

Summary
Synapse elimination occurs in development, plasticity and disease conditions. Although the importance of synapse elimination has been documented in many studies, the molecular mechanisms underlying this process remain to be understood. Here, using the development of C. elegans RME neurons as a model, we have uncovered the function of the apoptosis pathway in synapse elimination. We find that the conserved apoptotic cell death (CED) pathway and axonal mitochondria are required for elimination of transiently formed clusters of presynaptic components in RME neurons. The function of the CED pathway needs the activation of the actin filament severing protein, GSNL-1. Furthermore, we show that caspase CED-3 cleaves GSNL-1 at a conserved C-terminal region, and that the cleaved active form of GSNL-1 promotes its actin severing ability. Our data suggest that activation of the cell death pathway contributes to selective elimination of synapses through disassembly of actin filament network.
Keywords: CED-3, caspase, mitochondria, GSNL-1, gelsolin, RME neuron, synapse elimination, C. elegans
Graphical abstract
Introduction
The formation of precise connections between neurons is an essential step of brain development, starting from development of axons and dendrites, and followed by formation and elimination of synapses. In early development stages, excessive number of synapses are formed between neurons, and a portion of them are subsequently eliminated during the maturation of neural circuits to ensure the formation of correct connections (Huttenlocher et al., 1982; Kamiyama et al., 2006; Keller-Peck et al., 2001; Rosenthal and Taraskevich, 1977; Walsh and Lichtman, 2003; Zuo et al., 2005a). Although neuronal activity has been shown to play critical roles in synapse elimination, the molecular mechanisms of synapse elimination are still largely unknown (Flavell and Greenberg, 2008; Kakizawa et al., 2000; Nelson et al., 1993; Rabacchi et al., 1992; Thompson, 1983; Thompson, 1985; Zuo et al., 2005b).
The programmed-apoptosis cell death pathway removes extra cells during development, and eliminates unhealthy cells in diseases (Hyman and Yuan, 2012). More and more studies show that the apoptosis pathway also plays important roles in neural development and function. In Drosophila melanogaster and cultured hippocampal neurons, local activation of caspases promotes dendritic pruning (Erturk et al., 2014; Kuo et al., 2006; Williams et al., 2006). In olfactory sensory neurons and retinal ganglion cells the apoptosis pathway regulates axon guidance through cleavage of membrane-anchored semaphorin and MAP kinases (Campbell and Holt, 2003; Ohsawa et al., 2010). Caspases are also involved in learning and memory in zebra finch and mice (Huesmann and Clayton, 2006; Jiao and Li, 2011; Li et al., 2010). In long-term depression (LTD) local activation of caspase-3 mediates AMPA receptor internalization through cleavage of Akt (Li et al., 2010). Recently, several studies shed light on the function of apoptosis pathway in synapse elimination. Local activation of caspase-3 by mitochondrial dysfunction induces pruning of dendritic spines in cultured hippocampal neurons, and the spine density is increased in caspase-3 knock-out mice (Erturk et al., 2014). At the neuromuscular junction (NMJ), the activation of caspase-3 cleaves Dishevelled to promote the elimination of postsynaptic structures (Wang et al., 2014). However, Dishevelled appears to play moderate roles in other synapses (Luo et al., 2002), suggesting refinement of synapse connections in different types of synapses may involve other caspase targets. The elimination of synapses includes pruning of both presynaptic and postsynaptic structures. While many efforts have led to the dissection of the signaling pathways in regulation of postsynaptic structures, very few studies focus on refinement of presynaptic structures. Since presynaptic boutons can develop without postsynaptic signals (Murthy and De Camilli, 2003), it is reasonable to speculate that the elimination of presynaptic structures is an active process rather than the consequence of elimination of postsynaptic structures. Therefore, it is important to understand the regulatory mechanisms of elimination of presynaptic structures.
The filamentous actin (F-actin) is enriched at growth cones and synaptic regions, and regulation of actin dynamics is important for neural development (Luo, 2002). Polymerization and de-polymerization of actin filaments, upon stimulation by different guidance cues, regulates the formation and retraction of filopodia and lamellipodia as axons grow toward developmental targets (O'Donnell et al., 2009). In cultured hippocampal neurons, de-polymerization of F-actin in young synapses by latrunculin A triggers synapse loss (Zhang and Benson, 2001). F-actin assembly is also important for clustering synaptic vesicles around the active zone (Doussau and Augustine, 2000; Murthy and De Camilli, 2003). In addition, the Rho GTPase family including RhoA, Rac1 and Cdc42 modulate actin dynamics to instruct axonal growth and spine formation, growth, maintenance and retraction (Luo, 2002). In C. elegans, F-actin can bind with the presynaptic active zone proteins SYD-1 and SYD-2 through an adaptor protein NAB-1 (neurabin) to promote presynaptic assembly and axonal branching (Chia et al., 2014; Chia et al., 2012).
The actin-severing gelsolin/villin superfamily is a key regulator of actin filament assembly and disassembly. Most of gelsolin family proteins are cytoplasmic proteins and are regulated by calcium. Gelsolin proteins bind to the barbed ends of actin filaments to prevent monomer exchange and sever existing filaments (Silacci et al., 2004). In C. elegans there are three gelsolin related proteins: gsnl-1, viln-1 and fli-1, among which gsnl-1 is the most characterized. Unlike the conventional gelsolin proteins that have either three or six gelsolin-like domains, GSNL-1 has four gelsolin-like domains (Klaavuniemi et al., 2008). In vitro studies show that GSNL-1 can sever actin filaments and caps the barbed end in a calcium-dependent manner similar to that of conventional gelsolin proteins (Klaavuniemi et al., 2008). However the function of C. elegans gelsolin proteins in neural development remains unclear.
Here we show how the apoptosis pathway regulates activation of the gelsolin-like protein, GSNL-1, to instruct actin de-polymerization and to control the elimination of transient clusters of presynaptic components. We used a pair of C. elegans head motor neurons, RME dorsal (RMED) and ventral (RMEV) neurons, as our model. In an unbiased genetic screen, we uncovered a loss-of-function allele of ced-3 with strong defects in the localization of presynaptic components. CED-3 is the major apoptotic caspase in C. elegans and functional homolog of mammalian caspase 3 (Hyman and Yuan, 2012; Yuan et al., 1993). We also found that four core components of the C. elegans apoptosis pathway are all required for elimination of transient presynaptic components, and that axonal mitochondria are important for activating the CED pathway in this process. In the same genetic screen we identified a loss-of-function allele of gsnl-1 with similar phenotypes as ced-3(lf). Further studies showed that CED-3 cleaves GSNL-1 at a C-terminal conserved site to promote its F-actin severing ability and to promote disassembly of presynaptic components at improper locations. Therefore, our study has revealed the function of axonal mitochondria and the apoptosis pathway in elimination of presynaptic components in vivo, and identified a gelsolin protein as a downstream target of the caspase in elimination of presynaptic components. The conservation of the CED-3 cleavage site in GSNL-1 indicates this regulation may be a conserved mechanism for synapse elimination in other organisms.
Results
RME neurons transiently accumulate presynaptic components in dendritic-neurites
C. elegans head movement is controlled by four GABAergic RME neurons, RME dorsal (D), RME left (L), RME right (R) and RME ventral (V) (White et al., 1986). The somas of RME neurons are located close to the nerve ring, and their axons grow as a ring shaped bundle surrounding the pharyngeal region where they form en passant synapses with a subset of head muscles. RMED and RMEV extend two posterior dendritic neurites along the dorsal and ventral cords (Figure 1 A and B). Four RME neurons migrate to their final position during embryo development, but the development of those dorsal and ventral (D/V) neurites occurs after hatching, and their function remains unknown (Figure 1 B). Using synaptobrevin-GFP (SNB-1-GFP) to label presynaptic vesicles in RME neurons, we observed that at early larval stage (L1), in addition to their normal accumulation at the nerve ring, clusters of SNB-1-GFP also mis-accumulated at the end of D/V neurites and continued to add until late L1 stage (Figure 1 C and D). By L2 stage the mis-accumulated SNB-1-GFP puncta in D/V neurites started pruning, and were below detection in L3 or later stage animals (Figure 1 C). To verify that these fluorescent puncta might contain other presynaptic components, we further examined the localization of presynaptic active zone protein SYD-2/Liprin-α in different developmental stages (Yeh et al., 2005). As shown in Figure 1 E and F, SYD-2/Liprin-α formed puncta in D/V neurites at L1 stage, and those puncta were completely eliminated before L3 stage similar to those of the SNB-1-GFP maker. Using another marker for synaptic vesicles, RAB-3-mCherry, we also observed the presence of fluorescent punctate signals in L1 D/V neurites, and RAB-3-mCherry signal was completely co-localized with presynaptic active zone marker SYD-2 (Figure S1 A and B). To further verify our observation we tested whether the formation of SNB-1-GFP puncta in RMED/V neurites required syd-2 and unc-104, two molecules necessary for the formation of presynaptic vesicle clusters(Hall and Hedgecock, 1991; Zhen and Jin, 1999). As shown in Figure 1G, SNB-1-GFP puncta in D/V neurites were undetectable in L1 animals of loss-of-function mutants for syd-2 and unc-104. These analyses indicate that RMED/V neurons may transiently accumulate presynaptic components in their D/V neurites during development.
Figure 1. RMED and RMEV neurons form transient clusters of presynaptic components at dendritic D/V neurites during larval development.
