

Abstract
Background
The continued development of targeted therapeutics for cancer treatment has required the concomitant development of more expansive methods for the molecular profiling of the patient’s tumor. We describe the validation of the JAX Cancer Treatment Profile™ (JAX-CTP™), a next generation sequencing (NGS)-based molecular diagnostic assay that detects actionable mutations in solid tumors to inform the selection of targeted therapeutics for cancer treatment.
Methods
NGS libraries are generated from DNA extracted from formalin fixed paraffin embedded tumors. Using hybrid capture, the genes of interest are enriched and sequenced on the Illumina HiSeq 2500 or MiSeq sequencers followed by variant detection and functional and clinical annotation for the generation of a clinical report.
Results
The JAX-CTP™ detects actionable variants, in the form of single nucleotide variations and small insertions and deletions (≤50bp) in 190 genes in specimens with a neoplastic cell content of ≥10%. The JAX-CTP™ is also validated for the detection of clinically actionable gene amplifications.
Conclusions
There is a lack of consensus in the molecular diagnostics field on the best method for the validation of NGS-based assays in oncology, thus the importance of communicating methods, as contained in this report. The growing number of targeted therapeutics and the complexity of the tumor genome necessitates continued development and refinement of advanced assays for tumor profiling to enable precision cancer treatment.
Introduction
The increased prevalence of molecularly targeted cancer therapeutics has expanded the utility of multi-gene sequencing panels for detecting somatic mutations in cancers. Commonly used single-gene tests, such as for EGFR and BRAF, and small multiplexed “hotspot” panels1 detect very specific targetable mutations, but clinical research studies have led to an increasingly complex array of genomic alterations, either in isolation or in combinations, that influence sensitivity or resistance to targeted cancer therapeutics 2, 3. For example, TP53 mutations alone have been reported to increase progression-free survival upon bevacizumab treatment 4, but if a patient also has a KRAS mutation, the response to bevacizumab may be diminished or counter balanced 5. The development of next generation sequencing (NGS) and associated target sequence enrichment technologies has enabled the development of clinical cancer panels that detect molecular alterations in a large number of genes in a single multiplexed assay 6-8. This disruptive technology is the impetus for the healthcare shift from a one gene/one drug paradigm to a multi-gene/many drugs perspective 9.
While the number of molecular diagnostic laboratories that have developed cancer panel assays has quickly grown10, the analytical and post-analytical methods, as well as the approaches to validation, vary substantially, and no standard has been set. The validation of assays, such as ours that are designed to accurately detect variants at allele frequencies <10% across >1Mb of target sequence present significant challenges, and many different approaches have been utilized in similar assays 6, 7, 11-15. Communication and critique of the different approaches that have been utilized will help in the development of standard practice in the validation of such complex molecular diagnostics.
We describe the design and validation including the limit of detection, analytical sensitivity and specificity and accuracy of the JAX Cancer Treatment Profile™ (JAX-CTP™), an NGS-based assay for the detection of potentially clinically actionable alterations in 190 different genes (reportable range gene list in supplementary table S1) from formalin fixed paraffin embedded (FFPE) clinical specimens. “Actionable” is defined as genes with molecular alterations associated in peer-reviewed literature with a therapy approved for a diagnosis, approved in another diagnosis, or associated directly or by mechanism of action with an investigational drug. The JAX-CTP™ accurately detects single nucleotide polymorphisms (SNPs), small insertions and deletions (indels; up to 50-bp long) and gene-level amplifications (copy number variations (CNVs)) in clinical specimens with a sensitivity that is sufficient for samples with significant cellular heterogeneity. We have also developed an automated bioinformatic pipeline that ensures accurate and sensitive detection and clinical annotation of actionable mutations. The result is a comprehensive, clinically interpretable molecular profile of the patient tumor.
