

SUMMARY
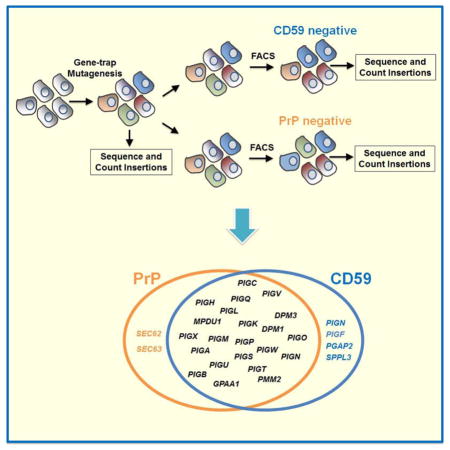
Glycophosphatidylinositol-anchored proteins (GPI-APs) play essential roles in physiology but their biogenesis and trafficking have not been systematically characterized. Here, we took advantage of the recently available haploid genetics approach to dissect GPI-AP pathways in human cells, using prion protein (PrP) and CD59 as model molecules. Our screens recovered a large number of common and, unexpectedly, specialized factors in the GPI-AP pathways. PIGN, PGAP2, and PIGF, which encode GPI anchor-modifying enzymes, were selectively isolated in the CD59 screen, suggesting that GPI anchor composition significantly influences the biogenesis of GPI-APs in a substrate-dependent manner. SEC62 and SEC63, which encode components of the ER targeting machinery, were selectively recovered in the PrP screen, indicating that they do not constitute a universal route for the biogenesis of mammalian GPI-APs. Together, these comparative haploid genetic screens demonstrate that, despite their similarity in overall architecture and subcellular localization, GPI-APs follow markedly distinct biosynthetic and trafficking pathways.
Graphical Abstract
INTRODUCTION
In eukaryotic cells, membrane proteins are usually manufactured in the endoplasmic reticulum (ER), where they fold into native conformations and receive posttranslational modifications (Walter and Ron, 2011). With the exception of ER-localized molecules, these proteins are exported from the ER and transported to their destined organelles (Bonifacino and Glick, 2004). Many mediators of membrane protein biogenesis and trafficking were identified by powerful genetic screens using model organisms, mainly the budding yeast Saccharomyces cerevisiae (Novick et al., 1980, Bankaitis et al., 1986). While these mediators are well conserved, membrane pathways in mammalian cells are significantly more complex, often exhibiting unique features not found in classic model organisms (Bryant et al., 2002).
Despite intensive efforts, genome-wide genetic screens in mammalian cells have been impeded by one critical barrier – the diploidy or aneuploidy of virtually all cultured mammalian cell lines. In these cultured cells, random mutagenesis usually only inactivates one copy of a gene, which seldom leads to obvious phenotypes at the cellular level. RNA interference (RNAi) has been instrumental in unraveling mammalian gene functions but is limited by incomplete gene silencing and substantial off-target effects (Sigoillot et al., 2012). Recently, multiple mammalian haploid cell lines – including tumor cells and pluripotent stem cells – were isolated (Kotecki et al., 1999, Carette et al., 2011b, Yang et al., 2012, Li et al., 2012, Leeb and Wutz, 2011). Since haploid cells contain only one copy of each gene, single mutations can abolish the expression of the gene and result in a null genotype. As a result, genetic screens can be performed in these haploid cells in a similar way as in yeasts.
In this work, we took advantage of the haploid genetics system to dissect membrane protein biogenesis and trafficking in human cells. We focused on a class of membrane-bound molecules – the glycophosphatidylinositol-anchored proteins (GPI-APs). Constituting 10–20% of membrane proteins, GPI-APs play essential roles in a range of biological processes (Nozaki et al., 1999, Orlean and Menon, 2007). Imbalances in their activities are associated with major forms of human disorder such as neurodegeneration and immunodeficiency (Fujita and Kinoshita, 2012, Bonnon et al., 2010, Mayor and Riezman, 2004). After translocation into the ER lumen, the protein moiety of the GPI-AP is covalently conjugated to the GPI anchor, a glycolipid structure spanning the lumenal/exoplasmic leaflet of the membrane bilayer. Subsequently, the mature GPI-AP is targeted to the cell surface where it remains attached to the membrane through its C-terminal GPI anchor (Orlean and Menon, 2007, Paulick and Bertozzi, 2008).
Using haploid genetics, we dissected the biosynthesis and trafficking of two unrelated human GPI-APs – the prion protein PrP and the immune molecule CD59. PrP is well known for its implications in prion diseases (Prusiner et al., 1998), whereas CD59 is a key regulator of complement-mediated cell lysis (Pettigrew et al., 2009, Yamashina et al., 1990). Our screens recovered a large number of factors required for both the PrP and CD59 pathways, most of which are involved in the synthesis of the GPI anchor. Unexpectedly, we isolated several genes that impact only one GPI-AP pathway but not the other. PIGN, PGAP2 and PIGF, which encode GPI anchor-modifying enzymes, were required for the CD59 pathway but not for the PrP pathway. Thus, GPI anchor composition can significantly influence the biogenesis of GPI-APs in a substrate-dependent manner. Sec62p and Sec63p (yeast homologues of SEC62 and SEC63), on the other hand, belong to the SRP-independent ER targeting machinery that is thought to regulate the entire class of GPI-APs in yeasts (Ast et al., 2013). However, we observed that in human cells SEC62 and SEC63 are dispensable for the biogenesis of all GPI-APs we examined except PrP, indicating that the SEC62-SEC63 pathway is not a universal requirement for GPI-APs. We further demonstrated that the signal sequence of PrP determines the engagement in the SEC62-SEC63 pathway. Together, these comparative haploid genetic screens demonstrate that, despite their similarity in overall architecture and subcellular localization, GPI-APs follow markedly distinct biosynthetic and trafficking pathways.
RESULTS
Haploid genetic screen of the PrP pathway
We built a mutant haploid cell library in which the haploid HAP1 cells were randomly mutagenized by gene-trap retroviral insertions (Fig. 1A). Gene-trap insertions create null versions of trapped genes by introducing a splice acceptor site within an intron or by causing frameshift mutations within an exon (Carette et al., 2009). We used fluorescence-activated cell sorting (FACS) to enrich for mutant HAP1 cells that exhibited strongly reduced surface expression of PrP (Fig. 1B). Since cell surface targeting represents the terminal step of the GPI-AP pathway, this quantitative sorting approach is expected to interrogate the entire biosynthetic and trafficking pathway of a GPI-AP. After three rounds of sorting, surface expression of PrP was largely abrogated in the enriched mutant cells (Fig. 1B). We divided the sorted mutant cells into two populations consisting of dim cells or dark cells (Fig. 1B), aiming to prevent downstream sequencing reactions from being overwhelmed by a small number of highly enriched genes.
Figure 1. Haploid genetic screen of the PrP pathway.