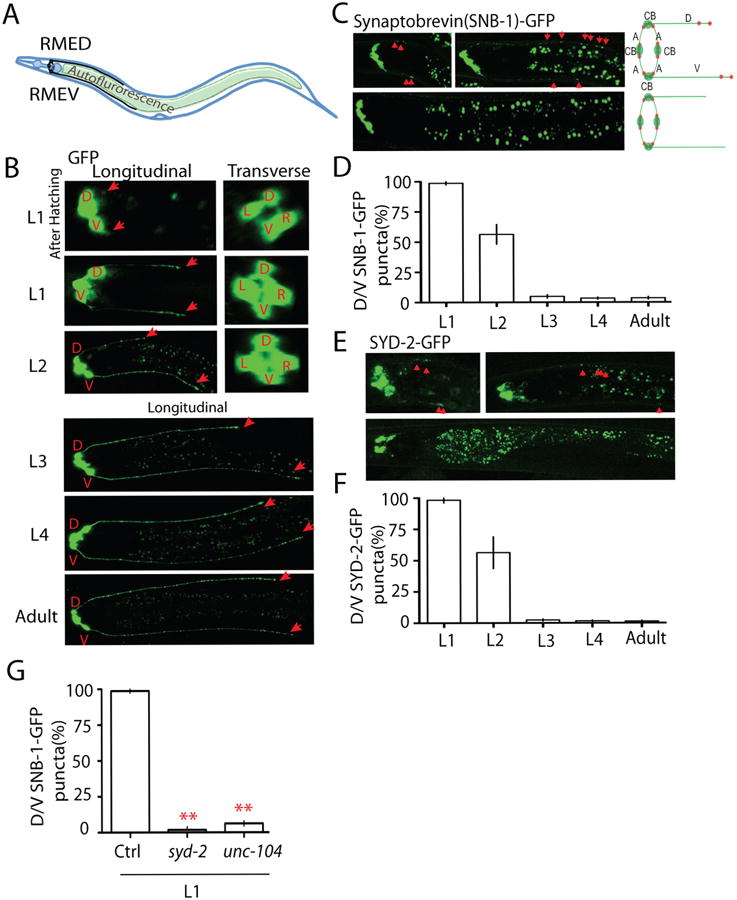
(A) Illustration of the morphology of RME neurons. RME neurons (black color) locate in the head region of C. elegans, and RME axons form a ring-shape bundle at the nerve ring region. RMED and RMEV grow two posterior neurites along the dorsal/ventral cords. When imaging RME neurons, the gut autofluorescence is often seen between the dorsal and ventral cords (B, C and E.). (B) The development of RME D/V neurites visualized by a marker expressing GFP in RME neurons (Punc-25∷GFP). The four RME neurons migrated to their final position before hatching, but the development of D/V neurites occurred after hatching. Red arrows label the end of D/V neurites. D: RMED, V: RMEV, L: RMEL, R: RMER. (C-F) RME D/V neurons form transient synapses at early developmental stages. At the first larval (L1) stage, synaptic vesicles and synaptic active zones are clustered at dorsal/ventral neurites. No synaptic vesicle and synaptic active zone puncta were observed in L3 worms. Representative images and schematics show the localization of synaptic vesicle puncta (Synaptobrevin (SNB-1)-GFP, C) and synaptic active zone puncta (SYD-2-GFP, E) in RME neurons. CB, cell bodies; A, axons; D/V, dorsal/ventral neurites; red dot, synaptic puncta. Red arrowheads highlight the transient clusters of presynaptic components. (D and F) Quantification of the percentage of animals with D/V puncta. (G) Quantification data show that loss-of-function in syd-2 and unc-104 causes absence of SNB-1-GFP puncta in D/V neurites of L1 animals. Experiments were performed at least 3 times, with N ≥80 animals each time. Data is shown as mean ± SD. Student test, ** P < .01. Scale bar, 10 μm.
To assess if some of the transient presynaptic clusters formed morphological synapses, we reconstructed the D/V neurite of RMED and RMEV in a wild type L1 animal (14 hours post-hatching) by serial section transmission electron microscopy (TEM). RMED and RMEV were identified based on their cell body locations and neurite trajectories. We traced the entire dorsal (D) and ventral (V) neurites from the cell bodies of each neuron. They both showed periodic swellings (Figure S2 A and B). Multiple cellular organelles, including mitochondria, microtubules, and vesicular structures were present in these swellings (Figure S2 A). Figure S2 B showed a complete series of EM images for one such swelling. However we did not detect mature presynaptic structures in the swelling regions (Figure S2 A and B). As a control, we observed the classic mature chemical synaptic structures with synaptic vesicle clusters and active zones in the neighboring DA and DB motor neuron axons. These results suggest that while both RME D/V branches contain swelling structures enriched with vesicular components at the L1 stage, and the fluorescent puncta from our reporters likely represent mis-localized or improperly formed clusters of presynaptic components during development. Since the formation and elimination of the fluorescent puncta happened at the same developmental stage in all animals, the development of RME D/V neurites is an appropriate system to study molecular mechanisms of elimination of synaptic components.
The cell death (CED) pathway regulates the elimination of presynaptic components
To understand what controls elimination of presynaptic components in RME neurons, we conducted a genetic screen and isolated mutants that retained SNB-1-GFP puncta in D/V neurites at the adult stage. One mutant uncovered in this screen, ju1056, showed synaptic vesicle cluster elimination defects in over 90% animals (Figure 2 A). ju1056 was mapped to chromosome IV, tightly linked to unc-30. After testing several candidate genes, we found that ju1056 failed to complement the loss-of-function (lf) allele of the C. elegans apoptotic gene ced-3(n717). Sequencing results showed that ju1056 causes a Glycine to Arginine point mutation in the conserved caspase domain of CED-3 (Figure S1 C and Table S1). Besides SNB-1-GFP phenotypes, we also noticed that about 30-40% animals have additional neurons in the head region (data no shown). To confirm the SNB-1-GFP elimination phenotype was not a secondary effect due to change of neuron numbers, we expressed ced-3 cDNA at low concentration only in RME neurons and quantified its rescue ability. As shown in Figure 2 B, expression of ced-3 in RME neurons fully rescued SNB-1-GFP elimination defects in ced-3(lf), suggesting ced-3 promotes elimination of presynaptic components through regulation of signals in RME neurons rather than through affecting cells surrounding RME neurons.
Figure 2. The apoptotic CED pathway is required for elimination of presynaptic components.
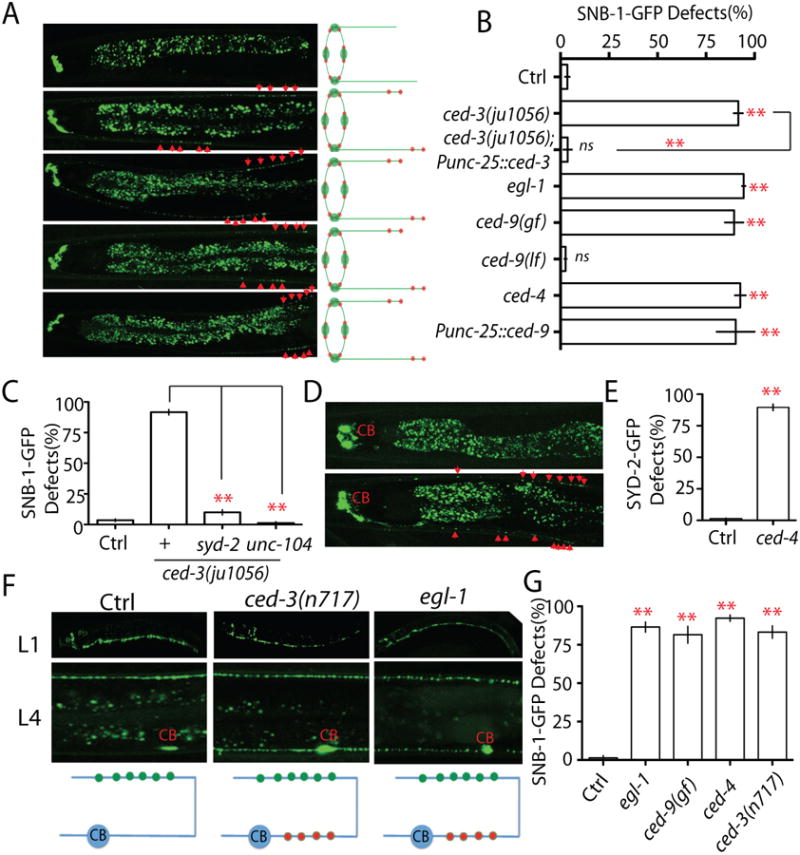
(A-B) Loss-of-function in the CED pathway prevents elimination of presynaptic components. (A) Images and schematics of the localization of synaptic vesicle puncta in wild type and mutant animals. (B) Quantification of SNB-1-GFP elimination defects (% animals) in different genotypes. Punc-25 promoter was used to express genes in RME neurons; unc-30(ju32) animals were used as background to eliminate unc-25 expression in the D neurons. The strain information is listed in Table S1 and Table S2. (C) Quantification data show that loss-of-function in syd-2 and unc-104 suppress SNB-1-GFP elimination defects in ced-3(lf) animals. (D-E) loss-of-function in ced-4 suppressed elimination of presynaptic active zones. (D) Images and schematics of the localization of active zone puncta (Punc-25∷SYD-2-GFP). (E) Quantification of active zone elimination defects. (F-G) The CED Pathway regulates synapse elimination in DD motor neutrons. (F) Images and schematics of the localization of synaptic puncta (Pflp-13-SNB-1∷GFP/juIs137) in wild type, ced-3(lf) and egl-1(lf) animals at L1 and L4 stages. CB, cell bodies; green dot, new synapses at the dorsal cord; red dot, synapses at the ventral cord. (G) Graph shows the quantification data of DD motor neuron synapse elimination defects in L4 animals. In Figure B, E and G, experiments were performed at least 3 times, with N ≥80 animals each time. For transgenic animals the results shown here are generated from at least three independent lines. Data is shown as mean ± SD. Student test, ** P < .01, ns: no significant difference. Scale bar, 10 μm. See also Supplemental Figure 1.