Materials and Methods
DNA Extraction
H&E slides are assessed for areas of high neoplastic cell content by a pathologist before macrodissection of FFPE specimens and require at least 50% tumor purity. The DNA is extracted with the QIAamp DNA FFPE Tissue Kit (Qiagen) from at least ten sections, each containing a 10 μm FFPE tumor section. The DNA quality is evaluated using the NanoDrop 2000 (Thermo Scientific) and run on an E-Gel EX Agarose Gel, 1% (Invitrogen). The DNA quantity is analyzed with the Qubit® Fluorometer (Life Technologies). The passing QC metrics to proceed to library preparation are the following: DNA yield > 200ng, OD260/280 > 1.4, and average MW > 400bp.
Library Preparation
Libraries are constructed using Agilent’s (Santa Clara, CA) 1 ug DNA sample preparation method (SureSelectXT Target Enrichment System for Illumina Paired-End Sequencing Library: Version 1.6, October 2013). Library yields were improved by a modification of the Agilent protocol to include the addition of an on-bead clean up method using a 20% PEG 8000/2.5 M NaCl solution. Solution hybridization is performed using an Agilent SureSelectXT custom designed bait. The cancer panel oligonucleotides or “baits” are 120 bp biotinylated RNA baits used to target 1.351 Mbp. Agilent’s standard SureSelectXT protocol is used for the hybridization with several modifications. The first modification affects the hybridization set-up, where a master mix is created containing “Hybridization Buffer” and “SureSelect Capture Library” in order to reduce pipette transfers leading to less evaporation at 65°C. Excessive evaporation will yield more off-target reads. The second modification is to use a plate-based method when washing the hybridized DNA. After the 16 - 24 hour incubation, the RNA bait – DNA hybrids are retrieved from the solution with streptavidin-coated magnetic beads, which are washed in bulk and distributed in a 96-well tall chimney PCR plate (Fisher Scientific). The off-target DNA is removed by washing each well 2 times with 250 μL “Wash 1 Buffer” at room temperature and 6 times with 200 μL “Wash 2 Buffer” at 65°C. The RNA is digested, leaving behind the target-captured DNA to be amplified with the addition of an 8 bp indexing barcode. The final library is then purified and quantitated by the Qubit®. The average fragment length is determined using the Agilent 2100 Bioanalyzer. All libraries are normalized to 2 nM and pooled for sequencing. DNA sequencing is performed on the Illumina (San Diego, CA) HiSeq 2500 or MiSeq, each with 150 bp, paired end sequencing.
Bioinformatic methods
The FASTQ files generated from CASAVA (version 1.8.0) are submitted to the Clinical Genomics Analytical (CGA) pipeline, developed at The Jackson Laboratory, comprised of tools (see below) to perform read quality assessment, alignment, and variant calling.
Read quality filtering and alignment
Reads are quality trimmed to remove low quality bases (Q < 30) from the 3’-end of reads, and reads with more than 30% low-quality (Q < 30) bases overall were filtered out. The resulting reads are aligned to the February 2009 release of the human reference genome (hg19) from UCSC using BWA-mem16 (http://bio-bwa.sourceforge.net/bwa.shtml).
Alignment post-processing
Duplicates are removed using Picard (http://picard.sourceforge.net) and the resulting alignments are further processed to minimize alignment artifacts (realignments around indels and base quality score recalibration from the GATK tool suite 17, 18).
SNV calling
Single nucleotide polymorphisms (SNPs) and short indels are called using Unified Genotyper tool from the GATK tool suite, and micro-indels (up to 50-bp in length) are called using Pindel19. Pindel’s true positive rate for detecting longer insertions is poor (true positive rate drops below 80% for insertions longer than 40% of the read-length19) hence, we restricted our analysis to micro-indels up to 50-bp in length.
CNV calling
Exon-level copy number variation (CNV) profiles of the tumor samples were assessed with CONTRA 20 using a normal baseline (comprised of 3 unrelated HapMap samples: NA12877, NA12878, and NA18507) as a control. The HapMap samples that went into constructing this baseline were also sequenced on the JAX-CTP™ to comparable coverage as the tumor samples, thus minimizing coverage- and technology-related biases. The statistical significance of the exon-level CNV calls was recalibrated using ConReg-R 21 to improve the false discovery rate estimates, and using these recalibrated p-values, the significance of CNV calls at the gene-level was assessed by Fisher’s method.