(A) Diagram showing the haploid genetic screen of the PrP pathway using the mutant HAP1 cell library. (B) Mutant HAP1 cells were labeled with anti-PrP antibodies and APC-conjugated anti-mouse antibodies. FACS was used to collect cells with the lowest fluorescence signal. After the third round of sorting, cells were divided into two populations – dim and dark – according to their fluorescence intensity. The dark population contained cells with log fluorescence intensity less than 10, while the dim population contained cells with log fluorescence intensity between 10 and 500. Genomic DNA samples from the dark and dim populations were combined in equal amounts and retroviral gene-trap insertions were mapped by deep sequencing.
Gene-trap retroviral insertions in the enriched HAP1 population were mapped by deep sequencing and compared to those in unselected mutagenized HAP1 cells (control population) to identify genes enriched for mutagenic gene-trap insertions. The significance of each gene hit was calculated using the Fisher’s exact test, and ranked according to the P value (Fig. 2 and Table S1). As expected, one of the most statistically significant hits identified in the screen was PRNP, the gene that encodes the PrP protein (Fig. 2). In addition, the screen recovered a large number of other factors including virtually all of the enzymes known to be essential to the synthesis of the GPI anchor (Fig. 2). Localized to the ER or the Golgi, these GPI synthesis enzymes sequentially build the GPI anchor on the membrane and ultimately transfer it to the protein moiety to form the mature GPI-AP (Ferguson et al., 2009, Tokunaga et al., 2014). Identification of PRNP and the known essential GPI synthesis genes indicates that the haploid screen is strikingly exhaustive. Moreover, no new genes encoding potential GPI synthetic or attachment factors were recovered, suggesting that the haploid genetic screen has reached saturation.
Figure 2. Hits from the haploid genetic screen of the PrP pathway.

The Y-axis represents the −Log10 of P values for the gene hits in the selected population as compared to a published unselected control (Jae et al., 2013) using Fisher’s exact test. We set a P value cutoff of 1×10−5 to account for multiple hypothesis testing. In addition, for genes with a P value less significant than 1×10−10, we only considered genes as hits if they also had strong bias for sense-strand intron insertions. The X-axis represents the chromosomal positions of the genes. Circle size is scaled according to the number of unique inactivating gene-trap insertions a gene received. Circles are colored according to functional groups. Dashed line indicates the cutoff of significance.
Haploid genetic dissection of the CD59 pathway and comparison with the PrP screen
To identify common and disparate components of GPI-AP pathways, we next performed a second haploid genetic screen to identify genes involved in the biogenesis of CD59, a GPI-AP unrelated to PrP (Yamashina et al., 1990, Hochsmann et al., 2014). Mutant cells deficient in CD59 surface expression were enriched using the same FACS sorting strategy as in the PrP screen, and their gene-trap insertions were mapped by deep sequencing. As expected, this haploid genetic screen identified the CD59 gene itself, as well as all the genes known to be critical to GPI anchor synthesis (Fig. 3 and Table S1).
Figure 3. Hits from the haploid genetic screen of the CD59 pathway.

The Y-axis represents the −Log10 of P values for the gene hits as described in Fig. 2. The X-axis represents the chromosomal positions of the genes. Circle size is scaled according to the number of unique inactivating gene-trap insertions a gene received. Circles are colored according to functional groups. Dashed line indicates the cutoff of significance.
In order to compare the relative importance of individual hits between the CD59 and PrP screens, unique gene-trap insertions in each dataset were normalized using the quantile method such that the numbers of unique mutagenic insertions could be compared across populations. The normalized datasets were then hierarchically clustered to generate a heatmap (Fig. 4A). As revealed by the heatmap, most of the genes common to both the PrP and CD59 pathways encode the essential components of the GPI synthesis and attachment machinery (Fig. 4A). Interestingly, we discovered that several genes – including PIGN, PGAP2, PIGF, SEC62, SEC63, and SPPL3 were only required for one GPI-AP pathway but not the other (Fig. 4B).
Figure 4. Comparison of the hits from the PrP and CD59 screens.

(A) Heatmap showing the gene hits from the PrP and CD59 screens. Unique gene-trap insertions of gene hits were quantile normalized and clustered using the Euclidean distance metric. Dashed turquoise lines represent the sample mean, and the solid turquoise lines represent each hit’s number of unique insertions relative to the sample mean. (B) Common and disparate genes recovered from the PrP and CD59 screens.
Several genes implicated in GPI anchor modifications – including PIGG, PGAP5, PIGY, DPM2 and PIGZ – were not recovered in our haploid genetic screens. PIGG and PGAP5 are involved in the addition and removal, respectively, of a side chain modification on the second mannose of the GPI anchor (Shishioh et al., 2005, Fujita et al., 2009). PIGY and DPM2, on the other hand, represent protein components of the GPI-N-acetylglucosaminyltransferase (Watanabe et al., 1998). Finally, PIGZ encodes an enzyme that transfers a fourth mannose to the maturing GPI anchor (Orlean and Menon, 2007). The absence of these genes in our screens suggests that they do not constitute central components of the GPI anchor biosynthetic pathway. In line with this notion, the expression of PIGZ appears to be restricted to neuronal and colon tissues, influencing only certain tissue-specific GPI-APs (Taron et al., 2004).
Together, these data demonstrate that GPI-AP pathways involve both common and substrate-specific factors.
GPI anchor modifications have disparate effects on GPI-AP pathways
One of the GPI anchor synthesis genes identified in both the PrP and CD59 screens is PIGP, which encodes a component of the GPI-N-acetylglucosaminyltransferase complex catalyzing the transfer of N-acetylglucosamine (GlcNAc) to phosphatidylinositol (PI) (Fig. S1A) (Orlean and Menon, 2007). We next used CRISPR/Cas9 to delete the PIGP gene in HAP1 cells. We observed that the surface expression of both PrP and CD59 was strongly reduced (Fig. S1B), confirming the results of the haploid genetic screens. These data also indicate that the targeting of mature PrP and CD59 to the cell surface is strictly dependent on the GPI anchor. The C-terminal hydrophobic GPI-conjugation sequence may also associate with membrane bilayers (Ast et al., 2013), but the conjugation sequence itself is clearly inadequate for properly targeting GPI-APs to the cell surface.
Three GPI anchor synthesis genes – PIGN, PGAP2, and PIGF – were only recovered in the CD59 screen. This is an unexpected finding because modifications conferred by these gene products are thought to constitute conserved steps of GPI anchor synthesis (Orlean and Menon, 2007, Fujita and Kinoshita, 2012). Next we used CRISPR/Cas9 to knock out the PIGN gene, which encodes an enzyme that transfers a phosphoethanolamine (EtNP) to the first mannose of the GPI anchor (Fig. S1A) (Hong et al., 1999). We observed that the surface expression of CD59, but not that of PrP, was markedly decreased in the PIGN null HAP1 cells (Fig S1C), confirming the results of the haploid genetic screens. Next we sought to expand this finding by examining CD55 and CD109, another two GPI-APs localized to the cell surface (Burge et al., 1981, Nicholson-Weller et al., 1981, Bizet et al., 2011). We observed that the surface level of CD109 was markedly reduced in PIGN null cells whereas the surface expression of CD55 was not significantly affected (Fig. S2).
