in UVW-NAT Cells: Determination of IC50
A day before the experiment, cells were plated in 24-well plates at a density of 8 × 104 cells/well/1 mL of the medium and incubated overnight at 37°C. The medium was removed and cells were washed twice with 0.5 mL of cold PBS. Fresh medium (0.5 mL), [125I]MIBG (3.7 kBq in 50 μL PBS), and 50 μL solutions of increasing concentrations of MIBG, FPOBBG, or FPOIBG were added (10−9 to 10−3 M final concentration), and the cells were incubated for 2 h at 37°C. At the end of the incubation, the cell culture supernatants were removed, and the cells were washed twice with cold PBS. Cells were lysed by incubation with 0.8 mL 1 N NaOH at 37°C for 10 min twice. Cell culture supernatants, lysed cells and input radioactivity standards were counted using a dual label program in an automated gamma counter. IC50 values were derived by the nonlinear regression of uptake versus concentration of the unlabeled compounds plot using GraphPad Prism software.
2.25. Paired-label biodistribution in normal mice
Animal studies were performed under guidelines established by the Duke University Institutional Animal Care and Use Committee. Three paired-label experiments were performed in normal Balb/c mice. Mice were injected via the tail vein with the following tracer pairs in 100 μL PBS: 1) 185 kBq of [125I]MIBG and 296 kBq of [18F]FPOIBG; 2) 185 kBq of [125I]MIBG and 296 kBq of [18F]FPOBBG; and 3) 129.5 kBq each of [131I]MIBG and [125I]FPOIBG. Groups of 5 animals were killed by an overdose of isoflurane and dissected at 1, 2, and 4 h. Organs of interest and blood were collected, blot-dried, weighed, and counted, together with injection standards in an automated dual-channel gamma counter for 125I and 18F radioactivity. Results are expressed as percentage of injected dose per gram of tissue (%ID/g). To determine the specificity of uptake, 5 mice were injected with DMI (10 mg per kg) in 100 μL PBS i.p. 30 min prior to radiotracer administration. One hour later, the mice were killed and tissue distribution was determined as described above. Differences in tissue radioactivity levels between the two co-administered agents was analyzed with a paired 2-tailed Student t-test using the Microsoft Excel program, with a p<0.05 considered to represent a statistically significant difference.
3. Results and Discussion
3.1. Chemistry
The synthesis of [18F]FPOIBG can be accomplished either from a precursor with an already built-in prosthetic moiety or by conjugation of a pre-labeled prosthetic agent such as 3-[18F]fluoropropyl 4-methylbenzenesulfonate with the phenol derivative. In this work, we followed the former approach, and 3-(4-(1,2-bis(tert-butoxycarbonyl)guanidino)methyl-2-iodo-phenoxy)propyl 4-methylbenzenesulfonate (6a) was synthesized as shown in Scheme 1. The cresol derivative 1a, synthesized following a reported procedure [32], was converted to 2a in 98.4% yield by treatment with (3-bromopropoxy)(tert-butyl)dimethylsilane. Benzylic bromination of 2a and subsequent guanidinylation of the resultant benzylic bromide [35] 3a rendered compound 4a in 24% yield for two steps. The TBDMS group in 4a was removed by treatment with TBAF, and the resultant alcohol 5a (81%) was converted to the tosylate derivative 6a using 1-(p-toluenesulfonyl)-3-methylimidazolinium triflate in 84% yield [36]. A standard of FPOIBG was synthesized as shown in Scheme 2. Iodo-cresol 1a was treated with 3-bromo-1-propanol to obtain alcohol 7 in 93% yield, which was subjected to deoxofluorination using an aminodisulfuorosulfinium tetrafluoroborate salt [34] to obtain the fluoro derivative 8 in 71% yield. The sequence of benzylic bromination (45%), guanidinylation (10%), and removal of Boc groups yielded the final product (74%).
Scheme 1.

Synthesis of tosylate precursor of FPOIBG and FPOBBG.
a) (3-Bromopropoxy)-(tert-butyl)-dimethylsilane, Kl, K2CO3, Acetonitrile b) NBS, benzoyl peroxide, Dichloromethane c) (Boc)2guanidine, Pot. tert-butoxide, THF d) TBAF/THF e) 1-Tosyl-3-methyl imidazolinium triflate, 1-methyl imidazole, THF
Scheme 2.

Synthesis of the standard of FPOIBG.
a) (3-Bromo-propanol, Kl, K2CO3, Acetonitrile
b)Triethylamine trihydrofluoride, Xtat-Fluor-E, Acetonitrile, c) NBS, benzoyl peroxide, Dichloromethane d) (Boc)2guanidine, Pot. tert-butoxide, THF e) TFA, triisopropylsilane, water
The results of imaging studies in rats, rabbits and non-human primates suggested that [18F]FPOBBG (LMI1195) was a promising agent for PET imaging of cardiac sympathetic neuronal function [31] and a phase I clinical evaluation of this agent has been reported recently [37]. Although our evaluation [18F]FPOIBG indicated it be not up to the par with MIBG (see below), we wondered whether its biological properties will be comparable to that of [18F]FPOBBG. For this reason, we synthesized the standard and a precursor of FPOBBG. Our methods for these syntheses were similar to those used for the iodo analogue and different from those reported by Purohit et al. [38]. The precursor 6b was synthesized following a path similar to that used for the synthesis of 6a but starting with commercially available bromo-cresol 1b (Scheme 1). The yield for 2b, 3b, 4b, 5b, and 6b was 53%, 77%, 77%, 90%, and 93%, respectively. As shown in Scheme 3, a standard of FPOBBG (13) was synthesized in two steps starting from 6b in 48% overall yield.