Quality Criteria (QC)
The passing QC metric for the clinical cancer panel is mean target coverage >300X. Variants from samples meeting this criterion were assessed for functional and clinical significance using genomic and therapeutic annotations from Genetic Variant Annotation (GVA), a molecular diagnostic tool from CollabRx (San Francisco, CA), as well as the in-house JAX Clinical Knowledgebase (CKB).
Results
Assay Description
Using the described analytical and post-analytical pipeline (Figure 1), the JAX-CTP™ is designed to identify mutations in 190 potentially clinically actionable genes from FFPE tumor specimens with an allele frequency as low as 10%. Slides are macrodissected to enrich for regions of high neoplastic cellularity followed by DNA preparation and QC. Using hybrid capture, the genes of interest are enriched and then sequenced on either the Illumina HiSeq or MiSeq sequencers. Following the generation of the high quality sequence data, SNPs, indels and CNVs are called using the JAX Clinical Genome Analytics (CGA) automated bioinformatic pipeline. Identified variants are then submitted for clinical curation using a combination of the in-house JAX Clinical Knowledgebase (CKB) and the external Genetic Variant Annotation (GVA) from CollabRx. Once clinically annotated, the variants are graded relative to their clinical utility for the specific tumor type and compiled into a clinical report to inform patient treatment.
Figure 1.
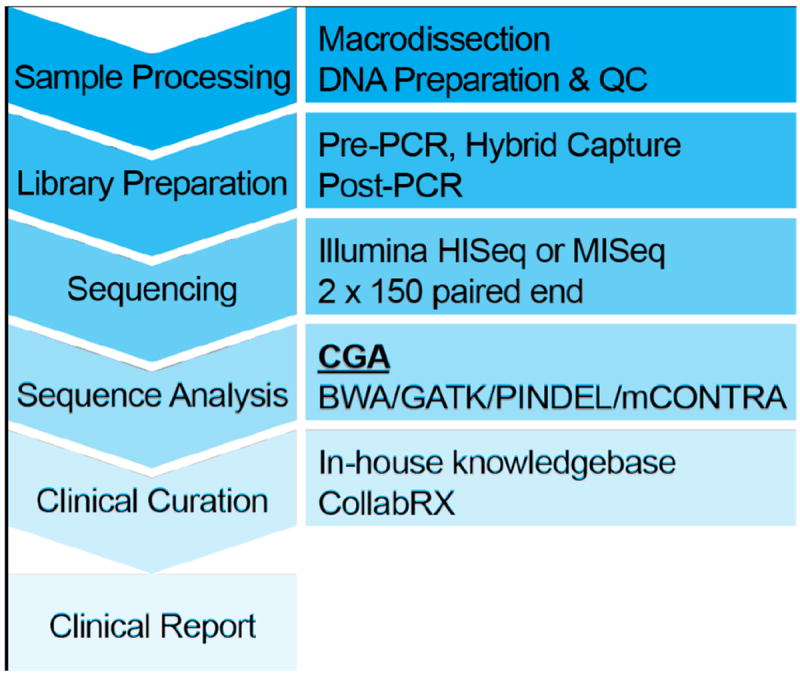
JAX-CTP™ workflow from sample receipt through clinical report generation.
Assay precision
Precision of the JAX-CTP™ assay was determined in terms of both repeatability (within-run precision) and reproducibility (between-run precision) using genomic DNA from 7 HapMap samples and 12 FFPE tumor specimens of the following tumor types: 4 colon, 2 ovarian, 2 endometrial, 1 prostate, 1 pancreatic, 1 breast, and 1 urothelial. Repeatability was assessed by concurrent replication of library preparation and simultaneous sequencing of all samples by the same technician using the same reagent lots and instruments, including the sequencer. Concordance was assessed by comparing the variant calls from the two technical replicates, with all samples meeting the acceptance threshold of at least 98% concordance.