CED-3 is one of the four core components of C. elegans apoptotic pathway that also includes the BH3 domain protein EGL-1, the anti-apoptotic Bcl-2 homolog CED-9, and the Apaf-1 like protein CED-4 (Hyman and Yuan, 2012). To test whether other members of this pathway are involved in elimination of presynaptic components, we quantified SNB-1-GFP defects in mutants altering functions of these proteins. We found that loss-of-function in pro-apoptotic genes, egl-1 and ced-4, and gain-of-function in the anti-apoptotic gene ced-9 induced elimination defects to a similar degree as ced-3(lf) mutants, while loss-of-function in ced-9 did not show any obvious defects (Figure 2 A and B). Further evidences showed that the formation of the SNB-1-GFP puncta in D/V neurites depended on key regulators of synapse formation: syd-2 and unc-104 (Figure 2 C). To evaluate whether the sizes of these SNB-1-GFP puncta were similar as those seen in normal synapses, we compared SNB-1-GFP puncta in RMED neurites with nearby synapses from DD motor neurons. We found that in all CED and other mutants described in this study, at the end of RMED neurites the SNB-1-GFP puncta were as similar size and fluorescence intensity as those in DD synapses (Figure S1 D). Quantification data also showed none of these mutants altered the overall expression level of SNB-1-GFP in RME neurons (Figure S1 E). Blockage of the CED pathway by loss-of-function in ced-4 also prevented the elimination of the presynaptic active zone marker SYD-2-GFP (Figure 2 D and E). These results demonstrate that the activation of the cell death pathway is required for elimination of synaptic components during development. To further support this conclusion, we suppressed the CED pathway by overexpression of the anti-apoptotic gene ced-9 cDNA in RME neurons. Unlike in CED mutants, we did not observe any additional neurons in CED-9 overexpressing transgenic lines (total 6 lines), but all these lines showed similar phenotypes to ced-3(lf) mutants, supporting that the elimination defects of CED mutants are due to inactivation of the CED pathway in RME neurons during development (Figure 2 B).
Elimination of synapses has been previously observed in DD motor neurons. DD neurons undergo remodeling during larval development, including elimination of transient synapses in the ventral cord and establishment of new synaptic connections in the dorsal cord (Hallam and Jin, 1998; White et al., 1978; White et al., 1986). DD neurons complete remodeling at L2 stage, eliminating all ventral synapses and forming dorsal synapses. Using a marker labeling DD neuron synapses, we found that the formation of dorsal synapses appeared to be normal in all CED mutants, but the elimination of ventral synapses was suppressed (Figure 2 F). About 80% of the CED mutant animals showed visible synapse in the ventral cord at L4 stage (Figure 2 F and G). But unlike in RME neurons, the synapse elimination defects dramatically decreased after L4 stage and were undetectable in two-day old adults (Figure S2 C), suggesting other pathways may act in parallel with the CED pathway in DD motor neurons. Nevertheless, these results show that the activation of the CED pathway is likely a common mechanism for synapse elimination in C. elegans. By examining DD neurons, we were also able to conclude that the observed CED mutant phenotypes are due to defects in synapse elimination but not defects in neuronal polarity. In L1 animals DD neurons extend neurites in both dorsal and ventral cords, but only establish synaptic connections in the ventral cord (Hallam and Jin, 1998). If the CED pathway were required for the establishment of neuronal polarity, we would expect to see synapses in both dorsal and ventral cords in CED mutants. But we did not observe any synaptic puncta in the dorsal cord of L1 mutant animals (Figure 2 F), supporting that the phenotypes in L4 worms are due to defects in synapse elimination. Taken together, our results demonstrate that the activation of the CED pathway plays an important role in synapse elimination in vivo.
Axonal mitochondria are required for synapse elimination
In cultured hippocampal neurons dysfunction of mitochondria can promote synapse elimination through activation of caspase-3 (Erturk et al., 2014). By examining a transgene marker expressing mitochondrial cytochrome C oxidase subunit-8 (Cox-8) Crimson fusion protein, we found that mitochondria are either at synaptic regions or very close to synapses in RMED/V neurites and DD motor neurons (Figure 2 A and S3 A), raising the possibility that mitochondria may also regulate synapse elimination in C. elegans. To test this hypothesis, we investigated the development of RME neurons in loss-of-function ric-7 (resistance to inhibitors of cholinesterase), which is essential for axonal localization of mitochondria in C. elegans (Rawson et al., 2014). We observed that loss-of-function in ric-7 blocked the distribution of mitochondria in D/V neurites, and displayed elimination phenotypes to a similar degree as CED mutants (Figure 3 A, B, C and D). Moreover, double mutants of ric-7 and CED genes did not enhance the elimination phenotypes, and loss-of-function in ced-9 can suppress ric-7(lf) back to control level (Figure 3 D, Figure S2 D), suggesting that axonal mitochondria can activate the CED pathway to regulate elimination of synaptic components in RME neurons. To address how axonal mitochondria affect the CED pathway, we generated transgene reporters labeling CED-3 in motor neurons and EGL-1 in RME neurons, and found that CED-3 and EGL-1 were enriched at synaptic regions and co-localized with mitochondria (Figure S3 B, C and F). Remarkably, loss-of-function in ric-7 completely blocked axonal/synaptic localization of CED-3 and CED-9 (Figure S3 D and E), indicating that the axonal/synaptic localization of CED-9 and CED-3 depends on the presence of mitochondria in axons. Previous studies showed that transport of mitochondria in axons is mediated by a motor protein unc-116 in motor neurons (Rawson et al., 2014). However, we did not observe any strong defects in mitochondrion distribution in RME neurons of unc-116(lf) animals, and overexpression the mitochondrion targeted kinesin (Kinesin-TOM7) did not rescue mitochondrion distribution or SNB-GFP elimination phenotypes in RME neurons of ric-7(lf) animals, suggesting mitochondria may use distinct motor proteins in different neurons (Figure S3 G, H and I). We next wanted to address whether the distribution of mitochondria in D/V neurites required the CED pathway. We found that disruption of the CED pathway did not affect mitochondria distribution, and loss-of-function in ced-9 did not restore D/V mitochondria in ric-7(lf) animals (Figure S4 A-E). Similar to the loss-of-function in the CED pathway, mutating ric-7 also prevented SNB-1-GFP elimination in DD motor neurons (Figure S5 A and B). In conclusion, our results show that axonal mitochondria act upstream of the CED pathway in elimination of presynaptic components.
Figure 3. Axonal mitochondria are required for synapse elimination in RME neurons.
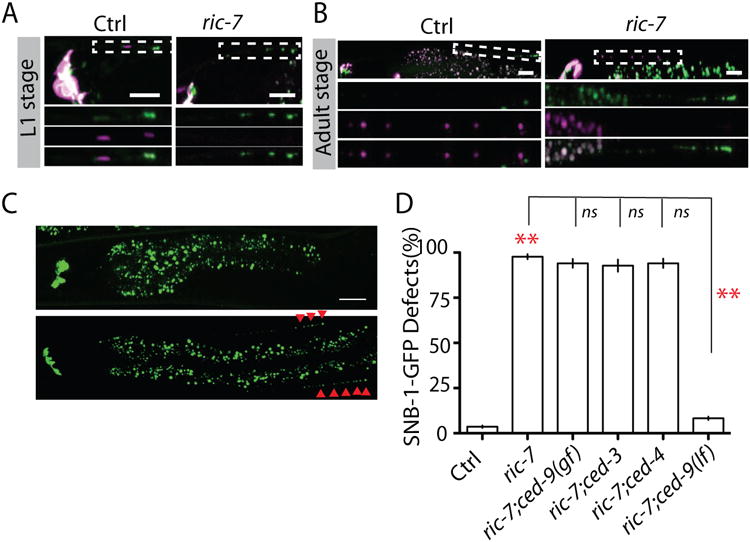
(A) Mitochondria, labeled by COX-8-Crimsion, are co-localized with SNB-1-GFP in D/V neurites of L1 animals, and this synaptic localization requires ric-7. (B) ric-7 plays an important role in D/V neurite localization of mitochondria to in adult animals. Images (C) and quantification (D) showed that loss-of-function in ric-7 prevents synapse elimination through CED-9. Experiments were performed at least 3 times, with N ≥80 animals each time. Data is shown as mean ± SD. Student test, ** P < .01. Scale bar, 10 μm. See also Supplemental Figure 2 and 3.