Scheme 3.

Synthesis of FPOBBG
a) TBAF/Acetonitrile b) Trifluoroacetic acid, tri-isopropylsilane, water
To derive the radioiodinated isotopologue of FPOIBG, we also synthesized a tin precursor (14) by stannylation of 10 under Stille conditions (Scheme 5). All novel compounds reported herein except 8 were characterized by 1H-NMR, and low and high resolution mass spectrometry. Only 1H-NMR and low resolution mass spectral data were obtained for 8 as it did not yield reasonable data with the DART machine, which we used for all high resolution analysis. This molecule is more suitable for EI mass spectrometry.
Scheme 5.
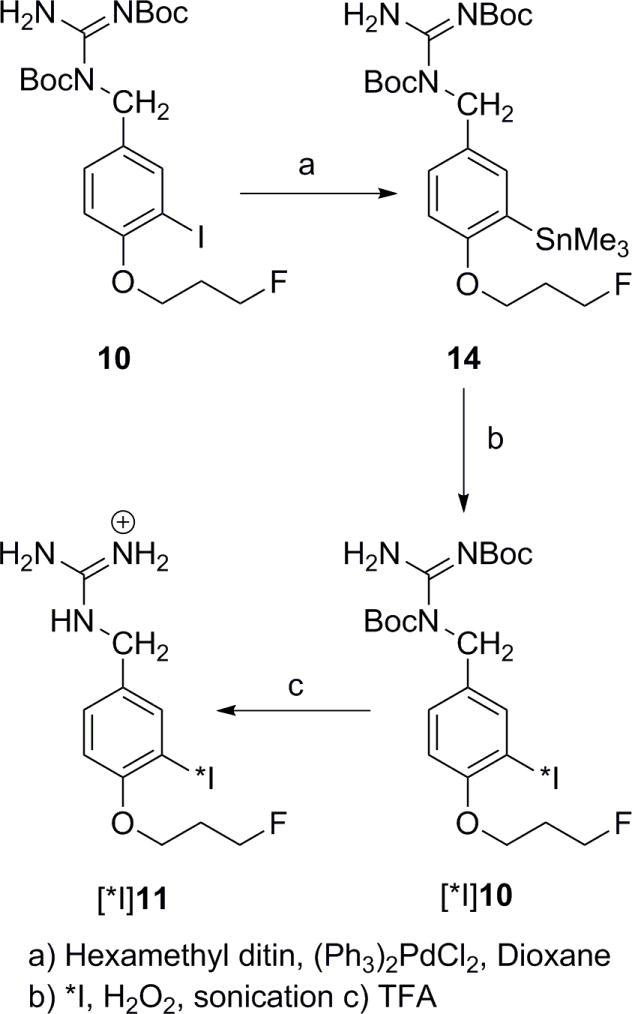
Synthesis of radioiodinated FPOIBG via its tin precursor.
a) Hexamethyl ditin, (Ph3)2PdCl2, Dioxane
b)*l, H2O2, sonication
3.2. Radiochemistry
Based on six runs of [18F]FPOIBG synthesis done under optimized conditions using the precursor 6a (Scheme 4), which involved a total synthesis of about 105 min, the percent of radioactivity injected onto the HPLC that eluted in the peak corresponding to [18F]10 was 39.9 ± 2.7. However, based on initial dried [18F]fluoride activity, the radioactivity present in the HPLC peak corresponded to a radiochemical yield of only 10.4 ± 0.7%. The overall radiochemical yield for the synthesis of [18F]FPOIBG ([18F]11) based on aqueous [18F]fluoride activity was 5.2 ± 0.5%. All of the yields given above are decay corrected to the respective starting 18F activity levels. In the case of [18F]FPOBBG, three runs were done and the total synthesis time was similar. The corresponding radiochemical yields for [18F]FPOBBG ([18F]13; Scheme 4) were 39.9 ± 2.7%, 8.6 ± 1.2%, and 5.1 ± 1.0%, respectively. Radiochemical purity was about 90% and 99% for FPOIBG and FPOBBG, respectively with specific activities estimated to be ≥111 TBq/mmol. Higher radiochemical yields might be obtainable by conducting the labeling reactions at a higher temperature for a shorter time. The formation of a slightly polar byproduct that eluted very close to [18F]10 in HPLC was seen in the [18F]FPOIBG synthesis, which could be reduced by running the reaction at 50°C rather than at higher temperatures. The first step of [18F]FPOBBG synthesis was likewise performed at 50°C. Another reason for the low radiochemical yields with both the iodo and bromo analogues is the formation of another byproduct in significant amounts that has a retention time (~15 min) on HPLC between that of 10 and 11 in the case of FPOIBG and 12 and 13 in the case of FPOBBG. Although not characterized, most likely this may be an intermediate in which one of the two Boc groups has been removed.
Scheme 4.