Reproducibility was assessed by replication of the library preparation and sequencing performed in the same lab with the same instruments, but by different technicians one week apart using different reagent lots. Concordance was assessed by comparing the variant calls from the two technical replicates, with all samples meeting the acceptance threshold of at least 98% concordance. Between the assessment of repeatability and reproducibility, each sample was processed in triplicate.
Limit of detection for SNPs and indels
Given intra-tumor heterogeneity and/or the presence of small numbers of tumor cells in a specimen, one needs to be able to reliably identify mutations across a spectrum of allele frequencies. The identification of low frequency mutations is particularly challenging since it is often difficult to distinguish true variants from sequencing errors. To evaluate our variant (SNPs and small indels) detection capability at different allele frequencies, we designed a titration experiment that involved mixing of two pairs of HapMap samples (NA18507 + NA12878 and NA12882 + NA18507) such that the minor allele was present at the following frequencies: 2.5%, 3.75%, 5%, 7.5%, 10%, 20%, 40%, 45%, 46.25% and 47.5%. The variants in these HapMap samples have been well characterized by multiple projects, including 1000 Genomes 22, Illumina Platinum Genomes (http://www.illumina.com/platinumgenomes/), and the Genome in a Bottle Consortium 23. We compared the variants called by us with those from these gold-standard resources to determine the limit of detection of our assay – i.e., the allele frequency at which a majority (defined as > 95%) of the true variants are called. At an allele frequency of 10%, > 98% of the variants for both HapMap mixtures were identified (See Supplemental Table S2), thus establishing the limit of detection of our assay as 10% for SNPs and indels.
Analytical Sensitivity, Specificity, and Accuracy of SNPs and indels
At/above the limit of detection of 10% (established above), we sought to determine the assay’s ability to (a) correctly call true mutations (sensitivity or true positive rate), (b) correctly identify wild-type loci as non-variant sites (specificity or true negative rate), and (c) correctly identify mutations called by an external CLIA-certified assay (accuracy). For this we used samples with (e.g., HapMap, HorizonDx) and without (e.g., 12 FFPE clinical tumor specimens sequenced at an external CLIA lab) known and/or validated mutations.
Sensitivity for the detection of SNPs and single-base indels was determined by sequencing the HorizonDX (Cambridge, UK) quantitative multiplex FFPE reference standard, which contains known clinically actionable mutations with minor allele frequencies from 1 to 33.5% (Table 1). All variants at/above our assay’s limit of detection (10%) were detected, indicating that the sensitivity for the detection of SNPs and single-base indels is 100%. Additionally, several variants below 10% were also detected. The only variants that were not detected by our assay were the EGFR T790M* variant present at 1% and EGFR L858R variant present at 3% frequency.
Table 1.
Validation of known SNVs and micro-indels
| Gene | Variant | True Allele Frequency | Estimated Allele Frequency |
|---|---|---|---|
|
| |||
| BRAF | V600E | 10.5% | 7.0% |
|
| |||
| cKIT | D816V | 10.0% | 11.0% |
|
| |||
| EGFR | L858R* | 3.0%* | - |
|
| |||
| EGFR | T790M* | 1.0%* | - |
|
| |||
| EGFR | G719S | 24.5% | 18.0% |
|
| |||
| KRAS | G13D | 15.0% | 16.0% |
|
| |||
| KRAS | G12D | 6.0% | 4.0% |
|
| |||