C. elegans gelsolin protein GSNL-1 is required for synapse elimination
Caspases are cysteine-aspartate proteases. Almost one thousand proteins have been predicted to be cleaved by caspases (Crawford and Wells, 2011). What is the downstream target of the caspase CED-3 in synapse elimination? To answer this question, we analyzed other mutants isolated in our genetic screen that resembled ced-3(lf) like phenotypes. One mutant, ju1061, caught our attention because it had strong SNB-1-GFP elimination defects, but unlike CED mutants, had no additional neurons, which suggests ju1061 affects a non-apoptotic gene required for elimination of presynaptic components. We mapped ju1061 to the center region of chromosome V, and analysis of whole genomic sequencing results showed that only a single gene, gsnl-1, was altered in the region of interest. gsnl-1 encodes a gelsolin-related protein that contains four conserved gelsolin domains (Klaavuniemi et al., 2008). ju1061 induces a nonsense mutation in the third gelsolin domain (Figure S5C and Table S1). We found about 90% of ju1061 adult animals showed SNB-1-GFP elimination defects, and expression of gsnl-1 genomic fragment in RME neurons rescued ju1061 phenotypes (Figure 4 A and B). A loss-of-function allele of gsnl-1, ok2979, displayed SNB-1-GFP and SYD-2-GFP elimination phenotypes similar to those in ju1061 (Figure 4 A and B, Figure S6 A and B). Loss-of-function in gsnl-1 could also suppress synapse elimination in DD motor neurons (Figure S7 A and B). Previous studies showed that GSNL-1 can sever actin filaments in vitro, and a conserved linker region between the first and second gesolin domain is required for its F-actin severing function (Klaavuniemi et al., 2008; Liu et al., 2010). We found that expression of GSNL-1 lacking this linker region in RME neurons failed to rescue ju1061 defects (Figure 4 B). These results lead us to conclude that ju1061 is a loss-of-function allele of gsnl-1, and that the function of gsnl-1 in SNB-1-GFP elimination relies on its F-actin severing activity. To further test if gsnl-1 can regulate F-actin de-polymerization in RME D/V neurites, we made a reporter strain expressing GFP-UtrCH to label F-actin. UtrCH is the calponin homology domain of F-actin binding protein utrophin, and has been shown to specifically bind F-actin in C. elegans (Chia et al., 2012). As shown in Figure 4 C, F-actin was enriched at the end of RME dorsal/ventral neurites where the transient synapses were formed in L1 animals, but no F-actin patch was observed in D/V neurites in L3 or later stage animals (Figure 4 C). This result is consistent with previous studies that the accumulation of F-actin is required for the formation of synapses (Chia et al., 2012). To test the function of gsnl-1 in regulation of F-actin in RME D/V neurites, we examined this marker in gsnl-1 loss-of-function background and found strong accumulation of F-actin at the end of D/V neurites in adult animals (Figure 4 C). In the DD motor neurons, we found that GFP-UtrCH was not only accumulated in axons, but also had strong signals in dendrites. As shown in Figure S6 D, in L1 animals DD neurons formed synapses at ventral cords, and dorsal processes functioned as dendrites, but GFP-UtrCH was accumulated in both ventral and dorsal cords. In L4 animals DD neurons have reversed their polarity to form synapses with dorsal muscles, and ventral processes served as dendrites to receive signals from other neurons. GFP-UtrCH had similar fluorescence intensity in dorsal and ventral cords in L4 animals (Figure S6 D, S6 F). We found that loss-of-function in gsnl-1 induced accumulation of enlarged GFP-UtrCH patches at un-eliminated transient synapses in ventral cords of L4 animals, and as a result the relative fluorescence intensity in ventral cords increased 50% (Figure S6 E and S6 F). These results suggest that de-polymerization of actin filaments in D/V neurites by gsnl-1 is important for elimination of transient synapses. Since loss-of-function in gsnl-1 has similar phenotypes as ced-3(lf), we next wanted to test whether the CED pathway could promote elimination of presynaptic components through activating GSNL-1.
Figure 4. gsnl-1 regulates actin dynamics in synapse elimination.
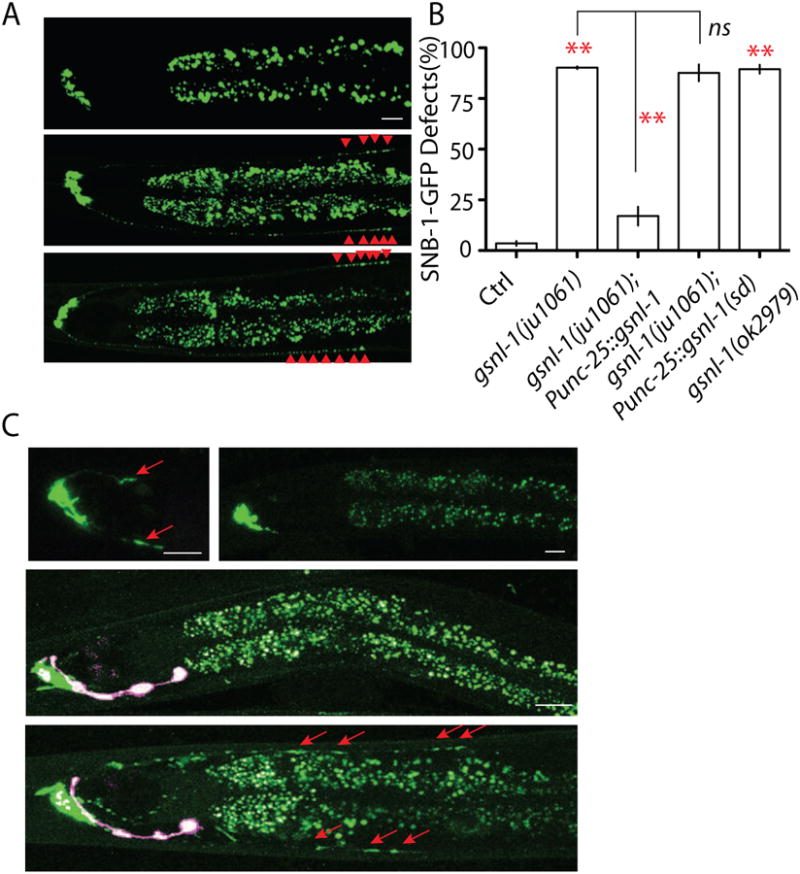
(A-B) the F-acting severing function of GSNL-1 is required for synapse elimination. (A) Images show two loss-of-function alleles of gsnl-1 both prevent the elimination of synapses in RME neurons. Red arrowheads highlight D/V synapses. (B) gsnl-1 acts cell- autonomously to regulate synapse elimination, and deletion of the linker region required for its severing function abolishes gsnl-1 functions in synapse elimination. (C) Loss-of-function in gsnl-1 induces mis-accumulation of actin filaments in D/V neurites. Polymerized F-actin is visualized by GFP-utrCH. F-actin accumulates in D/V neurites at the similar position of D/V synapses at L1 stage, and becomes undetectable in those D/V neurites in L3 and adult animals. In adult gsnl-1(lf) mutants, the strong F-actin signal can still be observed in D/V neurites (red arrows). The pink color neurons are AIY neurons from expression of co-injection marker Pttx-3-RFP. In Figure B, experiments were performed at least 3 times, with N ≥80 animals each time. For transgenic animals the results shown here are generated from at least three independent lines. Data is shown as mean ± SD. Student test, ** P < .01, ns: no significant difference. Scale bar, 10 μm. See also Supplemental Figure 4 and 5.
GSNL-1 is cleaved by CED-3
One way to address the genetic interactions between gsnl-1 and the CED pathway is to examine the double mutant phenotypes. If ced;gsnl-1 double mutants show stronger phenotypes than each single mutant, we can conclude that gsnl-1 and the CED pathway act in parallel during synapse elimination. If the double mutants do not enhance the single mutant phenotypes, they likely function in the same signaling pathway. As shown in Figure 5 A, double mutants of gsnl-1 with ced-3(lf), ced-9(gf) and ced-4(lf) exhibited the same percentage of animals with defects as single mutant animals. Moreover, we counted the number of SNB-1-GFP puncta in RMED/V neurites of animals with elimination defects, and found double mutants of ced-3;gsnl-1 did not change the number of SNB-1-GFP puncta (Figure S6 G). Due to the close genetic positions between gsnl-1 and egl-1 we were unable to analyze gsnl-1 egl-1 double mutant phenotypes. Nonetheless, our results support a conclusion that gsnl-1 acts in the CED pathway to regulate elimination of presynaptic components. De-polymerization of F-actin by GSNL-1 could act either upstream of EGL-1 or downstream of CED-3. In vitro studies showed the F-actin severing ability of GSNL-1 correlated with its protein level (Klaavuniemi et al., 2008). To test where is GSNL-1 in the CED pathway, we overexpressed gsnl-1 in the nervous system, and found increasing gsnl-1 expression levels partially suppressed ced-3(lf) phenotypes (Figure 5 B). More importantly the function of gsnl-1 in the CED pathway relied on its F-actin severing activity because deleting the linker region required for F-actin severing function from GSNL-1 totally abolished its suppression ability (Figure 5 B). Combining these results, we conclude that GSNL-1 acts downstream of CED-3, and that its activation is positively regulated by CED-3. One major mechanism by which caspases regulate downstream targets is to cleave proteins at cysteine-aspartate sites. To test whether CED-3 can cleave GSNL-1, we generated a reporter expressing N-terminal FLAG tagged GSNL-1 (yadIs10) in the nervous system. In wild type background, we detected two protein products around 50kDa and 40kDa (Figure 5 C). Since the predicted molecular weight for full length GSNL-1 is 52kDa, the 40kDa band is a product of cleavage of GSNL-1. Remarkably, loss-of-function in ced-3 strongly blocked the production of 40kDa band such that only about 10% GSNL-1 proteins were cleaved in ced-3 (lf), comparing to 45% in control animals (Figure 5 C and D). Now we know GSNL-1 acts downstream of, and can be cleaved by CED-3, what is the significance of this cleavage in synapse elimination?
Figure 5. GSNL-1 acts downstream of CED-3 and can be cleaved by CED-3.