We next generated PGAP2 knockout cells using CRISPR/Cas9 and examined how the mutation influences GPI-APs. PGAP2 encodes an enzyme that is required for re-acylation at the sn2 position with a saturated acyl chain (Hong et al., 1999). We observed that the surface level of CD59 was markedly reduced in PGAP2 null cells whereas the surface expression of PrP remained intact (Fig. S2), which is consistent with the results of the haploid genetic screens. Further analysis showed that the surface levels of both CD55 and CD109 were also significantly decreased in PGAP2 null cells (Fig. S2). Interestingly, the GPI-APs that are sensitive to PGAP2 knockout are different from those sensitive to PIGN knockout, suggesting that GPI modifications have markedly distinct effects on individual GPI-AP pathways.
Next we further characterized how GPI anchor modifications affect the surface targeting of GPI-APs. Here we focused on the trafficking of CD59 in PGAP2 null cells. Interestingly, while PGAP2 knockout strongly reduced the surface level of CD59, the total expression level of CD59 remained largely unchanged (Fig. S3A). Immunostaining showed that CD59 was retained in intracellular compartments in PGAP2 null cells (Fig. S3B). By contrast, CD59 was found mainly on the cell surface in WT cells, consistent with the flow cytometry data (Fig. S2). In PGAP2 null cells, substantial amounts of CD59 appeared to localize to a concentrated area characteristic of Golgi localization (Fig. S3B). Indeed, when the cells were treated with Brefeldin A, a Golgi-disrupting chemical, CD59 exhibited a more diffusive localization pattern (Fig. S3B). These data indicate that incomplete GPI anchor modification can result in intracellular retention of certain GPI-APs.
Together, these results demonstrate that, although critical to certain GPI-APs, the PIGN and PGAP2-conferred GPI modifications are not universal requirements for GPI-AP pathways. Our findings also indicate that the composition of the GPI anchor can significantly influence the trafficking of GPI-APs in a substrate-dependent manner.
SEC62 and SEC63 selectively regulate a subset of GPI-APs
Next we focused on the functional roles of SEC62 and SEC63, which were recovered in the PrP screen but not in the CD59 screen. SEC62 and SEC63 are poorly characterized molecules thought to regulate signal recognition particle (SRP)-independent protein translocation into the ER (Lang et al., 2012b, Lakkaraju et al., 2012, Mades et al., 2012). SEC63 has been shown to be required for the biosynthesis of subsets of membrane and secreted proteins, including PrP (Lang et al., 2012a, Lakkaraju et al., 2012). In yeasts, both Sec62p and Sec63p are thought to regulate the entire family of GPI-APs (Ast et al., 2013), which follow a SRP-independent route of ER targeting. However, PrP has been well established as a co-translational substrate in mammalian cells (Kim and Hegde, 2002). It is unclear whether PrP’s dependence on both SEC62 and SEC63 in mammalian cells is because PrP belongs to the class of GPI-APs, or if it is the result of protein-specific features. We used CRISPR/Cas9 to delete the SEC62 or SEC63 gene in HAP1 cells (Fig. S4A). We observed that the surface expression of PrP was strongly reduced in SEC62 or SEC63 null cells whereas the CD59 surface expression remained unchanged (Fig. 5A–B), confirming the findings of the haploid genetic screens.
Figure 5. SEC62 and SEC63 are required for the PrP pathway but are dispensable for the biogenesis of CD59, CD55 and CD109.

(A) SEC62, SEC63 or PRNP was deleted from HAP1 cells by pX330-based CRISPR/Cas9 and individual knockout clones were isolated. The surface levels of PrP in the indicated cells were measured by flow cytometry. (B) Surface levels of the indicated GPI-APs in WT and mutant HAP1 cells were measured by flow cytometry. To rescue gene expression, plasmids encoding SEC62 or SEC63 were transiently transfected into the knockout cells. (C) SEC62 or SEC63 was deleted from the diploid HCT116 cells by pX330-based CRISPR/Cas9. Surface levels of the indicated GPI-APs in the pooled knockout cells were measured by flow cytometry.
Sec62p and Sec63p are thought to regulate the entire family of GPI-APs in yeasts (Ast et al., 2013). However, our genetic screens indicate that this conclusion does not apply to human cells. To further investigate the roles of SEC62 and SEC63 in human cells, we examined the surface expression of CD55 and CD109. We observed that CD55 and CD109 were expressed normally on the cell surface in SEC62 or SEC63 null HAP1 cells, indicating that their biogenesis is also independent of SEC62 and SEC63 (Fig. 5B). We then characterized the functions of SEC62 and SEC63 in HCT116 cells, a diploid human cell line (Mueller et al., 2000). Similar to the results in HAP1 cells, the surface expression of PrP, but not CD59 or CD55, was compromised in SEC62 or SEC63 null HCT116 cells (Fig. 5C), indicating that our conclusions are not limited to haploid cells.
Our data demonstrate that, of all the GPI-APs examined in this study, PrP is the only one dependent on SEC62 and SEC63. Thus, rather than being universal for GPI-AP trafficking, the requirement of the SEC62-SEC63 pathway is restricted to a small subset of GPI-APs in human cells. Thus, mammalian GPI-AP pathways have diverged from their yeast counterparts and exhibit both SEC62-SEC63 dependent and independent mechanisms for ER targeting.
The signal sequence of PrP dictates the choice between ER targeting and cytosolic degradation
Next we sought to further characterize SEC62 and SEC63 in PrP biogenesis. It has been postulated that the requirement for the SEC62-SEC63 pathway is dependent on the intrinsic property of a signal sequence (Ng et al., 1996). If a signal sequence cannot be efficiently recognized by SRP, it is thought to engage the SEC62-SEC63 pathway for ER targeting (Kim and Hegde, 2002, Kim et al., 2002, Levine et al., 2005, Drisaldi et al., 2003, Orsi et al., 2006). Direct evidence for this possibility, however, is still lacking.
To assess the ER targeting and translocation of PrP, we took advantage of post-translational modifications that indicate distinct stages of its maturation (Fig. 6A–B) (Emerman et al., 2010b). In SEC62 or SEC63 null cells, all forms of PrP proteins were diminished (Fig. 6B), indicating that improperly targeted PrP proteins were degraded. To examine the functional role of the signal sequence, we replaced the signal sequence of PrP with that of prolactin (Prl-PrP, Fig. 6A), a secretory protein known to utilize the SRP-dependent ER targeting pathway (Kurzchalia et al., 1986). Strikingly, substitution with the prolactin signal sequence fully rescued the expression of PrP in SEC62 or SEC63 null cells (Fig. 6B). Moreover, Prl-PrP was properly glycosylated in SEC62 or SEC63 null cells (Fig. 6B), suggesting that both translocation and maturation were restored by the signal sequence substitution. PrP contains an internal hydrophobic domain that may also influence ER targeting (Hegde et al., 1998). However, when the hydrophobic domain was deleted, the mutant PrP protein (ΔHD-PrP) behaved similarly to WT PrP (Fig. 6B). These results indicate that the engagement of PrP in the SEC62-SEC63 pathway is solely determined by its signal sequence.