| NRAS | Q61K | 12.5% | 12.0% |
|
| |||
| PIK3CA | H1047R | 17.5% | 16.0% |
|
| |||
| PIK3CA | E545K | 9.0% | 13.0% |
|
| |||
| ALK | P1543S | 33.0% | 33.0% |
|
| |||
| APC | R2714C | 33.0% | 32.0% |
|
| |||
| ARID1A | P1562fs | 33.5% | 40.0% |
|
| |||
| BRCA2 | A1689fs | 33.0% | 35.0% |
|
| |||
| EP300 | K291fs | 8.0% | 6.0% |
|
| |||
| FBXW7 | G667fs | 33.5% | 32.0% |
|
| |||
| FGFR1 | P150L | 8.5% | 7.0% |
|
| |||
| FLT3 | S985fs | 10.5% | 8.0% |
|
| |||
| FLT3 | V197A | 11.5% | 9.0% |
|
| |||
| IDH1 | S261L | 10.0% | 8.0% |
|
| |||
| MET | V237fs | 6.5% | 5.0% |
|
| |||
| MLH1 | L323M | 8.5% | 5.0% |
|
| |||
| NF1 | L626fs | 7.5% | 5.0% |
|
| |||
| NF2 | P275fs | 8.0% | 4.0% |
|
| |||
| NOTCH1 | P668S | 31.5% | 31.0% |
|
| |||
| PDGFRA | G426D | 33.5% | 29.0% |
|
| |||
| EGFR | ΔE746 - A750 | 2.0%* | 1.0% |
| 5.0% | 2.5% | ||
| 10.0% | 8.5% | ||
| 20.0% | 14.9% | ||
| 30.0% | 21.9% | ||
| 50.0% | 42.3% | ||
|
| |||
| PMS2 | P246CfsStop3$ | N/A | 40% |
N/A: not available
Variants below the assay’s detection limit will not be reported
This variant is from a colon adenocarcinoma patient sample. All variants except this are from HorizonDx
The assay’s sensitivity for the detection of longer (15-bp) indels was assessed by mixing the HorizonDx FFPE reference standard containing a known EGFR exon 19 deletion (delE746-A750) at 50% frequency with a sample confirmed to be wild-type for EGFR to generate a series of samples with varying frequencies of the deletion mutation (0%, 5%, 10%, 20%, 30%, and 50%). The deletion was detected in all sample mixtures down to 5%.
Specificity for the detection of SNPs was determined using the gold-standard HapMap sample NA12878, which was analyzed using the JAX-CTP™. By calling all variants detected at a range of allele frequencies ≥5%, the false positive rate was determined (See Supplemental Table S3). At an allele frequency of ≥5%, the specificity is ≥99.5%.
Specificity for the detection of micro-indels was assessed by a PCR-based validation of 27 unique indels of length 4-45 bp present at ≥5% allele frequency across 41 FFPE tumor specimens and not present in HapMap control samples (NA12878, NA18507) (See Supplemental Table S4). All micro-indels were successfully genotyped with PCR, indicating that the specificity for micro-indel detection is 100%.
Accuracy of the JAX-CTP™ for the detection of SNPs and indels was determined by the parallel analysis of twelve clinical tumor specimens on the JAX-CTP™ and a CLIA-certified amplicon-based sequencing assay with 93 genes in common at an external laboratory (PacificDX, Pacific Diagnostics, Irvine, CA). The concordance for the detection of SNPs and indels was > 98% (See Supplemental Table S5). Additional assessment of the accuracy of indel detection was completed using the HorizonDX FFPE Quantitative Multiplex Reference Standard, which contains the 15bp deletion EGFR delE746-A750 at an allele frequency of 2%. Analysis of this sample was repeated five times with 100% accuracy in the detection of the deletion. Furthermore, we sequenced a colon adenocarcinoma patient sample with a previously validated germ-line micro-indel on the JAX-CTP™ to further assess accuracy of indel detection. This sample contained a heterozygous 6-bp deletion of AGGGGG and 11-bp insertion of CTTCACACACA between nucleotides 736 and 741 in exon 7 of the PMS2 gene, creating a frameshift change at codon 246 resulting in truncation of the PMS2 protein. We were able to detect this complex heterozygous micro-indel in this sample at a 40% allele frequency (Table 1), as depicted in Figure 2.
Figure 2.