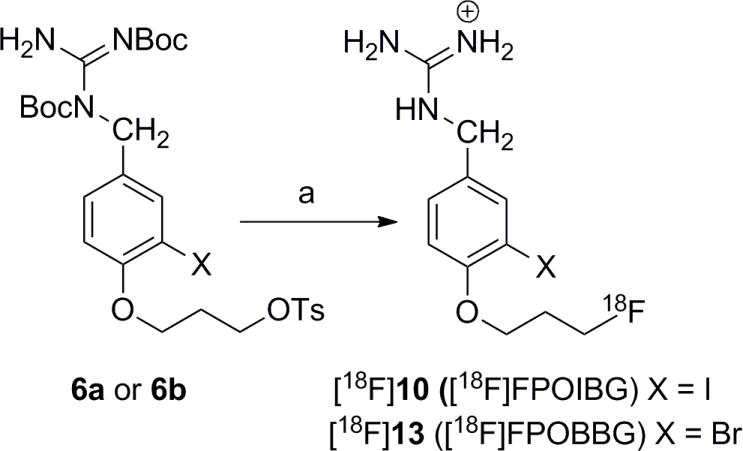
Synthesis of 18F-labeled FPOIBG and FPOBBG from the corresponding tosylate precursor.
a) i. [18F]Fluoride, Kryptofix, Acetonitrile. ii. Trifluoroacetic acid
To determine whether the lackluster behavior of [18F]FPOIBG was due to either a problem related to its synthesis or the nature of its catabolism under biological conditions, a radioiodinated version of FPOIBG was synthesized at a no-carrier-added level from its corresponding tin precursor (Scheme 5). In this case, the intermediate was deprotected in situ. The radiochemical yield for the synthesis of [125I]FPOIBG and [131I]FPOIBG from 14 in two steps was 44.6 ± 11.4% (n = 4) and 42.3 ± 3.0% (n =2), respectively. Contrary to the results obtained with the 18F-labeled compounds, yields for the radioiodinated compounds were compromised by a lack of complete deprotection. It should be noted that the conditions used for the deprotection reaction were not the same. Radiochemical purity of [125I]FPOIBG and [131I]FPOIBG was more than 95% with no detectable peaks present in the UV profile and thus their specific activity should be that of the radioiodide from which they were derived (81 GBq/mmol and 45 GBq/mmol, respectively).
3.3. In vitro uptake by NET-expressing cell lines
The uptake of [18F]FPOIBG was directly compared with that of [125I]MIBG in paired-label format in three NET-expressing cell lines in the absence and presence of the uptake-1 inhibitor desipramine (DMI), unlabeled MIBG, and FPOIBG. In the absence of blocking agents, the uptake of both tracers was considerably higher in UVW-NAT glioma cells (Figure 1) presumably due to higher NET expression in this cell line. Consistent with the results obtained with the current study, both SK-N-SH and SK-N-BE(2c) cell lines express comparable levels of NET and accumulated n.c.a. [131I]MIBG to a similar degree [39], and a two-fold higher uptake of MIBG in UVW-NAT cells compared to that in SK-N-BE(2c) [40]. However, in each cell line, the uptake of [18F]FPOIBG was significantly lower (p<0.05) than that seen for [125I]MIBG. In the absence of inhibitor, the uptake of [18F]FPOIBG was 10.2 ± 1.5%, 39.6 ± 13.4%, and 13.3 ± 2.5%, in SK-N-SH, UVW-NAT, and SK-N-BE(2c), respectively, while corresponding values for [125I]MIBG were 57.3 ± 8.1%, 82.7 ± 8.9%, and 66.3 ± 3.6%. Although [18F]FPOIBG uptake was lower, it was NET transporter-mediated as demonstrated by its considerable inhibition by the uptake-1 blocker DMI. Consistent with the above uptake results, while unlabeled FPOIBG blocked the uptake of [18F]FPOIBG to a same degree as by unlabeled MIBG, FPOIBG was not as effective as MIBG in blocking the uptake of [125I]MIBG.
Figure 1.
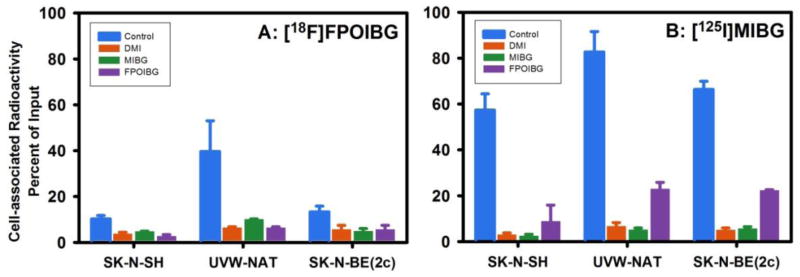
Paired-label uptake of [18F]FPOIBG (A) versus [125I]MIBG (B) in three NET-expressing cell lines in the presence and absence various uptake-1 blocking agents. Cells were incubated with the radiolabeled analogues and with or without the blocking agents at 37 °C for 2 h and cell-associated radioactivity determined as described in the text. Percent of total radioactivity (cell-associated plus supernatant) that was in cells is given on y-axis.
[18F]FPOIBG appeared to perform better with respect to uptake in SK-N-SH cells in vitro than the three fluoroalkyl MIBG analogues without an iodine in their structure; the uptake of best of the three compounds was 5.3% of the input dose [29]. On the other hand, the uptake of [18F]MFBG and [18F]PFBG, in SK-N-SH cells and NET-transduced C6 cells, although considerably lower than that seen for radioiodinated MIBG, was higher compared to that seen for [18F]FPOIBG [16, 17]. [18F]FIBG, the best 18F-labeled analogue of MIBG, demonstrated an uptake in SK-N-SH cells which was about 15% higher than that of the co-incubated n.c.a. [125I]MIBG [20].