Figure 6. The signal sequence of PrP dictates the choice between ER targeting and cytosolic degradation.

(A) Diagrams showing WT and mutant PrP proteins used in this study with key domains indicated. Red dots indicate the position of an HA tag in an unstructured domain of PrP. SS: signal sequence; HD: hydrophobic domain; GPI: C-terminal GPI-conjugating sequence. (B) Immunoblots showing the expression of WT and mutant PrP proteins. Plasmids encoding the indicated proteins were transiently expressed in WT and mutant HAP1 cells and were detected by immunoblotting using monoclonal anti-HA antibodies. Mutant HAP1 cells deficient in SEC62 or SEC63 were prepared as described in Figure 5. –CHO: the cytosolic/immature form; Mat: the fully glycosylated mature form; Comp: the fully glycosylated complexed form (Kretzschmar et al., 1986, Emerman et al., 2010a). (C) WT PrP was transiently expressed in WT or mutant HAP1 cells. The cells were treated with 100 nM of the proteasome inhibitor PS-341, 10 μM of the ERAD inhibitor Eeyarestatin I, or 1 μM of the ERAD inhibitor kifunensine for the indicated periods of time before analysis by immunoblotting. (D) Model illustrating the dual role of signal sequence in the PrP pathway. In WT cells, the signal sequence is recognized by SEC62-SEC63 and targeted to the ER. In the absence of SEC62-SEC63, the signal sequence directs PrP to the pQC pathway for degradation.
We observed that removal of the signal sequence from PrP (ΔSS-PrP) selectively stabilized an unglycosylated form of PrP (Fig. 6B), indicating that the signal sequence is required for PrP degradation in the SEC62 or SEC63 null cells. Improperly targeted membrane proteins can be degraded via two distinct pathways. First, they can be directly degraded in the cytosol by the proteasome through a pre-emptive quality control (pQC) pathway (Kang et al., 2006, Orsi et al., 2006). Alternatively, a substrate may first enter the ER and then be degraded through ER-associated degradation (ERAD) (Ashok and Hegde, 2008). To distinguish between these possibilities, we treated the cells with the proteasome inhibitor PS-341. We observed that PS-341 treatment strongly stabilized the cytosolic form of PrP in SEC62 or SEC63 null cells. PS-341 also moderately increased the levels of the cytosolic species of PrP in WT cells (Fig. 6C), consistent with the notion that a fraction of PrP is naturally mis-targeted and degraded in the cytosol (Rane et al., 2004). Next we inhibited ERAD using Eeyarestatin I, an inhibitor of the p97 ATPase (Wang et al., 2008), and kifunensine, an inhibitor of alpha-mannosidase (Elbein et al., 1990, Tokunaga et al., 2000). In contrast to PS-341, neither Eeyarestatin I nor kifunensine stabilized any form of PrP (Fig. 6C), indicating that PrP was not targeted to the ER prior to degradation. In agreement with this finding, PS-341 did not stabilize the ER-localized species of PrP (Fig. 6C). Thus, in SEC62 or SEC63 null cells, PrP is degraded mainly by the cytosolic pQC pathway.
In SEC62 or SEC63 null cells, small amounts of PrP proteins remained (Fig. 6B). These remaining proteins appeared to be properly glycosylated (Fig. 6B). Indeed, they were fully resistant to endoglycosidase H (Endo H) treatment (Fig. S4B), indicating that these proteins had acquired mature glycosylation. These data are in agreement with the notion that PrP also engages in the SRP-dependent ER targeting pathway (Rane et al., 2008). The vast majority of PrP molecules, however, are dependent on the SEC62-SEC63 pathway for targeting to the ER membrane.
Together, these results demonstrate that the signal sequence determines the engagement of PrP in the SEC62-SEC63 ER targeting pathway. When the SEC62-SEC63 pathway is blocked, the same signal sequence directs PrP to the cytosolic pQC pathway for degradation.
DISCUSSION
Our comparative haploid genetic screens provide comprehensive views of the biogenesis and trafficking pathways of PrP and CD59, two molecules that have attracted substantial attention due to their disease implications. The PrP and CD59 pathways share many common components, most of which are known to be involved in the synthesis of the GPI anchor. The most interesting insights, however, came from the specialized factors identified only in one GPI-AP pathway but not in the other. The recovery of these specialized factors is unexpected because all GPI-APs are thought to follow similar biosynthetic and trafficking routes (Tokunaga et al., 2014). After all, despite their diverse functions, GPI-APs are similarly tethered to the exoplasmic face of the plasma membrane.
Three of the specialized factors we recovered are GPI-modifying enzymes encoded by PIGN, PGAP2, and PIGF. The PIGN-encoded enzyme catalyzes the addition of EtNP side chain to the first alpha 1,4-linked mannose of the GPI anchor. The enzyme encoded by PGAP2, on the other hand, is required for re-acylation at the sn2 position with a saturated acyl chain, which likely plays a role in stabilizing CD59 at the cell surface (Hong et al., 1999). PIGF is required for the stabilization of another EtNP transferase known as PIGO (Inoue et al., 1993, Shishioh et al., 2005), which adds EtNP to the third mannose of the GPI anchor. The recovery of these three genes in the CD59 screen but not in the PrP screen suggests that GPI-APs exhibit markedly distinct sensitivity to alterations in the GPI anchor. These data are also consistent with the findings of a previous study using a chemical inhibitor of PIGN (Hong et al., 1999). Thus, the GPI anchor does not act as a passive tether for GPI-APs. Instead, its composition can significantly influence the biogenesis and stability of GPI-APs in a substrate-dependent manner.
Another two specialized factors – SEC62 and SEC63 – were recovered only in the PrP screen but not in the CD59 screen. Yeast studies suggested that the SEC62-SEC63 ER targeting pathway is the universal route for GPI-APs (Ast et al., 2013). However, of all the mammalian GPI-APs we examined, only PrP is dependent on the SEC62-SEC63 pathway, suggesting that SEC62 and SEC63 only regulate a small subset of mammalian GPI-APs. Thus, we conclude that the requirement for SEC62-SEC63 is determined on an individual protein basis, instead of including the entire class of GPI-APs.