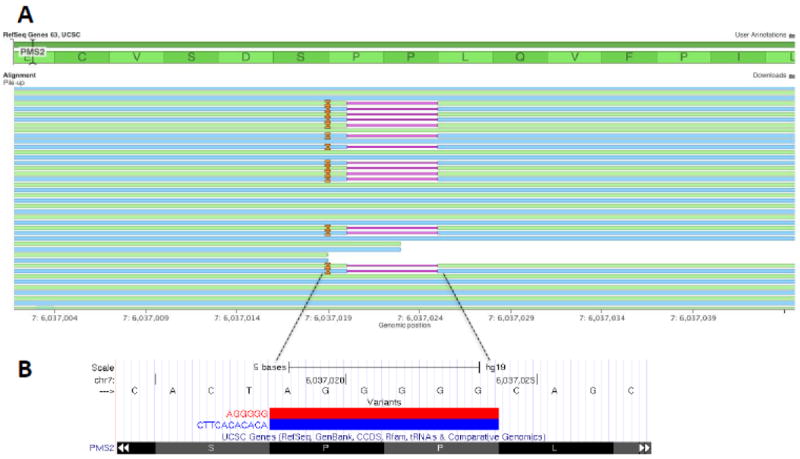
Depiction of a complex indel (a heterozygous deletion of AGGGGG and insertion of CTTCACACACA) in PMS2 gene from a colon adenocarcinoma patient sample: (A) Pile up of reads at the locus: Deletion represented by a solid pink horizontal line and Insertion by a solid orange vertical line. (B) UCSC Genome Browser track showing the two alleles at this locus.
Limit of detection for CNVs
The limit of detection for CNVs at different levels of tumor purity was assessed by mixing the DNA from two FFPE samples (1218_GES14_00876_CGACACAC_L002 – Lung Squamous cell carcinoma, SS_13_15281_GES14_00880_GACAGTGC_L002 – Colon Adenocarcinoma) with a HapMap control sample (NA12878) at different proportions to produce samples with 75%, 50%, and 25% tumor purity (See Supplemental Table S6). Additionally, these two FFPE samples were CNV profiled using the NanoString (Seattle, WA) nCounter® technology at an external laboratory. Using CNV calls from NanoString (for forty common genes) as the benchmark, we assessed the limit of detection for CNVs – i.e., the copy number at which a majority (defined as > 95%) of the true variants are called. At a tumor purity of ≥ 75%, the assay is able to detect 100% of CNVs of copy number ≥ 5, and at a tumor purity of ≥ 50%, 100% of CNVs of copy number ≥ 6 are detected. Given our tumor purity requirement of 50% for acceptable FFPE specimens, the limit of detection for CNVs is copy number 6. We were not able to assess the limit of detection for deletions, as the tested samples did not include any homozygous deletions. Samples containing homo- and heterozygous deletions are currently being sought.
Analytical Sensitivity, Specificity and Accuracy of CNVs
In addition to the two FFPE samples used for limit of detection determination above, six FFPE clinical samples that were sequenced with the JAX-CTP™ were CNV profiled using the NanoString nCounter® technology. At/above the limit of detection of copy number 6, the assay’s sensitivity, specificity, and accuracy (see section “Sensitivity, Specificity, and Accuracy of SNPs and indels” for definitions) for CNV detection were assessed using CNV calls from NanoString (for forty common genes) as the benchmark (See Supplemental Table S6). At a copy number of six and above, the sensitivity was 100% and the specificity was 99.4%. The current dataset does not allow for the assessment of sensitivity and specificity for the detection of a homozygous deletion. An example of the concordance between the two independent technologies is highlighted in Figure 3 in a lung squamous cell carcinoma specimen. This example demonstrates the concordance in the detection of a high-level amplification (> 80 copies) in CCND1, and low-intermediate amplifications (> 5 copies) in KDR, FGFR1, and WHSC1L1. (Figure 3).
Figure 3.