As mentioned above, Yu et al. have demonstrated excellent NET-specific myocardial uptake of [18F]FPOBBG (LMI1195) in three different animal models [31]. Although the uptake of [18F]FPOIBG in our in vitro assays was not as high as that of MIBG, it seemed prudent to compare its behavior to that of [18F]FPOBBG, which is very similar in structure. To investigate this, uptake of [18F]FPOBBG and [125I]MIBG was compared using the same in vitro assay conditions utilized to compare [18F]FPOIBG and [125I]MIBG. As shown in Figure 2, the uptake of [18F]FPOBBG was significantly lower (p<0.05) than that of [125I]MIBG in all three cell lines; however, it was considerably higher than that seen in the previous assays with [18F]FPOIBG. To facilitate comparison, the uptake of the 18F-labeled analogue was normalized to the values obtained for [125I]MIBG from each experiment. For [18F]FPOIBG, the specific uptake (total minus that in the presence of DMI) was 12%, 44% and 13% that of [125I]MIBG in SK-N-SH, UVW-NAT and SK-N-BE(2c) cells, respectively, while the corresponding values for [18F]FPOBBG were 41%, 83%, and 47%, clearly demonstrating that iodine-for-bromine substitution had a deleterious effect on NET-mediated uptake in these cell lines.
Figure 2.
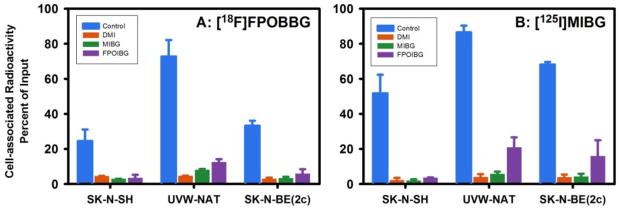
Paired-label uptake of [18F]FPOBBG (A) versus [125I]MIBG (B) in three NET-expressing cell lines in the presence and absence various uptake-1 blocking agents. Cells were incubated with the radiolabeled analogues and with or without the blocking agents at 37 °C for 2 h and cell-associated radioactivity determined as described in the text. Percent of total radioactivity (cell-associated plus supernatant) that was in cells is given on y-axis.
To investigate whether the lower uptake of [18F]FPOIBG in NET-expressing cells in vitro was not due to any potential problem related to its radiosynthesis or catabolism of label, its 125I-labeled isotopologue [125I]FPOIBG was synthesized and its uptake compared to that of [125I]MIBG in the three NET-expressing cell lines. As shown in Figure 3, the uptake of [125I]FPOIBG was 31–53% of [131I]MIBG, consistent with the results obtained with the 18F-labeled isotopologue. These results confirm that the affinity of FPOIBG for NET is inherently lower than that of MIBG.
Figure 3.
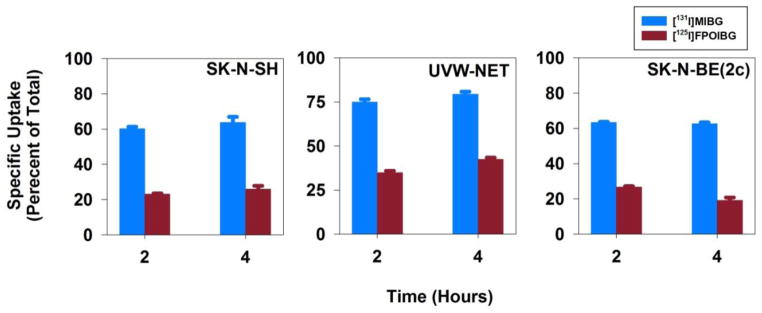
Paired-label uptake of [125F]FPOIBG (A) versus [131I]MIBG (B) in three NET-expressing cell lines. Cells were incubated with the radiolabeled analogues and with or without DMI at 37 °C for 2 and 4 h and cell-associated radioactivity determined as described in the text. Percent of total radioactivity (supernatant plus cell-associated) that was in the cells was determined from assays both in the presence or absence of DMI. Difference of these two (specific uptake) is given on y-axis.
3.4. IC50 determination
IC50 values obtained from competitive inhibition experiments using UVW-NAT cells for MIBG, FPOBBG, and FPOIBG were 1.5 ± 1.1, 11.0 ± 2.9, and 20.0 ± 0.1 μM respectively. These data suggest that while FPOIBG has some affinity for the NET, compared with FPOBBG, it is a weaker analogue of MIBG.