We discovered that the engagement in the SEC62-SEC63 pathway is solely determined by the N-terminal signal sequence of PrP. In yeasts, GPI-APs appear to possess weak signal sequences that may direct them to the SRP-independent ER targeting pathway (Ast et al., 2013). However, the signal sequences of human PrP, CD59 and CD109 are very similar in both length and hydrophobicity (Fig. S5), suggesting that signal sequence strength alone cannot account for the engagement in the SEC62-SEC63 ER targeting pathway. Instead, the ER targeting mechanism is likely determined by an intrinsic property of each signal sequence, dependent on not only the length and hydrophobicity but also composition and location of the sequence. We further showed that the signal sequence plays dual role in the PrP pathway. In WT cells, the signal sequence directs the PrP protein to the SEC62-SEC63 ER targeting pathway. In the absence of SEC62-SEC63, the signal sequence directs PrP to the cytosolic pQC pathway for degradation, consistent with the notion that the prolonged exposure of any unchaperoned hydrophobic sequence in the cytosol can target mislocalized proteins to the proteasome for degradation (Hessa et al., 2011). Thus, it is the dynamic competition of ER targeting and cytosolic pQC that determines the outcome of PrP biogenesis. We expect that this mechanism represents a common feature for other SEC62-SEC63 dependent substrates.
Finally, our screens identified SPPL3 in the CD59 pathway. Like signal peptide peptidase (SPP), SPPL3 is an aspartyl intramembrane cleaving protease (I-CLiP). SPPL3 is evolutionarily conserved but its physiological importance remains unknown (Voss et al., 2013). The identification of SPPL3 in the CD59 pathway suggests that SPPL3 plays important roles in membrane protein biogenesis, and its function cannot be substituted by other SPP or SPP-like proteases.
Together, these results suggest that GPI-APs can follow divergent biosynthetic and trafficking pathways in spite of their similarity in overall architecture and subcellular localization. In addition to providing insights into GPI-AP pathways, our results also demonstrate that comparative haploid genetics constitutes a powerful platform for systematic dissection of membrane pathways. Haploid genetic screens present two key advantages. First, the expression of a gene is fully eliminated when the gene-trap retroviral insertion occurs in the exons or early introns of the gene. Second, gene-trap insertions can be precisely mapped to the human genome such that haploid genetics does not suffer the off-target effects observed in RNAi or CRISPR/Cas9. Like in other genetic systems, redundant or essential genes are not recovered in haploid genetic screens. For example, no molecular chaperones or vesicle budding/fusion regulators were recovered in our screens although they are known to be essential to membrane trafficking pathways (Bonifacino and Glick, 2004). Nevertheless, our findings pave the way for further interrogating these pathways through complementary approaches such as biochemical reconstitution. Recently, several haploid pluripotent stem cells were isolated (Leeb and Wutz, 2011, Li et al., 2012, Yang et al., 2012, Elling et al., 2011). Since pluripotent stem cells can be programmed into various cell types, haploid genetic screens can be performed to investigate cell type-specific pathways. Altogether, these haploid genetic systems will likely play important roles in uncovering novel features of mammalian membrane biology.
EXPERIMENTAL PROCEDURES
Cell culture and reagents
HAP1 cells were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM, Hyclone, #SH30380) supplemented with 10% fetal bovine serum (FBS, Sigma, #F2442) and penicillin/streptomycin (Sigma, #P4333). The 293T cells (ATCC, #CRL-3216) were grown in Dulbecco’s Modified Eagles Medium (DMEM, Sigma, #D5671) supplemented with 20% FBS. HCT116 cells were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin. We obtained Brefeldin A from Sigma (#B7651), Eeyarestatin I from Tocris Bioscience (#3922), Kifunensine from Enzo Life Sciences (#BML-S114), and PS-341 from LC laboratories (# NC9669075). Endo H (#P0702S) and PNGase F (#P0704S) were both obtained from New England BioLabs.
Generation of the mutant HAP1 cell library
We generated the mutant HAP1 cell library using a previously described procedure with minor modifications (Carette et al., 2009). Briefly, gene-trap retroviruses were produced by transfecting six T175 flasks of 293T cells with a cocktail of plasmids including pGT-GFP0, pGT-GFP1, pGT-GFP2, pAdVAntage (Promega, #E1711), pGAL and pCMV-VSVG using TurboFectin (Origene, #TF81001). The viruses were collected 40 and 50 hours after transfection, and concentrated in a Beckman SW 28 rotor at 25,000 RPM for 1.5 hours. Viral pellets were resuspended in 200 μl PBS overnight at 4 °C. HAP1 cells (1.5×108) were spin-infected in the presence of 8 μg/mL protamine sulfate at two 12-hour intervals in 12-well plates. Cells were plated at 1.5×106 cells per well with 1.5x viral concentrate (by flask surface area) and centrifuged at 900g for 1.5 hours at room temperature in a Thermo Legend RT+ centrifuge. The multiplicity of infection (MOI) was kept at 1.0 or lower based on GFP fluorescence.
Fluorescence-activated cell sorting (FACS)
HAP1 cells were detached from culture flasks using Accutase (Innovative Cell Technologies, #AT 104) to preserve surface antigens and labeled with anti-PrP (eBiosciences, #14-9230) or anti-CD59 (eBiosciences, #17-0596) antibodies followed by incubation with allophycocyanin (APC)-conjugated anti-mouse secondary antibodies (eBiosciences, #17-4015). The labeled cells were sorted using a MoFlo cell sorter (Beckman Coulter) and the data were analyzed using the FlowJo software.
Deep sequencing of retroviral gene-trap insertion
Genomic DNA was extracted from HAP1 cells using a QIAamp DNA Mini Kit (Qiagen, #51304). Linear PCR was performed using 2 μg of genomic DNA as template and the following primer: 5′-Biotin-GGTCTCCAAATCTCGGTGGAAC-3′. After 125 cycles of amplification, the linear PCR products were purified using Dynabeads (M280 Biotin Binder, Life Technologies, #11047). On-bead ligation of a 5′ phosphorylated, 3′ ddC linker was performed using Circligase II (Epicentre, #CL9021K). The product was purified and used as a template for PCR to add Illumina adapter sequences I (5′-AATGATACGGCGACCACCGAGATCTGATGGTTCTCTAGCTTGCC-3′) and II (5′-CAAGCAGAAGACGGCATACGA-3′). PCR products from 6 individual PCR reactions were pooled using a Qiagen Spin column, and sequenced by the Illumina HiSeq 2000 sequencing system using a custom primer recognizing the 5′ end of the LTR (5′-CTAGCTTGCCAAACCTACAGGTGGGGT CTTTCA-3′).
Bioinformatic analysis of retroviral gene-trap insertions
FASTQ files were preprocessed to filter duplicate reads using custom scripts. FASTQ files containing unique sequences were aligned to the human genome (hg19) using Bowtie software v0.12.08 (Langmead et al., 2009). The 50 bp FASTQ sequences were trimmed from their 3′ ends to a length of 35 bp, and were aligned in “--best mode” allowing one mismatch. Reads with more than one genomic alignment were suppressed. Aligned sequences were intersected with gene tables obtained from the UCSC genome browser containing either exons or introns using BEDTools software v2.17.0 (Quinlan and Hall, 2010). Unique insertions per gene were counted for exons and for introns. The total numbers of sense and antisense insertions within introns were counted for each gene. For Fisher’s exact test, we used a published control (Carette et al., 2011a), as well as a homemade control, with no differences observed in results. The heatmap was generated by normalizing unique insertion counts per gene using the quantile method in R. Normalized values were used as input for heatmap.2 (package ‘gplots’) run in R. We developed custom scripts in Python using NumPy, Pandas, SciPy, and matplotlib modules for downstream data analysis, statistics, and visualization.