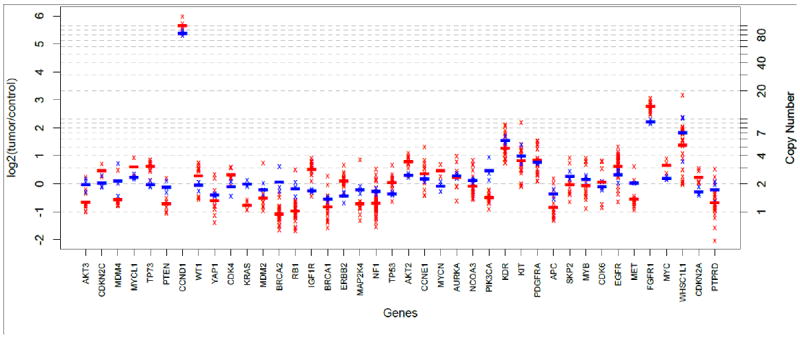
Comparison of copy-number profiles from JAX-CTP™ with NanoString for a squamous cell carcinoma patient sample. Red and blue crosses represent exon (or probe) level log ratios measured by JAX-CTP™ and NanoString respectively. Red and blue lines represent the averages of the exon (or probe) level log ratios measured by JAX-CTP™ and NanoString respectively.
Similar to SNPs and indels, accuracy for CNVs was determined by comparing calls from CTP with those from an external CLIA-certified assay for 12 FFPE clinical samples. The concordance for the detection of CNVs was > 96% (See Supplemental Table S8).
Discussion
With the continued development of targeted therapeutics for cancer, there is an expanding need for molecular diagnostic tests that provide a broad mutational spectrum. Clinical research studies continue to demonstrate the impact of mutations in multiple pathways and show how those interact to cause sensitivity or resistance to both chemotherapeutic and targeted therapies3, 24, 25. The JAX-CTP™ is designed to identify mutations in 190 potentially actionable genes across multiple cancer relevant pathways (figure 4) to facilitate the selection of the appropriate therapeutic strategy.
Figure 4.
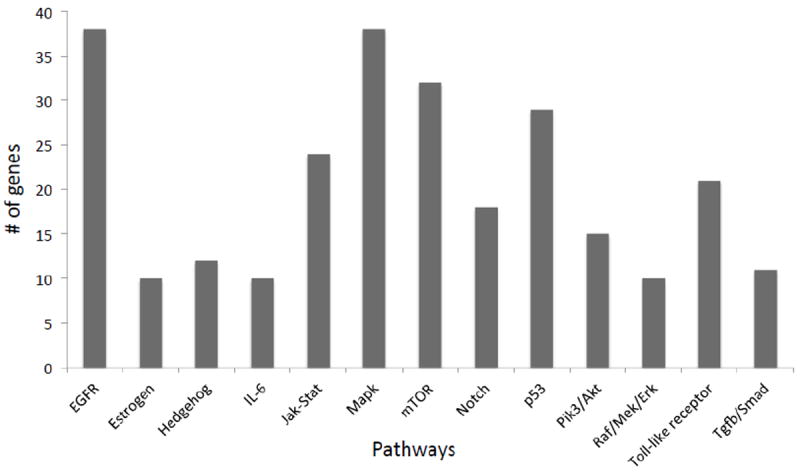
Each bar represents the number of genes (Y axis) of the JAX-CTP™ within each of the described biological pathways (X axis).
Implementation of rapidly evolving analytical molecular technologies and associated bioinformatic and curation methodologies in a clinical setting require a thoughtful validation plan to assess the accuracy, sensitivity, specificity, limit of detection, and precision of the assay. An additional complication is that there is not yet a consensus in the field on the approach to validation or the types of samples that should be utilized. As summarized in Table 2, we have described our approach to analytically validating a hybrid-capture based targeted sequencing assay of DNA from FFPE tumor specimens.
Table 2.