3.5. Biodistribution in vivo
Three paired-label experiments were performed in normal mice in order to compare the tissue distribution of [18F]FPOIBG, [125I]FPOIBG, and [18F]FPOBBG to that of [125/131I]MIBG. Biodistribution data for accumulation in heart, an organ known to exhibit NET-mediated uptake, and selected other tissues are summarized in Figure 4 ([18F]FPOIBG vs. [125I]MIBG), Figure 5 ([18F]FPOBBG vs. [125I]MIBG), and Figure 6 ([125I]FPOIBG vs. [131I]MIBG). As shown in Figure 4, the myocardial uptake of [18F]FPOIBG at the three time points studied was only about 72–74% of that observed for [125I]MIBG, and pretreatment of mice with DMI did not significantly reduce heart uptake (DMI, 13.2 ± 3.3% ID/g; controls, 13.9 ± 1.5% at 1 h, data not shown). In contrast, and in agreement with previous data [41, 42], heart uptake of [125I]MIBG was reduced to 57% of control values when mice were pretreated with DMI. Similarly, the uptake of [125I]FPOIBG was 41–61% of that seen for [131I]MIBG at 1–4 h (Figure 6). As seen with [18F]FPOIBG, the myocardial uptake of [125I]FPOIBG was not reduced by DMI pretreatment (DMI, 17.8 ± 1.8% ID/g; controls, 17.0 ± 3.4% at 1 h) whereas DMI reduced [131I]MIBG uptake in the same mice by 51%. In contrast to the radiolabeled FPOIBG analogues, heart uptake of [18F]FPOBBG (21.1 ± 5.6% ID/g, 20.1 ± 2.4% ID/g, and 16.5 ± 2.3% ID/g at 1 h, 2 h and 4 h, respectively) was slightly higher than that of co-injected [125I]MIBG (18.7 ± 7.7% ID/g, 16.1 ± 1.4% ID/g, and 10.2 ± 0.9% ID/g at 1 h, 2 h and 4 h, respectively) at all three time points (Figure 5). In this experiment, DMI pretreatment reduced heart uptake of [18F]FPOBBG and [125I]MIBG to 73% and 69%, respectively, of the controls. It is not clear why DMI did not block the uptake of [18F]FPOIBG in heart in vivo while it did inhibit its accumulation in NET-expressing tumor cells in vitro. These biodistribution results, like those from the cell uptake studies, confirm that substitution of iodine for bromine in FPOBBG compromises its ability to interact with NET in a manner similar to MIBG. It should be pointed out that the uptake of MIBG and its analogues in neuronal cells is mainly mediated by a sodium- and energy-dependent, specific Uptake-1 mechanism; however, extraneuronal Uptake-2 as well as passive diffusion also contributes [43–45]. Furthermore, the role of the Uptake-1 and Uptake-2 mechanisms is species-dependent. In humans as well as in rabbits and nonhuman primates, cardiac uptake of MIBG is facilitated by Uptake-1, whereas Uptake-1 is absent in the rat heart [43, 44]. Thus, for assessing cardiac sympathetic neuronal function by imaging, an agent that predominantly reflects Uptake-1 is preferred. This is not necessarily the case for imaging and therapy of neuroendocrine tumors [46, 47].
Figure 4.

Paired label uptake of [18F]FPOIBG (green) and [125I]MIBG (maroon) in selected tissues in normal mice. Values are %ID/gram, mean ± SD for 5 replicates each. *Indicates the differences are not statistically significant.
Figure 5.

Paired label uptake of [18F]FPOBBG (green) and [125I]MIBG (maroon) in selected tissues in normal mice. Values are %ID/gram, mean ± SD for 5 replicates each. *Indicates the differences are not statistically significant.
Figure 6.

Paired label uptake of [125I]FPOIBG (green) and [131I]MIBG (maroon) in selected tissues in normal mice. Values are in %ID/gram (for thyroid it is %ID/organ), mean ± SD for 5 replicates each. *Indicates the differences are not statistically significant.
Two other tissues of note are bone and liver. As is frequently observed with 18F-labeled compounds, especially those with the fluorine attached to a sp3 carbon, the bone uptake of both 18F-labeled compounds was high, substantially higher compared to that of [125I]MIBG. The bone uptake of 18F activity from both [18F]FPOIBG and [18F]FPOBBG were similar at 1 h; however, the bone uptake of radioactivity from [18F]FPOBBG decreased with time while that from [18F]FPOIBG increased such that at 4 h, the bone uptake of [18F]FPOIBG was about 4-fold higher than that of [18F]FPOBBG. This suggests that [18F]FPOIBG is considerably less inert to defluorination in vivo than [18F]FPOBBG. On the other hand, thyroid uptake, an indicator of deiodination, for [125I]FPOIBG was relatively low and comparable to that observed with [131I]MIBG. Hepatic uptake of both 18F-labeled compounds was considerably lower than that of co-injected [125I]MIBG. Although the reason for this difference is not known, it does not reflect defluorination of these molecules because lower liver uptake was also observed with [125I]FPOIBG.
MIBG contains an iodine substituent and thus it is enigmatic why, contrary to our expectations, FPOIBG behaved poorly compared with its bromine-substituted analogue, FPOBBG. With regard to bromine-substituted MIBG analogues, [76Br]MBBG has been evaluated in PC-12 rat pheochromocytoma cells in vitro, isolated rat hearts ex vivo, and animal models in vivo [14, 48–50]. The neuronal uptake of MIBG is mediated by two mechanisms—active and passive (see above). The myocardial uptake of [76Br]MBBG was reduced to a significantly higher degree (64 ± 3%) than that of [125I]MIBG (42 ± 4%) by DMI pretreatment, leading the authors to suggest that MBBG may be less dependent on the passive mechanism for its neuronal uptake [48]. In another study, the biodistribution of [76Br]MBBG was compared with that of [123I]MIBG in mice bearing PC-12 rat pheochromocytoma xenografts [50]. Uptake of [76Br]MBBG in both tumor and heart at 8 h was about 4-fold higher than that for [123I]MIBG. However, it should be pointed out that this study was done in single label format, and while [76Br]MBBG was synthesized at a no-carrier-added level, the [125I]MIBG was of low specific activity (~370 GBq/mmol). Furthermore, [76Br]MBBG was administered intravenously while [125I]MIBG was injected intraperitoneally. Finally, in a paired-label study comparing the uptake of [77Br]MBBG and [125I]MIBG in PC-12 cells, [125I]MIBG accumulation was higher at all time points; on the other hand, in vivo uptake in NET-expressing tissues—tumor, heart and adrenals—was slightly higher for the bromo analogue [14]. Taken together, the results from the above studies do not conclusively demonstrate that substitution of bromine for iodine in MIBG results in a substantive advantage with regard to NET-mediated uptake, and thus the inferiority of [18F]FPOIBG compared with [18F]FPOBBG is unexpected. The differential uptake observed for the three tracers both in vitro and in vivo could have been due to the differences in the molar amounts used for the studies and not due to pharmacological differences. It should be pointed out that preparations of high specific activity were used in our studies. Furthermore, IC50 values indicate that the pharmacological property of FPOIBG is indeed inferior to that of FPOBBG and MIBG. The results obtained from our study with the two 18F-labeled analogues suggest that the evaluation of compounds wherein the iodine/bromine in FPOIBG/FPOBBG is replaced with chlorine, fluorine, or even hydrogen, may be warranted.