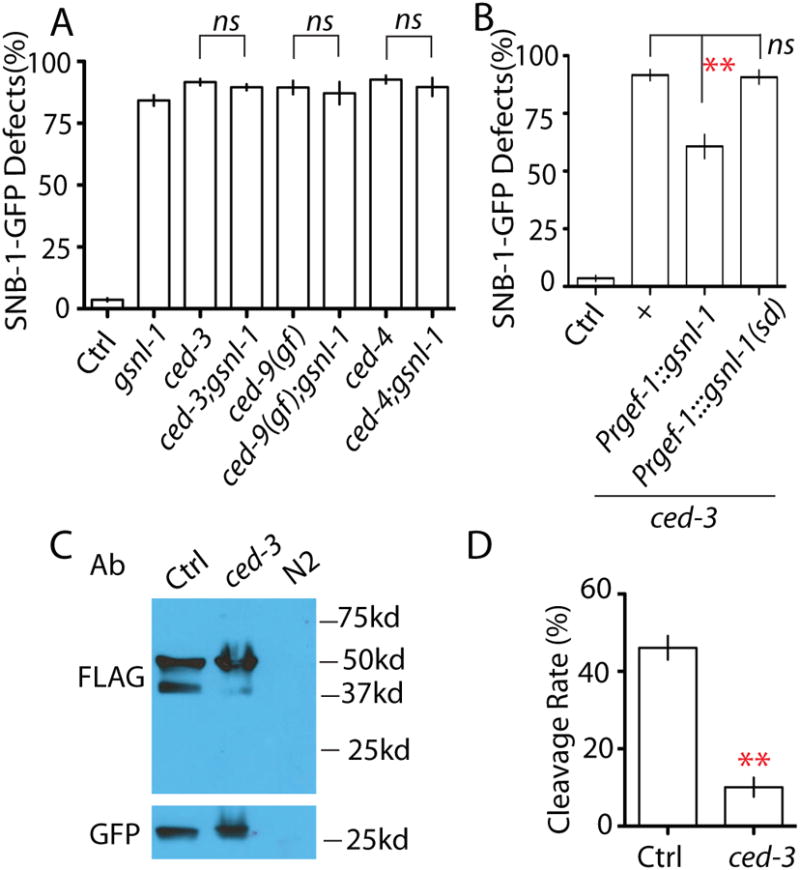
(A) gsnl-1 acts in the same pathway with CED genes. Double mutants of gsnl-1 and CED genes have similar degree of SNB-1-GFP elimination defects as single mutants. (B) The F-actin severing function of gsnl-1 is required for synapse elimination induced by activation of the cell death pathway. Overexpression of gsnl-1 can partially suppress ced-3 phenotypes in RME neurons, and deleting the linker region required for severing function abolishes its suppression ability on ced-3(lf). (C-D) CED-3 can cleave GSNL-1. (C) Western blotting analysis shows loss of function in ced-3 suppresses the cleavage of GSNL-1. FLAG antibodies recognize the full length (50kd) and the cleaved (40kd) GSNL-1. No signal was detected in wild type N2 worms without the transgene. Pttx-3∷gfp was used as a co-injection marker for gsnl-1 transgene, and GFP was served as a loading control in western blot. (D) Quantification of cleavage rate of GSNL-1. Five independent experiments were performed, and band intensity was measured using ImageJ. The cleavage rate was calculated by: P40 Intensity/(P50 intensity+ P40 intensity) × 100%. In Figure A and B, experiments were performed at least 3 times, with N ≥80 animals each time. For transgenic animals the results shown here are generated from at least three independent lines. Data is shown as mean ± SD. Student test, ** P < .01, ns: no significant difference.
The CED pathway promotes F-actin disassembly through cleavage of GSNL-1 at a conserved C-terminal region
GSNL-1 can be cleaved by CED-3 to a fragment around 40kDa at the C-terminal, indicating the cleavage site is between amino acids 350 and 400. Using online caspase substrate prediction server CASVM (http://www.casbase.org/casvm/index.html), we found one conserved potential caspase site in the region of interest (Figure 6 A). To test whether CED-3 can cleave GSNL-1 at this site, we generated transgenes expressing mutated GSNL-1 proteins without this cut site or changing the critical aspartate to alanine, and found that both mutations totally blocked the cleavage of GSNL-1 (Figure 6 B). We next asked whether the cleavage by CED-3 is required for the gsnl-1 function. We compared the rescue ability of wild type and the CED-3 cleavage site deleted gsnl-1. The results showed that compared to wild type gsnl-1, deletion of the CED-3 cleavage site strongly reduced the rescuing ability of gsnl-1, indicating the importance of CED-3 cleavage in regulation of gsnl-1 activation (Figure 6 C). The partial rescue of gsnl-1(lf) by the CED-3 cleavage site deleted gsnl-1 suggests other mechanisms, for example binding with other F-actin regulators, may also regulate gsnl-1 activation (Figure 6 C). To further confirm the cleaved GSNL-1 is the active form, we expressed P40, a truncated GNSL-1 protein representing the cleavage product by CED-3, in ced-3(lf) animals. As shown in Figure 6 D, overexpression of P40 suppressed ced-3(lf) much better than that of full length GSNL-1. Consistent with this observation, deletion of the CED-3 site or mutating the critical aspartate to alanine in GSNL-1 totally abolished its suppression ability on ced-3(lf) phenotypes (Figure 6 D). Previous in vitro studies (Klaavuniemi et al., 2008) and our results here show one major function of GSNL-1 is to sever actin filaments. To test whether the CED pathway was involved in regulation of F-actin, we compared F-actin distribution in control and mutant animals. We found that ced-4(lf), egl-1(lf), ric-7(lf) and ced-9(gf) induced mis-accumulation of F-actin in RMED/V neurites in all animals observed (≥100 animals for each genotypes) (Figure 6 E and Figure S7 C, D, E). Similar as we observed in gsnl-1(lf) animals, F-actin formed enlarged patches in dorsal processes of DD neurons in L4 animals of mutants affecting each members of the CED pathway and ric-7 (Figure S7 F and G). Furthermore, the effect of CED mutants and ric-7(lf) could be completely suppressed by expression of P40, the active form GSNL-1, in all animals observed (≥70 animals for each genotypes) (Figure 6 E and Figure S7 C, D, E). Taken together, our results demonstrate that the cleavage of GSNL-1 at the conserved C-terminal site by CED-3 promotes its activation and triggers F-actin de-polymerization to eliminate synaptic components during development.
Figure 6. CED-3 cleaves GSNL-1 at a C-terminal conserved site to promote its F-actin severing ability.
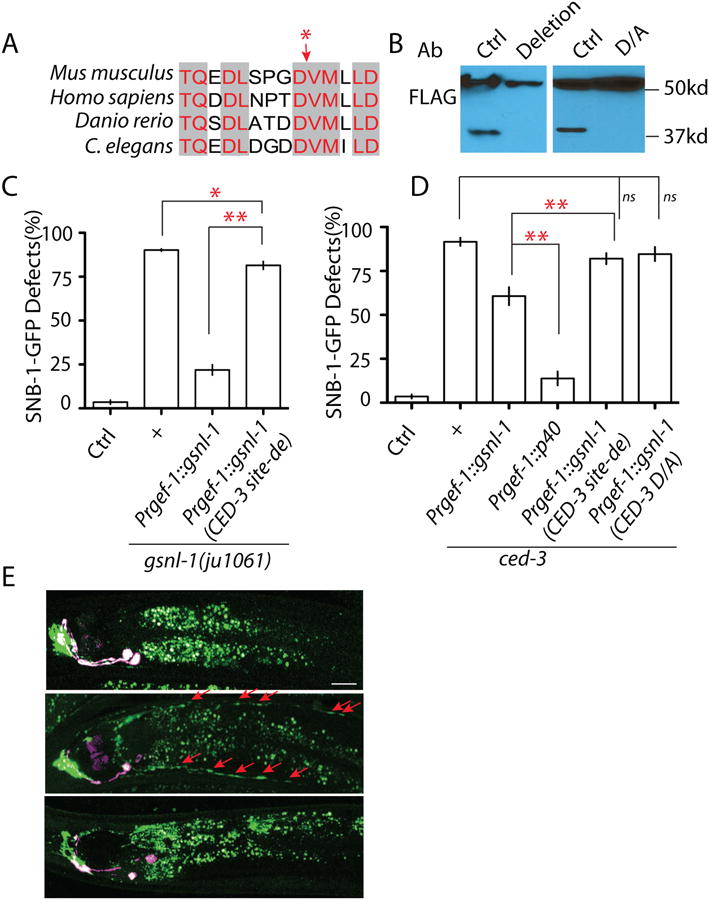
(A) Sequence analysis identifies a conserved candidate caspase cleavage site at the region where cleavage can produce a 40kb band in C. elegans. NCBI Reference Sequence: Danio rerio XP_003198825.1; Mus musculus (gene: Advillin) XP_006513165.1; Homo sapiens (Gene: Advillin) NP_006567.3. The red star points the potential cut site. (B) Deletion of the five core amino acids “DDVMI” (deletion) or mutating the aspartate (“DDVMI”) to alanine (“DAVMI”) (D/A) in this region blocks the cleavage of GSNL-1. (C) Deletion of the CED cleavage site strongly affects gsnl-1 functions. Expression of gsnl-1 lacking the CED cleavage site only shows weak rescue ability, while wild type gsnl-1 rescues mutant phenotypes in over 90% animals (D) Expression of the cleaved GSNL-1 can bypass the requirement of ced-3 in synapse elimination. Expression of P40 almost totally suppresses ced-3 phenotypes, while un-cleavable GSNL-1 fails to suppress ced-3 phenotypes. (E) Loss-of-function in the CED pathway induces mis-accumulation of F-actin in D/V neurites, and overexpression of P40 can suppress the effect of ced-4(lf). In Figure C and D, experiments were performed at least 3 times, with N ≥80 animals each time. For transgenic animals the results shown here are generated from at least three independent lines. Data is shown as mean ± SD. Student test, ** P < .01, * P<.05, ns: no significant difference.