Genome editing using CRISPR/Cas9
To prepare CRISPR/Cas9 genome-editing constructs, guide sequences were designed according to previously published protocols (Ran et al., 2013). The following guide sequences were used in this study with protospacer adjacent motifs (PAMs) underlined:
-
PIGP:
5′-TACAGTACTTTACCTCGTGTGGG-3′
-
PIGN:
5′-GGTCATGTAGCTCTGATAGCTGG-3′
-
PGAP2:
5′-TGGTGAAGCGGAGCCGTACCAGG-3′
-
SEC62:
5′-CCACCAATATGATGGGTCACCGG-3′
-
SEC63:
5′-TCCATTCTTCTTATAGTCTATGG-3′
Double stranded oligonucleotides containing the guide sequences were subcloned into the pX330 or pLentiCRISPR vectors (Ran et al., 2013). To knock out a gene using the pX330 vector, CRISPR/Cas9 plasmids were transiently transfected into HAP1 or HCT116 cells along with pRetroSuper-Puro (OligoEngine, #VEC-PRT-0002) at 1:1 ratio using Fugene HD (Promega, #E2311). On the following day, the cells were selected with 1 μg/ml puromycin for 2 days before the cells were harvested for downstream analysis. To delete a gene using the pLentiCRISPR vector, we used 293T cells to generate viruses using the same packaging plasmids used to produce gene-trap viruses. The viruses were concentrated as described above, and target cells were spin-infected with 1.5-2X viral titer according to plate surface area. Two days following infection, the cells were treated with 1 μg/ml puromycin for at least one week prior to downstream analysis.
In gene rescue experiments, plasmids encoding the genes were obtained from the Functional Genomics Facility at the University of Colorado Cancer Center and transiently transfected into the cells using Fugene HD. These plasmids were originally generated by the Center for Cancer Systems Biology (Yang et al., 2011).
Immunoblotting
Cells grown in 24-well plates were lysed in SDS sample buffer and the samples were resolved on 8% Bis-Tris SDS-PAGE. Endogenous SEC62 and SEC63 were detected using rabbit polyclonal anti-SEC62 antibodies (Sigma, #SAB1303608) and mouse polyclonal anti-SEC63 antibodies (Sigma, #SAB1407122), respectively. CD59 was detected using rabbit polyclonal anti-CD59 antibodies (Abcam, #ab69084). Plasmids expressing HA-tagged PrP proteins were previously described (Hegde et al., 1998, Kim and Hegde, 2002, Rane et al., 2004, Chakrabarti and Hegde, 2009). Transiently expressed PrP proteins were probed using mouse monoclonal anti-HA antibodies (Covance/BioLegend, #MMS-101P). Monoclonal anti-α-tubulin (#14-4502-82) and anti-vinculin (#14-9777-80) antibodies were both obtained from eBiosciences. Horseradish peroxidase-conjugated secondary antibodies were obtained from Sigma (#A6154 and A6782).
Immunostaining
Cells were fixed by 4% paraformaldehyde (Sigma, #P6148) and permeabilized using PBS containing 0.1% Triton X-100 (Fisher, #BP151). After blocking with 10% BSA (Fisher, #BP1600), CD59 was stained using rabbit polyclonal anti-CD59 antibodies (Abcam, #ab69084) and goat Alexa Fluor 488-conjugated secondary antibodies (Life Technologies, #A-11008). Images were acquired on a Carl Zeiss 3i Marianas spinning disk confocal microscope and processed using ImageJ.
Supplementary Material
Highlights.
Haploid genetic screen probes biogenesis pathways for GPI-anchored proteins.
GPI anchor composition influences trafficking of GPI-anchored proteins.
Mammalian GPI-anchored proteins are targeted to the ER via two distinct routes.
Acknowledgments
We thank Dr. Thijn Brummelkamp for providing HAP1 cells and gene-trap plasmids, Dr. Ramanujan Hegde for providing PrP-related plasmids, Yan Ouyang for technical assistance, Dr. Yuming Han for assistance with flow cytometry, Dr. Jolien Tyler for assistance with confocal imaging, and Dr. Joe Heimiller for discussions on bioinformatics. We are grateful to Drs. Peter Orlean and Erik Snapp for insightful comments on the manuscript. We thank the Functional Genomics Facility at the University of Colorado Cancer Center for providing the expression plasmids of SEC62 and SEC63. This work was supported by the NIH grant GM102217 (JS), a Pew Scholar Award (JS), and a seed grant from the Cancer League of Colorado (AT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ASHOK A, HEGDE RS. Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol Biol Cell. 2008;19:3463–76. doi: 10.1091/mbc.E08-01-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AST T, COHEN G, SCHULDINER M. A network of cytosolic factors targets SRP-independent proteins to the endoplasmic reticulum. Cell. 2013;152:1134–1145. doi: 10.1016/j.cell.2013.02.003. [DOI] [PubMed] [Google Scholar]
- BANKAITIS VA, JOHNSON LM, EMR SD. Isolation of yeast mutants defective in protein targeting to the vacuole. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:9075–9. doi: 10.1073/pnas.83.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIZET AA, LIU K, TRAN-KHANH N, SAKSENA A, VORSTENBOSCH J, FINNSON KW, BUSCHMANN MD, PHILIP A. The TGF-beta co-receptor, CD109, promotes internalization and degradation of TGF-beta receptors. Biochim Biophys Acta. 2011;1813:742–53. doi: 10.1016/j.bbamcr.2011.01.028. [DOI] [PubMed] [Google Scholar]
- BONIFACINO JS, GLICK BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–66. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- BONNON C, WENDELER MW, PACCAUD JP, HAURI HP. Selective export of human GPI-anchored proteins from the endoplasmic reticulum. Journal of cell science. 2010;123:1705–15. doi: 10.1242/jcs.062950. [DOI] [PubMed] [Google Scholar]
- BRYANT NJ, GOVERS R, JAMES DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–77. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- BURGE J, NICHOLSON-WELLER A, AUSTEN KF. Isolation of C4-binding protein from guinea pig plasma and demonstration of its function as a control protein of the classical complement pathway C3 convertase. J Immunol. 1981;126:232–5. [PubMed] [Google Scholar]