4. Conclusion
A method was developed for the synthesis of [18F]FPOIBG essentially in a single step from the corresponding tosylate precursor. Contrary to expectations, in vitro studies in NET-expressing cancer cell lines and tissue distribution measurements in mice indicated that this tracer and its radioiodinated isotopologue were sub-optimal MIBG analogues, with properties inferior to that of FPOBBG, a compound recently reported. Given that [18F]FPOBBG possesses more favorable characteristics for NET imaging than [18F]FPOIBG, analogues wherein the iodine in FPOIBG is replaced with chlorine, fluorine or hydrogen might be worth considering.
Acknowledgments
Technical assistance of Ms. Donna Affleck and Dr. Xiao-Guang Zhao is greatly appreciated. The authors thank Drs. Rob Mairs and Mathias Tesson, University of Glasgow (Glasgow, Scotland) for providing the NET-expressing cell lines UVW-NAT and SK-N-BE(2c), and Marek Pruszynski for running the mass spectra of some samples synthesized herein. This work was supported by a seed grant from Duke Cancer Institute and Grant CA42324 from the National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vaidyanathan G. Meta-iodobenzylguanidine and analogues: chemistry and biology. Q J Nucl Med Mol Img. 2008;52:351–68. [PubMed] [Google Scholar]
- 2.Wieland DM, Brown LE, Tobes MC, Rogers WL, Marsh DD, Mangner TJ, et al. Imaging the primate adrenal medulla with [123I] and [131I] meta-iodobenzylguanidine: concise communication. J Nucl Med. 1981;22:358–64. [PubMed] [Google Scholar]
- 3.Teresinska A. Metaiodobenzylguanidine scintigraphy of cardiac sympathetic innervation. Nucl Med Rev Cent East Eur. 2012;15:61–70. doi: 10.5603/nmr-18732. [DOI] [PubMed] [Google Scholar]
- 4.Sisson JC, Yanik GA. Theranostics: evolution of the radiopharmaceutical meta-iodobenzylguanidine in endocrine tumors. Sem Nucl Med. 2012;42:171–84. doi: 10.1053/j.semnuclmed.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Amartey JK, Al-Jammaz I, Lambrecht RM. An efficient batch preparation of high specific activity. Appl Radiat Isot. 2001;54:711–4. doi: 10.1016/s0969-8043(00)00340-7. [DOI] [PubMed] [Google Scholar]
- 6.Vaidyanathan G, Affleck DJ, Alston KL, Zalutsky MR. A tin precursor for the synthesis of no-carrier-added [*I]MIBG and [211At]MABG. J Label Compd Radiopharm. 2007;50:177–82. [Google Scholar]
- 7.Carrasquillo JA, Pandit-Taskar N, O’Donoghue JA, Humm JL, Zanzonico P, Smith-Jones PM, et al. 124I-huA33 antibody PET of colorectal cancer. J Nucl Med. 2011;52:1173–80. doi: 10.2967/jnumed.110.086165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doubrovin MM, Doubrovina ES, Zanzonico P, Sadelain M, Larson SM, O’Reilly RJ. In vivo imaging and quantitation of adoptively transferred human antigen-specific T cells transduced to express a human norepinephrine transporter gene. Cancer Res. 2007;67:11959–69. doi: 10.1158/0008-5472.CAN-07-1250. [DOI] [PubMed] [Google Scholar]
- 9.Seo Y, Gustafson WC, Dannoon SF, Nekritz EA, Lee CL, Murphy ST, et al. Tumor dosimetry using [124I]m-iodobenzylguanidine microPET/CT for [131I]m-iodobenzylguanidine treatment of neuroblastoma in a murine xenograft model. Mol Img Biol. 2012;14:735–42. doi: 10.1007/s11307-012-0552-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brader P, Kelly KJ, Chen N, Yu YA, Zhang Q, Zanzonico P, et al. Imaging a genetically engineered oncolytic vaccinia virus (GLV-1h99) using a human norepinephrine transporter reporter gene. Clin Cancer Res. 2009;15:3791–801. doi: 10.1158/1078-0432.CCR-08-3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fonge H, Leyton JV. Positron emission tomographic imaging of iodine 124 anti-prostate stem cell antigen-engineered antibody fragments in LAPC-9 tumor-bearing severe combined immunodeficiency mice. Mol Img. 2013;12:191–202. [PubMed] [Google Scholar]
- 12.Koehler L, Gagnon K, McQuarrie S, Wuest F. Iodine-124: a promising positron emitter for organic PET chemistry. Molecules. 2010;15:2686–718. doi: 10.3390/molecules15042686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verel I, Visser GW, van Dongen GA. The promise of immuno-PET in radioimmunotherapy. J Nucl Med. 2005;46 (Suppl 1):164S–71S. [PubMed] [Google Scholar]
- 14.Watanabe S, Hanaoka H, Liang JX, Iida Y, Endo K, Ishioka NS. PET imaging of norepinephrine transporter-expressing tumors using 76Br-meta-bromobenzylguanidine. J Nucl Med. 2010;51:1472–9. doi: 10.2967/jnumed.110.075465. [DOI] [PubMed] [Google Scholar]
- 15.Raffel DM, Chen W, Jung YW, Jang KS, Gu G, Cozzi NV. Radiotracers for cardiac sympathetic innervation: transport kinetics and binding affinities for the human norepinephrine transporter. Nucl Med Biol. 2013;40:331–7. doi: 10.1016/j.nucmedbio.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garg PK, Garg S, Zalutsky MR. Synthesis and preliminary evaluation of para- and meta-[18F]fluorobenzylguanidine. Nucl Med Biol. 1994;21:97–103. doi: 10.1016/0969-8051(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Huang R, Pillarsetty N, Thorek DL, Vaidyanathan G, Serganova I, et al. Synthesis and evaluation of 18F-labeled benzylguanidine analogs for targeting the human norepinephrine transporter. Eur J Nucl Med Mol Img. 2014;41:322–32. doi: 10.1007/s00259-013-2558-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Huang R, Cheung NK, Guo H, Zanzonico PB, Thaler HT, et al. Imaging the norepinephrine transporter in neuroblastoma: a comparison of [18F]-MFBG and 123I-MIBG. Clin Cancer Res. 2014;20:2182–91. doi: 10.1158/1078-0432.CCR-13-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaidyanathan G, Affleck DJ, Zalutsky MR. (4-[18F]fluoro-3-iodobenzyl)guanidine, a potential MIBG analogue for positron emission tomography. J Med Chem. 1994;37:3655–62. doi: 10.1021/jm00047a022. [DOI] [PubMed] [Google Scholar]