Discussion
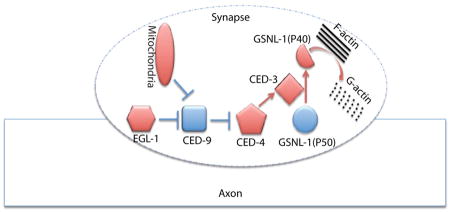
Using the development of C. elegans RME neurons, we investigated the molecular mechanisms of synapse elimination. During development RME D/V neurons form transient clusters of presynaptic components in D/V neurites at L1 stage, and those clusters are eliminated before L3 stage. In EM studies, we uncovered RMED/V neurites contained swelling structures enriched with vesicles, but the nature of these swellings is unknown. Among many possibilities, one could be that they might represent immature synapses, as our analyses of presynaptic markers showed strong correlation with these swellings. In C. elegans, the DD motor neurons form synapses at the ventral cord at the L1 stage, and those synapses are eliminated in the L2 stage. Although the SNB-1-GFP puncta in RME D/V dendrites may not represent mature synapses, the consistent phenotypes of multiple fluorescent reporters from DD motor neurons and RME neurons supports that the apoptotic CED pathway can regulate some aspects of synapse elimination in vivo. Taking advantage of the powerful genetic tools in C. elegans, we further uncovered the important roles of axonal mitochondria and F-actin severing protein GSNL-1 in elimination of presynaptic materials. Finally, we demonstrated that the caspase CED-3 cleaves GSNL-1 at a conserved site to promote GSNL-1 activation. Therefore, our working model is that during development some internal cues trigger the local activation of the CED pathway through mitochondria to activate GSNL-1 by protein cleavage, and the activated GSNL-1 promotes disassembly of the F-actin network at synaptic regions to induce elimination of synaptic components (Figure 7).
Figure 7. A working model for regulation of mitochondria-CED-GSNL-1 in synapse elimination.
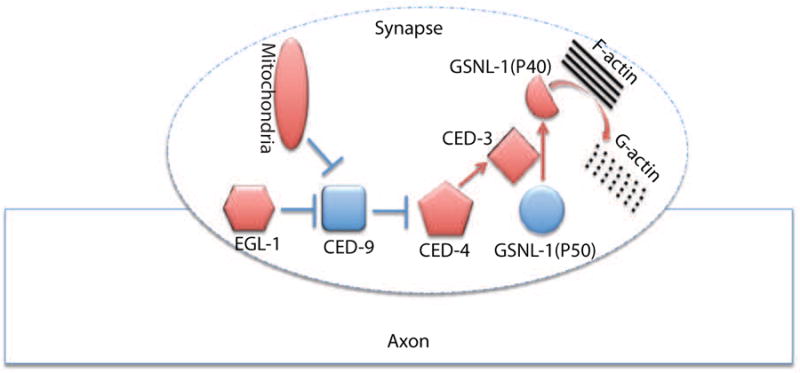
Axonal mitochondria activate the CED pathway to promote the cleavage of GSNL-1, and then the cleaved GSNL-1(P40) disassembles F-actin network to eliminate synapses.
Cell death and synapse elimination
It has long been known that the activation of apoptosis pathway triggers cell death (Hyman and Yuan, 2012). Previous studies (Erturk et al., 2014; Wang et al., 2014) and our results here show that activation of the apoptosis pathway can promote synapse elimination without loss of neurons during development. The question is how neurons restrict the activation of the caspase pathway only at synaptic regions to prevent cell death from happening. Actin is a ubiquitous component of all types of cells, and is involved in almost all biological processes. It is not a surprise that as a major F-actin severing protein family gelsolin proteins play important roles in regulation of cell death. The first study linked gelsolin with apoptosis is in cultured Jurkat cells, in which overexpression of gelsolin inhibits the activation of CPP32 protease (caspase-3) to prevent cell death induced by treatment of anti-FAS antibodies (Ohtsu et al., 1997). Since then gelsolin has been shown to inhibit apoptosis through directly binding with caspase-9 and caspase-3, preventing mitochondrial membrane potential loss and cytochrome C release, blocking mitochondrial voltage-dependent anion channel and depolymerization of F-actin (Azuma et al., 2000; Harms et al., 2004; Koya et al., 2000; Kusano et al., 2000). Activation of gelsolin has also been reported to play anti-apoptosis roles in human epidermoid cancer cells, oncogenic K-ras mutation cell lines, thermal injury and neural loss (Boccellino et al., 2004; Harms et al., 2004; Klampfer et al., 2004; Zhang et al., 2011). However increase of gelsolin activity failed to prevent apoptosis in lymphocyte and fas antibody-induced liver failure in vivo (Leifeld et al., 2006; Posey et al., 2000). Nevertheless, gelsolin proteins can play anti-apoptotic roles in caspase mediated cell death. In our studies, we found that activation of the apoptosis pathway promotes GSNL-1 activation through cleavage at the C-terminal conserved site. It is possible that the activated GSNL-1 generated by local activation of the CED pathway can inhibit further activation of caspases, thereby restricting the activation of apoptosis pathway only at certain subcellular regions to prevent cell death from happening.
Calcium, caspases, gelsolin and synapse elimination
In the mammalian cortex, synaptic elimination starts from the late postnatal period until the end of life. In multiple regions of cortex studies showed blockage of NMDA receptors prevents the loss of synapses in vivo (Ohno et al., 2010; Rabacchi et al., 1992). In long-tem-depression (LTD), the shrinkage of dendritic spines relies on the activation of NMDA receptors (Zhou et al., 2004). In neurodegenerative diseases, synapse loss happens in a NMDA receptor dependent manner (Milnerwood and Raymond, 2010). Calcium channels such as voltage dependent calcium channels and TRP channels are also found to be important for synapse elimination (Gibson et al., 2008; Hashimoto et al., 2011). These studies support the essential role of calcium in regulation of synapse elimination. In the C. elegans CED pathway CED-4 is an Apaf-1-like protein, and contains two EF-hand calcium binding domains (Yuan and Horvitz, 1992). In axon regeneration, elevation of calcium concentration upon injury can activate CED-4 to promote axonal regrowth (Pinan-Lucarre et al., 2012). The second gelsolin domain of GSNL-1 can change its conformation upon binding with calcium to regulate its F-actin severing ability (Liu et al., 2011). These findings suggest that change in local calcium concentration either by neuronal activity or ER/mitochondrial release may modulate the strength of the CED-GSNL-1 pathway in synapse elimination.
Caspases and gelsolin
Gelsolin was one of the first caspase substrates identified using in vitro assays (Kothakota et al., 1997). The fragmentation of gelsolin by caspases contributes to the morphological change of apoptotic cells (Kothakota et al., 1997). Neuronal cleavage of gelsolin is reported in Alzheimer's disease, spinal cord injury, ischemia and burn injury (Ji et al., 2009; Matsushita et al., 2000; Springer et al., 1999; Zhang et al., 2013). However these studies have mainly focused on the apoptotic roles of caspase-mediated gelsolin cleavage in the nervous system. During aging and in disease conditions, neurodegeneration often happens in a “dying back” manner that starts from abnormal pruning of pre-synaptic and post-synaptic structures, and then the degeneration progresses along axons and dendrites, and finally the “dying signal” induces the loss of neurons (Adalbert and Coleman, 2012; Wang et al., 2012). Several studies showed that prevention of the early degeneration events at synapses and axons can protect neurons from death (Ferri et al., 2003; George and Griffin, 1994; Kaneko et al., 2006; Meyer zu Horste et al., 2011). Although our findings highlight the role of CED-GSNL-1 regulation in synapse elimination during development, the same mechanism could also be applied to synapse elimination in aging or disease conditions, and may provide a new angle for understanding the function of cell death pathway in those processes.
Synapse elimination and axon regeneration
In our experiments, we expressed GSNL-1 in all neurons and detected cleavage of GSNL-1, suggesting the CED-GSNL-1 regulation may also function in other processes. Recent studies in C. elegans revealed the important role of CED-4 and CED-3 in axon regeneration. Using mechanosensory ALM neurons as a model, Pinan-Lucarre and colleagues showed that axons sprout out filopodia-like processes shortly after injury, and loss-of-function in both ced-4 and ced-3 blocks the formation of those filopodia structures and eventually suppresses regeneration (Pinan-Lucarre et al., 2012). The filopodia structure in regenerating axons is similar to that in growth cones during axon pathfinding. Extensive studies show that actin dynamics is the major cause for filopodia growth and retraction in axon guidance (Luo, 2002). More interestingly, the caspase-mediated gelsolin cleavage is observed after spinal cord injury (Springer et al., 1999). Therefore it is possible that in axon regeneration CED-3 may also cleave GSNL-1 to regulate F-actin assembly and filopodia formation after injury. One key regulator for axon regeneration in C. elegans is DLK-1 (Hammarlund and Jin, 2014). Genetic studies showed CED-3 acts in the same pathway with DLK-1 in regeneration (Pinan-Lucarre et al., 2012). DLK-1 is required both for the re-initiation of growth cones and elongation of axons. Regulation of microtubule dynamics by the DLK-1 pathway has been shown to be important for the elongation of regenerating axons (Ghosh-Roy et al., 2012). But promoting microtubule assembly can only partially rescue dlk-1 phenotypes, suggesting additional components are required in early period of regeneration (Ghosh-Roy et al., 2012). It will be interesting to test whether the CED-3 mediated actin regulation is controlled by DLK-1 during rebuilding of growth cones after injury.