- CARETTE JE, GUIMARAES CP, VARADARAJAN M, PARK AS, WUETHRICH I, GODAROVA A, KOTECKI M, COCHRAN BH, SPOONER E, PLOEGH HL, BRUMMELKAMP TR. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- CARETTE JE, GUIMARAES CP, WUETHRICH I, BLOMEN VA, VARADARAJAN M, SUN C, BELL G, YUAN B, MUELLNER MK, NIJMAN SM, PLOEGH HL, BRUMMELKAMP TR. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nature biotechnology. 2011a;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARETTE JE, RAABEN M, WONG AC, HERBERT AS, OBERNOSTERER G, MULHERKAR N, KUEHNE AI, KRANZUSCH PJ, GRIFFIN AM, RUTHEL G, DAL CIN P, DYE JM, WHELAN SP, CHANDRAN K, BRUMMELKAMP TR. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011b;477:340–3. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRABARTI O, HEGDE RS. Functional depletion of mahogunin by cytosolically exposed prion protein contributes to neurodegeneration. Cell. 2009;137:1136–1147. doi: 10.1016/j.cell.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRISALDI B, STEWART RS, ADLES C, STEWART LR, QUAGLIO E, BIASINI E, FIORITI L, CHIESA R, HARRIS DA. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–43. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- ELBEIN AD, TROPEA JE, MITCHELL M, KAUSHAL GP. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem. 1990;265:15599–605. [PubMed] [Google Scholar]
- ELLING U, TAUBENSCHMID J, WIRNSBERGER G, O’MALLEY R, DEMERS SP, VANHAELEN Q, SHUKALYUK AI, SCHMAUSS G, SCHRAMEK D, SCHNUETGEN F, VON MELCHNER H, ECKER JR, STANFORD WL, ZUBER J, STARK A, PENNINGER JM. Forward and reverse genetics through derivation of haploid mouse embryonic stem cells. Cell Stem Cell. 2011;9:563–74. doi: 10.1016/j.stem.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMERMAN AB, ZHANG ZR, CHAKRABARTI O, HEGDE RS. Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Molecular biology of the cell. 2010a;21:4325–4337. doi: 10.1091/mbc.E10-09-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMERMAN AB, ZHANG ZR, CHAKRABARTI O, HEGDE RS. Compartment-restricted biotinylation reveals novel features of prion protein metabolism in vivo. Mol Biol Cell. 2010b;21:4325–37. doi: 10.1091/mbc.E10-09-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FERGUSON MAJ, KINOSHITA T, HART GW. Glycosylphosphatidylinositol Anchors. In: VARKI A, CUMMINGS RD, ESKO JD, FREEZE HH, STANLEY P, BERTOZZI CR, HART GW, ETZLER ME, editors. Essentials of Glycobiology. 2. Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]
- FUJITA M, KINOSHITA T. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochimica et biophysica acta. 2012;1821:1050–1058. doi: 10.1016/j.bbalip.2012.01.004. [DOI] [PubMed] [Google Scholar]
- FUJITA M, MAEDA Y, RA M, YAMAGUCHI Y, TAGUCHI R, KINOSHITA T. GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell. 2009;139:352–65. doi: 10.1016/j.cell.2009.08.040. [DOI] [PubMed] [Google Scholar]
- HEGDE RS, MASTRIANNI JA, SCOTT MR, DEFEA KA, TREMBLAY P, TORCHIA M, DEARMOND SJ, PRUSINER SB, LINGAPPA VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–34. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- HESSA T, SHARMA A, MARIAPPAN M, ESHLEMAN HD, GUTIERREZ E, HEGDE RS. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature. 2011;475:394–7. doi: 10.1038/nature10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOCHSMANN B, DOHNA-SCHWAKE C, KYRIELEIS HA, PANNICKE U, SCHREZENMEIER H. Targeted therapy with eculizumab for inherited CD59 deficiency. The New England journal of medicine. 2014;370:90–2. doi: 10.1056/NEJMc1308104. [DOI] [PubMed] [Google Scholar]
- HONG Y, MAEDA Y, WATANABE R, OHISHI K, MISHKIND M, RIEZMAN H, KINOSHITA T. Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. J Biol Chem. 1999;274:35099–106. doi: 10.1074/jbc.274.49.35099. [DOI] [PubMed] [Google Scholar]
- INOUE N, KINOSHITA T, ORII T, TAKEDA J. Cloning of a human gene, PIG-F, a component of glycosylphosphatidylinositol anchor biosynthesis, by a novel expression cloning strategy. J Biol Chem. 1993;268:6882–5. [PubMed] [Google Scholar]
- JAE LT, RAABEN M, RIEMERSMA M, VAN BEUSEKOM E, BLOMEN VA, VELDS A, KERKHOVEN RM, CARETTE JE, TOPALOGLU H, MEINECKE P, WESSELS MW, LEFEBER DJ, WHELAN SP, VAN BOKHOVEN H, BRUMMELKAMP TR. Deciphering the Glycosylome of Dystroglycanopathies Using Haploid Screens for Lassa Virus Entry. Science. 2013 doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG SW, RANE NS, KIM SJ, GARRISON JL, TAUNTON J, HEGDE RS. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM SJ, HEGDE RS. Cotranslational partitioning of nascent prion protein into multiple populations at the translocation channel. Molecular biology of the cell. 2002;13:3775–3786. doi: 10.1091/mbc.E02-05-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM SJ, MITRA D, SALERNO JR, HEGDE RS. Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev Cell. 2002;2:207–17. doi: 10.1016/s1534-5807(01)00120-4. [DOI] [PubMed] [Google Scholar]
- KOTECKI M, REDDY PS, COCHRAN BH. Isolation and characterization of a near-haploid human cell line. Exp Cell Res. 1999;252:273–80. doi: 10.1006/excr.1999.4656. [DOI] [PubMed] [Google Scholar]
- KRETZSCHMAR HA, STOWRING LE, WESTAWAY D, STUBBLEBINE WH, PRUSINER SB, DEARMOND SJ. Molecular cloning of a human prion protein cDNA. Dna. 1986;5:315–24. doi: 10.1089/dna.1986.5.315. [DOI] [PubMed] [Google Scholar]
- KURZCHALIA TV, WIEDMANN M, GIRSHOVICH AS, BOCHKAREVA ES, BIELKA H, RAPOPORT TA. The signal sequence of nascent preprolactin interacts with the 54K polypeptide of the signal recognition particle. Nature. 1986;320:634–6. doi: 10.1038/320634a0. [DOI] [PubMed] [Google Scholar]
- LAKKARAJU AKK, THANKAPPAN R, MARY C, GARRISON JL, TAUNTON J, STRUB K. Efficient secretion of small proteins in mammalian cells relies on Sec62-dependent posttranslational translocation. Molecular biology of the cell. 2012;23:2712–2722. doi: 10.1091/mbc.E12-03-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANG S, BENEDIX J, FEDELES SV, SCHORR S, SCHIRRA C, SCHAUBLE N, JALAL C, GREINER M, HASSDENTEUFEL S, TATZELT J, KREUTZER B, EDELMANN L, KRAUSE E, RETTIG J, SOMLO S, ZIMMERMANN R, DUDEK J. Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J Cell Sci. 2012a;125:1958–69. doi: 10.1242/jcs.096727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANG S, BENEDIX J, FEDELES SV, SCHORR S, SCHIRRA C, SCHÄUBLE N, JALAL C, GREINER M, HASSDENTEUFEL S, TATZELT J, KREUTZER B, EDELMANN L, KRAUSE E, RETTIG J, SOMLO S, ZIMMERMANN R, DUDEK J. Different effects of Sec61α, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. Journal of cell science. 2012b;125:1958–1969. doi: 10.1242/jcs.096727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B, TRAPNELL C, POP M, SALZBERG SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEEB M, WUTZ A. Derivation of haploid embryonic stem cells from mouse embryos. Nature. 2011;479:131–4. doi: 10.1038/nature10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEVINE CG, MITRA D, SHARMA A, SMITH CL, HEGDE RS. The efficiency of protein compartmentalization into the secretory pathway. Mol Biol Cell. 2005;16:279–91. doi: 10.1091/mbc.E04-06-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI W, SHUAI L, WAN H, DONG M, WANG M, SANG L, FENG C, LUO GZ, LI T, LI X, WANG L, ZHENG QY, SHENG C, WU HJ, LIU Z, LIU L, WANG XJ, ZHAO XY, ZHOU Q. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature. 2012;490:407–11. doi: 10.1038/nature11435. [DOI] [PubMed] [Google Scholar]