- 20.Vaidyanathan G, Affleck DJ, Zalutsky MR. Validation of 4-[18F]fluoro-3-iodobenzylguanidine as a positron-emitting analog of MIBG. J Nucl Med. 1995;36:644–50. [PubMed] [Google Scholar]
- 21.Chun JH, Lu S, Lee YS, Pike VW. Fast and high-yield microreactor syntheses of ortho-substituted [18F]fluoroarenes from reactions of [18F]fluoride ion with diaryliodonium salts. J Org Chem. 2010;75:3332–8. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichiishi N, Brooks AF, Topczewski JJ, Rodnick ME, Sanford MS, Scott PJ. Copper-catalyzed [18F]fluorination of (mesityl)(aryl)iodonium salts. Org Lett. 2014;16:3224–7. doi: 10.1021/ol501243g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cardinale J, Ermert J, Humpert S, Coenen HH. Iodonium ylides for one-step, no-carrier-added radiofluorination of electron rich arenes, exemplified with 4-(([F-18] fluorophenoxy)-phenylmethyl) piperidine NET and SERT ligands. Rsc Adv. 2014;4:17293–9. [Google Scholar]
- 24.Kamlet AS, Neumann CN, Lee E, Carlin SM, Moseley CK, Stephenson N, et al. Application of palladium-mediated 18F-fluorination to PET radiotracer development: overcoming hurdles to translation. PloS one. 2013;8:e59187. doi: 10.1371/journal.pone.0059187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee E, Hooker JM, Ritter T. Nickel-mediated oxidative fluorination for PET with aqueous [18F] fluoride. J Am Chem Soc. 2012;134:17456–8. doi: 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, et al. A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science. 2011;334:639–42. doi: 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tredwell M, Preshlock SM, Taylor NJ, Gruber S, Huiban M, Passchier J, et al. A general copper-mediated nucleophilic 18F fluorination of arenes. Angewandte Chemie. 2014;53:7751–5. doi: 10.1002/anie.201404436. [DOI] [PubMed] [Google Scholar]
- 28.Jang KS, Jung YW, Sherman PS, Quesada CA, Gu G, Raffel DM. Synthesis and bioevaluation of [18F]4-fluoro-m-hydroxyphenethylguanidine ([18F]4F-MHPG): a novel radiotracer for quantitative PET studies of cardiac sympathetic innervation. Bioorg Med Chem Lett. 2013;23:1612–6. doi: 10.1016/j.bmcl.2013.01.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee BC, Paik JY, Chi DY, Lee KH, Choe YS. Potential and practical adrenomedullary PET radiopharmaceuticals as an alternative to m-iodobenzylguanidine: m-(omega[18F]fluoroalkyl)benzylguanidines. Bioconjugate Chem. 2004;15:104–11. doi: 10.1021/bc034115e. [DOI] [PubMed] [Google Scholar]
- 30.Lee H, Inbasekaran MN, Wieland DM, Sherman PS, Fisher SJ, Mangner TJ, et al. Development of a kit-form analog of metaiodobenzylguanidine. J Nucl Med. 1986;27:256–67. [PubMed] [Google Scholar]
- 31.Yu M, Bozek J, Lamoy M, Guaraldi M, Silva P, Kagan M, et al. Evaluation of LMI1195, a novel 18F-labeled cardiac neuronal PET imaging agent, in cells and animal models. Circulation Cardiovas Img. 2011;4:435–43. doi: 10.1161/CIRCIMAGING.110.962126. [DOI] [PubMed] [Google Scholar]
- 32.Reddy KR, Venkateshwar M, Maheswari CU, Kumar PS. Mild and efficient oxy-iodination of alkynes and phenols with potassium iodide and tert-butyl hydroperoxide. Tetrahedron Lett. 2010;51:2170–3. [Google Scholar]
- 33.Vaidyanathan G, White BJ, Zalutsky MR. Propargyl 4-[18F]fluorobenzoate: A putatively more stable prosthetic group for the fluorine-18 labeling of biomolecules via click chemistry. Curr Radiopharm. 2009;2:63–74. doi: 10.2174/1874471010902010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beaulieu F, Beauregard LP, Courchesne G, Couturier M, LaFlamme F, L’Heureux A. Aminodifluorosulfinium tetrafluoroborate salts as stable and crystalline deoxofluorinating reagents. Org Lett. 2009;11:5050–3. doi: 10.1021/ol902039q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaidyanathan G, Zalutsky MR. A new route to guanidines from bromoalkanes. J Org Chem. 1997;62:4867–9. [Google Scholar]
- 36.Oconnell JF, Rapoport H. 1-(Benzenesulfonyl)-3-methylimidazolium and 1-(para-toluenesulfonyl)-3-methylimidazolium triflates - efficient reagents for the preparation of arylsulfonamides and arylsulfonates. J Org Chem. 1992;57:4775–7. [Google Scholar]
- 37.Sinusas AJ, Lazewatsky J, Brunetti J, Heller G, Srivastava A, Liu YH, et al. Biodistribution and radiation dosimetry of LMI1195: first-in-human study of a novel 18F-labeled tracer for imaging myocardial innervation. J Nucl Med. 2014;55:1445–51. doi: 10.2967/jnumed.114.140137. [DOI] [PubMed] [Google Scholar]
- 38.Purohit A, Harris T, Radeke HS, Robinson SP, Yu M, Casebier DS. Ligands for imaging cardiac innervation. 2008/083056 A2 World intelectual property organization WO.