Experimental Procedures
C. elegans genetics
We maintained C. elegans strains on NGM plates at 20 - 22.5°C as described by Brenner (Brenner, 1974). All transgenes and strains are described in Tables S1 and S2. We use juIs1 (Punc-25∷SNB-1-GFP) (Hallam and Jin, 1998) in unc-30 (ju32) background (Table S1) to visualize RME neurons presynaptic terminals, and use juIs137 (Pflp-13∷SNB-1-GFP) (Abrams et al., 2008) to visualize DD motor neuron synapses. hpIs3 (Punc-25∷SYD-2-GFP) (Yeh et al., 2009) was used to visualize RME presynaptic active zones. unc-30 (ju32) was isolated by Mei Zhen and Y.J. in an early screen for synapse formation defects, and contains a G to A mutation in the 3′ splice acceptor of the last intron. ced-3(ju1056) and gsnl-1(ju1061) were isolated from the strain CZ15720 unc-30(ju32) juIs1(Punc-25∷SNB-1-GFP) after ethyl methane sulfonate mutagenesis.
Fluorescence microscopy
We scored fluorescent reporters in live animals using a Zeiss Axio Imager 2 microscope equipped with Chroma HQ filters. For quantification of RME neuron synapse elimination defects, at least three independent experiments were performed and total 200-400 one-day old adults were analyzed. DD motor neuron phenotypes were analyzed at the L1, L4 and two-day adult stage. Confocal images were collected at animals immobilized in 1% 1-phenoxy-2-propanol (TCI America, Portland, OR) in M9 buffer using a Zeiss LSM700 confocal microscope. Pictures shown in the figures are Z-stack images (1 μm/section).
Statistical analysis
We analyzed our data using one-tailed Student's t test in Graphpad Prism (GraphPad Software, La Jolla, CA).
Protein analysis
For expression studies in C. elegans, we used a integrated transgene (yadIs10) expressing FLAG-GSNL-1 driven by a pan-neuronal promoter Prgef-1 (PNYL99). Synchronous animals were collected at L2 stages, and protein lysis in SDS sample buffer containing 1 mM DTT were obtained by freeze-throw 20-50 times in dry ice/ethanol and 37°C water bath, and then denatured by heating to 95 for 5 minutes. Blots were probed with rabbit anti-Flag antibodies (Sigma, F1804), or a mouse anti-GFP monoclonal antibody (Sigma, G1544), and visualized with Amersham HRP-conjugated anti-rabbit or anti-mouse secondary antibodies at 1:5000 (Amersham) using the SuperSignal West Femto kit (Pierce, Rockford, IL).
DNA constructs and generation of transgenes
All DNA expression constructs were made using Gateway cloning technology (Invitrogen). Sequences of the final clones were confirmed. The information for each construct is in Table S2. The primer sequences are available upon request.
Transgenic animals were generated following standard procedures (Mello et al., 1991). In general, plasmid DNAs of interest were used at 1-50 ng/μl with the co-injection marker Pttx-3-RFP or Pttx-3-GFP at 50 ng/μl. For each construct, 3 to 10 independent transgenic lines were analyzed. Table S2 lists the genotypes and DNA constructs for the transgenes.
Electron microscopy
L1 worms were fixed by high pressure freezing followed by freeze substitution as previously described (Hung et al., 2013; Rostaing et al., 2004; Stigloher et al., 2011). The worm was cut into 70nm-thick serial sections. Images were taken on an FEI Tecnai 20 electron microscope with an AMT 16000 digital camera at 1.4nm/pixel for reconstruction of the posterior processes, and 0.3nm/pixel for the images shown of the neurite swellings. Images were stitched and aligned using TrakEM2 (Cardona et al., 2012). RMED and RMEV were identified based on their cell body position (Sulston et al., 1983) and the trajectory of their neurites (White et al., 1986), and were reconstructed volumetrically within TrakEM2.
Supplementary Material
Acknowledgments
We thank Drs. Mei Zhen, Aravi Samuel and Jeff Lichtman for the EM analysis. Some strains used in this study were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Dr. Yuji. Kohara for cDNAs, Dr. Erik Jorgensen for Kinesin-TOM7 plasmid, Dr. Joachim Kurth for Apoliner reporter, Dr. Mei Zhen for the active zone reporter and contribution in the isolation of unc-30(ju32). We thank our lab members for comments on the manuscript. The C. elegans L1 EM project is supported by Human frontier science program (HFSP RGP0051/2014 to A. S., J. L. and M. Z.). S.C is supported by NIH Grant 5T32GM007491 awarded to the Albert Einstein College of Medicine. B.M. is supported by CIHR grants MOP 123250 and MOP 74530. Y. J. is an Investigator of the Howard Hughes Medical Institute. Part of the work was supported by grants from NIH (R01 NS035546 to Y. J.; K99NS076646 to D. Y.). The Yan lab is supported by R00 award (NS076646) from the National Institute Of Neurological Disorders and Stroke.
Footnotes
Contributions: D.Y. and L.M. designed and performed the experiments. B. M., S. C. and M. N. carried out the EM analysis. B. M. and M. Z. analyzed the EM data. A.W. did all western blot analysis. The genetic screen for RME development was carried out by D.Y. when he was a postdoctoral fellow in Y.J. lab. L.M. and D.Y. wrote the manuscript.
Competing financial interests: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams B, Grill B, Huang X, Jin Y. Cellular and molecular determinants targeting the Caenorhabditis elegans PHR protein RPM-1 to perisynaptic regions. Developmental dynamics : an official publication of the American Association of Anatomists. 2008;237:630–639. doi: 10.1002/dvdy.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adalbert R, Coleman MP. Axon pathology in age-related neurodegenerative disorders. Neuropathology and applied neurobiology. 2012 doi: 10.1111/j.1365-2990.2012.01308.x. [DOI] [PubMed] [Google Scholar]
- Azuma T, Koths K, Flanagan L, Kwiatkowski D. Gelsolin in complex with phosphatidylinositol 4,5-bisphosphate inhibits caspase-3 and -9 to retard apoptotic progression. The Journal of biological chemistry. 2000;275:3761–3766. doi: 10.1074/jbc.275.6.3761. [DOI] [PubMed] [Google Scholar]
- Boccellino M, Giuberti G, Quagliuolo L, Marra M, D'Alessandro AM, Fujita H, Giovane A, Abbruzzese A, Caraglia M. Apoptosis induced by interferon-alpha and antagonized by EGF is regulated by caspase-3-mediated cleavage of gelsolin in human epidermoid cancer cells. Journal of cellular physiology. 2004;201:71–83. doi: 10.1002/jcp.20058. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron. 2003;37:939–952. doi: 10.1016/s0896-6273(03)00158-2. [DOI] [PubMed] [Google Scholar]
- Chia PH, Chen B, Li P, Rosen MK, Shen K. Local F-actin network links synapse formation and axon branching. Cell. 2014;156:208–220. doi: 10.1016/j.cell.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia PH, Patel MR, Shen K. NAB-1 instructs synapse assembly by linking adhesion molecules and F-actin to active zone proteins. Nature neuroscience. 2012;15:234–242. doi: 10.1038/nn.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford ED, Wells JA. Caspase substrates and cellular remodeling. Annual review of biochemistry. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- Doussau F, Augustine GJ. The actin cytoskeleton and neurotransmitter release: an overview. Biochimie. 2000;82:353–363. doi: 10.1016/s0300-9084(00)00217-0. [DOI] [PubMed] [Google Scholar]
- Erturk A, Wang Y, Sheng M. Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:1672–1688. doi: 10.1523/JNEUROSCI.3121-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Sanes JR, Coleman MP, Cunningham JM, Kato AC. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Current biology : CB. 2003;13:669–673. doi: 10.1016/s0960-9822(03)00206-9. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annual review of neuroscience. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George R, Griffin JW. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Experimental neurology. 1994;129:225–236. doi: 10.1006/exnr.1994.1164. [DOI] [PubMed] [Google Scholar]
- Ghosh-Roy A, Goncharov A, Jin Y, Chisholm AD. Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Developmental cell. 2012;23:716–728. doi: 10.1016/j.devcel.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA. TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron. 2008;57:746–759. doi: 10.1016/j.neuron.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Hallam SJ, Jin Y. lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature. 1998;395:78–82. doi: 10.1038/25757. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Jin Y. Axon regeneration in C. elegans. Current opinion in neurobiology. 2014;27C:199–207. doi: 10.1016/j.conb.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms C, Bosel J, Lautenschlager M, Harms U, Braun JS, Hortnagl H, Dirnagl U, Kwiatkowski DJ, Fink K, Endres M. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Molecular and cellular neurosciences. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Tsujita M, Miyazaki T, Kitamura K, Yamazaki M, Shin HS, Watanabe M, Sakimura K, Kano M. Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9987–9992. doi: 10.1073/pnas.1101488108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesmann GR, Clayton DF. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron. 2006;52:1061–1072. doi: 10.1016/j.neuron.2006.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR, de Courten C, Garey LJ, Van der Loos H. Synaptogenesis in human visual cortex--evidence for synapse elimination during normal development. Neuroscience letters. 1982;33:247–252. doi: 10.1016/0304-3940(82)90379-2. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nature reviews Neuroscience. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- Ji L, Chauhan A, Wegiel J, Essa MM, Chauhan V. Gelsolin is proteolytically cleaved in the brains of individuals with Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2009;18:105–111. doi: 10.3233/JAD-2009-1127. [DOI] [PubMed] [Google Scholar]