- MADES A, GOTTHARDT K, AWE K, STIELER J, DÖRING T, FÜSER S, PRANGE R. Role of human sec63 in modulating the steady-state levels of multi-spanning membrane proteins. PloS one. 2012;7:e49243. doi: 10.1371/journal.pone.0049243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAYOR S, RIEZMAN H. Sorting GPI-anchored proteins. Nature reviews Molecular cell biology. 2004;5:110–20. doi: 10.1038/nrm1309. [DOI] [PubMed] [Google Scholar]
- MUELLER S, CADENAS E, SCHONTHAL AH. p21WAF1 regulates anchorage-independent growth of HCT116 colon carcinoma cells via E-cadherin expression. Cancer research. 2000;60:156–63. [PubMed] [Google Scholar]
- NG DT, BROWN JD, WALTER P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol. 1996;134:269–78. doi: 10.1083/jcb.134.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLSON-WELLER A, BURGE J, AUSTEN KF. Purification from guinea pig erythrocyte stroma of a decay-accelerating factor for the classical c3 convertase, C4b,2a. J Immunol. 1981;127:2035–9. [PubMed] [Google Scholar]
- NOVICK P, FIELD C, SCHEKMAN R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–15. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- NOZAKI M, OHISHI K, YAMADA N, KINOSHITA T, NAGY A, TAKEDA J. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab Invest. 1999;79:293–9. [PubMed] [Google Scholar]
- ORLEAN P, MENON AK. Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J Lipid Res. 2007;48:993–1011. doi: 10.1194/jlr.R700002-JLR200. [DOI] [PubMed] [Google Scholar]
- ORSI A, FIORITI L, CHIESA R, SITIA R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J Biol Chem. 2006;281:30431–8. doi: 10.1074/jbc.M605320200. [DOI] [PubMed] [Google Scholar]
- PAULICK MG, BERTOZZI CR. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry. 2008;47:6991–7000. doi: 10.1021/bi8006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETTIGREW HD, TEUBER SS, GERSHWIN ME. Clinical significance of complement deficiencies. Ann N Y Acad Sci. 2009;1173:108–23. doi: 10.1111/j.1749-6632.2009.04633.x. [DOI] [PubMed] [Google Scholar]
- PRUSINER SB, SCOTT MR, DEARMOND SJ, COHEN FE. Prion protein biology. Cell. 1998;93:337–48. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- QUINLAN AR, HALL IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAN FA, HSU PD, WRIGHT J, AGARWALA V, SCOTT DA, ZHANG F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANE NS, KANG SW, CHAKRABARTI O, FEIGENBAUM L, HEGDE RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Developmental cell. 2008;15:359–370. doi: 10.1016/j.devcel.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANE NS, YONKOVICH JL, HEGDE RS. Protection from cytosolic prion protein toxicity by modulation of protein translocation. Embo j. 2004;23:4550–9. doi: 10.1038/sj.emboj.7600462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHISHIOH N, HONG Y, OHISHI K, ASHIDA H, MAEDA Y, KINOSHITA T. GPI7 is the second partner of PIG-F and involved in modification of glycosylphosphatidylinositol. J Biol Chem. 2005;280:9728–34. doi: 10.1074/jbc.M413755200. [DOI] [PubMed] [Google Scholar]
- SIGOILLOT FD, LYMAN S, HUCKINS JF, ADAMSON B, CHUNG E, QUATTROCHI B, KING RW. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012;9:363–6. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TARON BW, COLUSSI PA, WIEDMAN JM, ORLEAN P, TARON CH. Human Smp3p adds a fourth mannose to yeast and human glycosylphosphatidylinositol precursors in vivo. J Biol Chem. 2004;279:36083–92. doi: 10.1074/jbc.M405081200. [DOI] [PubMed] [Google Scholar]
- TOKUNAGA F, BROSTROM C, KOIDE T, ARVAN P. Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J Biol Chem. 2000;275:40757–64. doi: 10.1074/jbc.M001073200. [DOI] [PubMed] [Google Scholar]
- TOKUNAGA M, KOKUBU C, MAEDA Y, SESE J, HORIE K, SUGIMOTO N, KINOSHITA T, YUSA K, TAKEDA J. Simulation and estimation of gene number in a biological pathway using almost complete saturation mutagenesis screening of haploid mouse cells. BMC Genomics. 2014;15:1016. doi: 10.1186/1471-2164-15-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOSS M, SCHRODER B, FLUHRER R. Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochimica et biophysica acta. 2013;1828:2828–39. doi: 10.1016/j.bbamem.2013.03.033. [DOI] [PubMed] [Google Scholar]
- WALTER P, RON D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- WANG Q, LI L, YE Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem. 2008;283:7445–54. doi: 10.1074/jbc.M708347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATANABE R, INOUE N, WESTFALL B, TARON CH, ORLEAN P, TAKEDA J, KINOSHITA T. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. EMBO J. 1998;17:877–85. doi: 10.1093/emboj/17.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMASHINA M, UEDA E, KINOSHITA T, TAKAMI T, OJIMA A, ONO H, TANAKA H, KONDO N, ORII T, OKADA N, et al. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1990;323:1184–9. doi: 10.1056/NEJM199010253231707. [DOI] [PubMed] [Google Scholar]
- YANG H, SHI L, WANG BA, LIANG D, ZHONG C, LIU W, NIE Y, LIU J, ZHAO J, GAO X, LI D, XU GL, LI J. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell. 2012;149:605–17. doi: 10.1016/j.cell.2012.04.002. [DOI] [PubMed] [Google Scholar]
- YANG X, BOEHM JS, SALEHI-ASHTIANI K, HAO T, SHEN Y, LUBONJA R, THOMAS SR, ALKAN O, BHIMDI T, GREEN TM, JOHANNESSEN CM, SILVER SJ, NGUYEN C, MURRAY RR, HIERONYMUS H, BALCHA D, FAN C, LIN C, GHAMSARI L, VIDAL M, HAHN WC, HILL DE, ROOT DE. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659–61. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.