- 39.Carlin S, Mairs RJ, McCluskey AG, Tweddle DA, Sprigg A, Estlin C, et al. Development of a real-time polymerase chain reaction assay for prediction of the uptake of meta-[131I]iodobenzylguanidine by neuroblastoma tumors. Clin Cancer Res. 2003;9:3338–44. [PubMed] [Google Scholar]
- 40.McCluskey AG, Boyd M, Ross SC, Cosimo E, Clark AM, Angerson WJ, et al. [131I]meta-iodobenzylguanidine and topotecan combination treatment of tumors expressing the noradrenaline transporter. Clin Cancer Res. 2005;11:7929–37. doi: 10.1158/1078-0432.CCR-05-0982. [DOI] [PubMed] [Google Scholar]
- 41.Vaidyanathan G, Shankar S, Affleck DJ, Welsh PC, Slade SK, Zalutsky MR. Biological evaluation of ring- and side-chain-substituted m-iodobenzylguanidine analogues. Bioconjugate Chem. 2001;12:798–806. doi: 10.1021/bc010032r. [DOI] [PubMed] [Google Scholar]
- 42.Vaidyanathan G, Welsh PC, Vitorello KC, Snyder S, Friedman HS, Zalutsky MR. A 4-methyl-substituted meta-iodobenzylguanidine analogue with prolonged retention in human neuroblastoma cells. Eur J Nucl Med Mol Img. 2004;31:1362–70. doi: 10.1007/s00259-004-1596-8. [DOI] [PubMed] [Google Scholar]
- 43.Higuchi T, Yousefi BH, Kaiser F, Gartner F, Rischpler C, Reder S, et al. Assessment of the 18F-labeled PET tracer LMI1195 for imaging norepinephrine handling in rat hearts. J Nucl Med. 2013;54:1142–6. doi: 10.2967/jnumed.112.104232. [DOI] [PubMed] [Google Scholar]
- 44.Yu M, Bozek J, Kagan M, Guaraldi M, Silva P, Azure M, et al. Cardiac retention of PET neuronal imaging agent LMI1195 in different species: impact of norepinephrine uptake-1 and -2 transporters. Nucl Med Biol. 2013;40:682–8. doi: 10.1016/j.nucmedbio.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Degrado TR, Zalutsky MR, Vaidyanathan G. Uptake mechanisms of meta-[123I]iodobenzylguanidine in isolated rat heart. Nucl Med Biol. 1995;22:1–12. doi: 10.1016/0969-8051(94)00084-w. [DOI] [PubMed] [Google Scholar]
- 46.Streby KA, Shah N, Ranalli MA, Kunkler A, Cripe TP. Nothing but NET: A review of norepinephrine transporter expression and efficacy of 131I-mIBG therapy. Ped Blood Cancer. 2015;62:5–11. doi: 10.1002/pbc.25200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutgers M, Gubbels AA, Hoefnagel CA, Voute PA, Smets LA. A human neuroblastoma xenograft model for [131I]-metaiodobenzylguanidine (MIBG) biodistribution and targeted radiotherapy. Prog Clin Biol Res. 1991;366:471–8. [PubMed] [Google Scholar]
- 48.Valette H, Loc’h C, Mardon K, Bendriem B, Merlet P, Fuseau C, et al. Bromine-76-metabromobenzylguanidine: a PET radiotracer for mapping sympathetic nerves of the heart. J Nucl Med. 1993;34:1739–44. [PubMed] [Google Scholar]
- 49.Raffel D, Loc’h C, Mardon K, Maziere B, Syrota A. Kinetics of the norepinephrine analog [76Br]-meta-bromobenzylguanidine in isolated working rat heart. Nucl Med Biol. 1998;25:1–16. doi: 10.1016/s0969-8051(97)00141-8. [DOI] [PubMed] [Google Scholar]
- 50.Clerc J, Mardon K, Galons H, Loc’h C, Lumbroso J, Merlet P, et al. Assessing intratumor distribution and uptake with MBBG versus MIBG imaging and targeting xenografted PC12-pheochromocytoma cell line. J Nucl Med. 1995;36:859–66. [PubMed] [Google Scholar]