- Jiao S, Li Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron. 2011;70:758–772. doi: 10.1016/j.neuron.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa S, Yamasaki M, Watanabe M, Kano M. Critical period for activity-dependent synapse elimination in developing cerebellum. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:4954–4961. doi: 10.1523/JNEUROSCI.20-13-04954.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama T, Yoshioka N, Sakurai M. Synapse elimination in the corticospinal projection during the early postnatal period. Journal of neurophysiology. 2006;95:2304–2313. doi: 10.1152/jn.00295.2005. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Peck CR, Walsh MK, Gan WB, Feng G, Sanes JR, Lichtman JW. Asynchronous synapse elimination in neonatal motor units: studies using GFP transgenic mice. Neuron. 2001;31:381–394. doi: 10.1016/s0896-6273(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Klaavuniemi T, Yamashiro S, Ono S. Caenorhabditis elegans gelsolin-like protein 1 is a novel actin filament-severing protein with four gelsolin-like repeats. The Journal of biological chemistry. 2008;283:26071–26080. doi: 10.1074/jbc.M803618200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Oncogenic Ras promotes butyrate-induced apoptosis through inhibition of gelsolin expression. The Journal of biological chemistry. 2004;279:36680–36688. doi: 10.1074/jbc.M405197200. [DOI] [PubMed] [Google Scholar]
- Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LT. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, Kuzumaki N. Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. The Journal of biological chemistry. 2000;275:15343–15349. doi: 10.1074/jbc.275.20.15343. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron. 2006;51:283–290. doi: 10.1016/j.neuron.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, Tsujimoto Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene. 2000;19:4807–4814. doi: 10.1038/sj.onc.1203868. [DOI] [PubMed] [Google Scholar]
- Leifeld L, Fink K, Debska G, Fielenbach M, Schmitz V, Sauerbruch T, Spengler U. Anti-apoptotic function of gelsolin in fas antibody-induced liver failure in vivo. The American journal of pathology. 2006;168:778–785. doi: 10.2353/ajpath.2006.050323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jo J, Jia JM, Lo SC, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Kanzawa N, Ono S. Calcium-sensitive activity and conformation of Caenorhabditis elegans gelsolin-like protein 1 are altered by mutations in the first gelsolin-like domain. The Journal of biological chemistry. 2011;286:34051–34059. doi: 10.1074/jbc.M111.237404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Klaavuniemi T, Ono S. Distinct roles of four gelsolin-like domains of Caenorhabditis elegans gelsolin-like protein-1 in actin filament severing, barbed end capping, and phosphoinositide binding. Biochemistry. 2010;49:4349–4360. doi: 10.1021/bi100215b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annual review of cell and developmental biology. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, He X, Wynshaw-Boris A, Xiong WC, Lu B, Mei L. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron. 2002;35:489–505. doi: 10.1016/s0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- Matsushita K, Wu Y, Qiu J, Lang-Lazdunski L, Hirt L, Waeber C, Hyman BT, Yuan J, Moskowitz MA. Fas receptor and neuronal cell death after spinal cord ischemia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:6879–6887. doi: 10.1523/JNEUROSCI.20-18-06879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer zu Horste G, Miesbach TA, Muller JI, Fledrich R, Stassart RM, Kieseier BC, Coleman MP, Sereda MW. The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration. Neurobiology of disease. 2011;42:1–8. doi: 10.1016/j.nbd.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends in neurosciences. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Murthy VN, De Camilli P. Cell biology of the presynaptic terminal. Annual review of neuroscience. 2003;26:701–728. doi: 10.1146/annurev.neuro.26.041002.131445. [DOI] [PubMed] [Google Scholar]
- Nelson PG, Fields RD, Yu C, Liu Y. Synapse elimination from the mouse neuromuscular junction in vitro: a non-Hebbian activity-dependent process. Journal of neurobiology. 1993;24:1517–1530. doi: 10.1002/neu.480241106. [DOI] [PubMed] [Google Scholar]
- O'Donnell M, Chance RK, Bashaw GJ. Axon growth and guidance: receptor regulation and signal transduction. Annual review of neuroscience. 2009;32:383–412. doi: 10.1146/annurev.neuro.051508.135614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno T, Maeda H, Murabe N, Kamiyama T, Yoshioka N, Mishina M, Sakurai M. Specific involvement of postsynaptic GluN2B-containing NMDA receptors in the developmental elimination of corticospinal synapses. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15252–15257. doi: 10.1073/pnas.0906551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa S, Hamada S, Kuida K, Yoshida H, Igaki T, Miura M. Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13366–13371. doi: 10.1073/pnas.0910488107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu M, Sakai N, Fujita H, Kashiwagi M, Gasa S, Shimizu S, Eguchi Y, Tsujimoto Y, Sakiyama Y, Kobayashi K, Kuzumaki N. Inhibition of apoptosis by the actin-regulatory protein gelsolin. The EMBO journal. 1997;16:4650–4656. doi: 10.1093/emboj/16.15.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinan-Lucarre B, Gabel CV, Reina CP, Hulme SE, Shevkoplyas SS, Slone RD, Xue J, Qiao Y, Weisberg S, Roodhouse K, et al. The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in Caenorhabditis elegans. PLoS biology. 2012;10:e1001331. doi: 10.1371/journal.pbio.1001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey SC, Martelli MP, Azuma T, Kwiatkowski DJ, Bierer BE. Failure of gelsolin overexpression to regulate lymphocyte apoptosis. Blood. 2000;95:3483–3488. [PubMed] [Google Scholar]
- Rabacchi S, Bailly Y, Delhaye-Bouchaud N, Mariani J. Involvement of the N-methyl D-aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science. 1992;256:1823–1825. doi: 10.1126/science.1352066. [DOI] [PubMed] [Google Scholar]
- Rawson RL, Yam L, Weimer RM, Bend EG, Hartwieg E, Horvitz HR, Clark SG, Jorgensen EM. Axons degenerate in the absence of mitochondria in C. elegans. Current biology : CB. 2014;24:760–765. doi: 10.1016/j.cub.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JL, Taraskevich PS. Reduction of multiaxonal innervation at the neuromuscular junction of the rat during development. The Journal of physiology. 1977;270:299–310. doi: 10.1113/jphysiol.1977.sp011953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silacci P, Mazzolai L, Gauci C, Stergiopulos N, Yin HL, Hayoz D. Gelsolin superfamily proteins: key regulators of cellular functions. Cellular and molecular life sciences : CMLS. 2004;61:2614–2623. doi: 10.1007/s00018-004-4225-6. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nature medicine. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- Thompson W. Synapse elimination in neonatal rat muscle is sensitive to pattern of muscle use. Nature. 1983;302:614–616. doi: 10.1038/302614a0. [DOI] [PubMed] [Google Scholar]
- Thompson WJ. Activity and synapse elimination at the neuromuscular junction. Cellular and molecular neurobiology. 1985;5:167–182. doi: 10.1007/BF00711091. [DOI] [PubMed] [Google Scholar]
- Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. The Journal of cell biology. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Chen F, Fu XQ, Ding CS, Zhou L, Zhang XH, Luo ZG. Caspase-3 cleavage of dishevelled induces elimination of postsynaptic structures. Developmental cell. 2014;28:670–684. doi: 10.1016/j.devcel.2014.02.009. [DOI] [PubMed] [Google Scholar]
- White JG, Albertson DG, Anness MA. Connectivity changes in a class of motoneurone during the development of a nematode. Nature. 1978;271:764–766. doi: 10.1038/271764a0. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. Local caspase activity directs engulfment of dendrites during pruning. Nature neuroscience. 2006;9:1234–1236. doi: 10.1038/nn1774. [DOI] [PubMed] [Google Scholar]
- Yeh E, Kawano T, Ng S, Fetter R, Hung W, Wang Y, Zhen M. Caenorhabditis elegans innexins regulate active zone differentiation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:5207–5217. doi: 10.1523/JNEUROSCI.0637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Kawano T, Weimer RM, Bessereau JL, Zhen M. Identification of genes involved in synaptogenesis using a fluorescent active zone marker in Caenorhabditis elegans. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:3833–3841. doi: 10.1523/JNEUROSCI.4978-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Horvitz HR. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development. 1992;116:309–320. doi: 10.1242/dev.116.2.309. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Zhang QH, Chen Q, Kang JR, Liu C, Dong N, Zhu XM, Sheng ZY, Yao YM. Treatment with gelsolin reduces brain inflammation and apoptotic signaling in mice following thermal injury. Journal of neuroinflammation. 2011;8:118. doi: 10.1186/1742-2094-8-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QH, Li JC, Dong N, Tang LM, Zhu XM, Sheng ZY, Yao YM. Burn injury induces gelsolin expression and cleavage in the brain of mice. Neuroscience. 2013;228:60–72. doi: 10.1016/j.neuroscience.2012.10.013. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen M, Jin Y. The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature. 1999;401:371–375. doi: 10.1038/43886. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005a;46:181–189. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Yang G, Kwon E, Gan WB. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 2005b;436:261–265. doi: 10.1038/nature03715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.