Summary of validation methodologies and results
| Molecular Alteration | Samples Used | Validation Parameter | QC Result |
|---|---|---|---|
|
| |||
| SNPs, Indels | Titration of 2 HAPMAP samples | Limit of Detection | ≥ 10% AF |
|
| |||
| CNVs | 2 FFPEs | Limit of Detection | ≥ 6 copies at 50% neoplastic content |
| HAPMAP (NA12878) | |||
|
| |||
| SNPs, Indels | HorizonDX | Sensitivity | 100% at ≥ 10% AF |
|
| |||
| ≥ 10 bp Indels | HorizonDX + titration with EGFR delE746-A750 | Sensitivity | 100% at 5% AF |
|
| |||
| CNVs | 8 FFPEs | Sensitivity | 100% at ≥ 6 copies |
|
| |||
| SNPs | HAPMAP NA12878 | Specificity | 99.5% at ≥ 5% AF |
|
| |||
| Indels | 41 FFPEs | Specificity | 100% at ≥ 5% AF (confirmed by PCR) |
|
| |||
| CNVs | 8 FFPEs | Specificity | 99.4% at ≥ 6 copies |
|
| |||
| SNPs, Indels | 12 FFPEs | Accuracy | 98% concordance |
| Cross-reference lab | |||
|
| |||
| CNVs | 12 FFPEs | Accuracy | 96% concordance |
| Cross-reference lab | |||
A particular challenge was to define an appropriate validation method for the assessment of specificity of microindel detection. Attaining samples that have very specific actionable mutations such as EGFR exon 19 deletion (ΔE746 - A750) is easily accomplished and is useful for the assessment of sensitivity and limit of detection, but this does not address the specificity of an assay. It is also not appropriate to generalize the detection of this one mutation to the detection of other potentially actionable mutations across the reportable range. Therefore, we independently verified 27 unique indels of length 4-45 bp present at ≥5% allele frequency across 41 FFPE tumor specimens and not present in HapMap control samples (NA12878, NA18507). There was a 100% concordance, demonstrating that the wet and dry lab methodologies we have developed achieve very high specificity in detecting this important class of mutations.
The JAX-CTP™ utilizes the latest state-of-the-art methodologies for the detection and clinical annotation of potentially actionable mutations in tumors in a clinical setting. With the recent commercialization of the Illumina HiSeq X Ten, which has made whole genome analysis significantly more affordable and the innovation and improvements in long read single molecule sequencing technologies (Pacific BioSciences, Oxford Nanopores and others), there is little doubt that the clinically relevant genome will move far beyond the protein coding sequences that are the primary focus of current targeted methodologies. Diagnostics that can accommodate the greater complexity of whole genome based clinical assays will be developed and adopted, building on the work that we and others have taken to fully vet the utility of next generation sequencing technologies in the clinic.
Supplementary Material
Highlights.
Using NGS, the JAX-CTP™ detects potentially actionable mutations in 190 genes.
The JAX-CTP™ accurately detects SNP’s, small indels and gene-level CNV’s.
The JAX-CTP™ accurately detects variants at a 10% allele frequency.
DNA is analyzed from macrodissected FFPE tumor specimens
Acknowledgments
We thank the following for their contributions to the development of the JAX-CTP™ and the preparation of this manuscript; Dr. Xiaoan Ruan, Mr. David Walton, Mr. Glen Beane, Ms. Jennifer Bourne, and Mr. Mark Wanner. Research reported in this publication was partially supported by the National Cancer Institute under institutional award number P30 CA034196.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, Kanagal-Shamanna R, Greaves WO, Medeiros LJ, Aldape KD, Luthra R. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. The Journal of molecular diagnostics : JMD. 2013;15:607–22. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DB, Dahlman KH, Knol J, Gilbert J, Puzanov I, Means-Powell J, Balko JM, Lovly CM, Murphy BA, Goff LW, Abramson VG, Crispens MA, Mayer IA, Berlin JD, Horn L, Keedy VL, Reddy NM, Arteaga CL, Sosman JA, Pao W. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. The oncologist. 2014;19:616–22. doi: 10.1634/theoncologist.2014-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran B, Brown AM, Bedard PL, Winquist E, Goss GD, Hotte SJ, Welch SA, Hirte HW, Zhang T, Stein LD, Ferretti V, Watt S, Jiao W, Ng K, Ghai S, Shaw P, Petrocelli T, Hudson TJ, Neel BG, Onetto N, Siu LL, McPherson JD, Kamel-Reid S, Dancey JE. Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. International journal of cancer Journal international du cancer. 2013;132:1547–55. doi: 10.1002/ijc.27817. [DOI] [PubMed] [Google Scholar]
- 4.Said R, Hong DS, Warneke CL, Lee JJ, Wheler JJ, Janku F, Naing A, Falchook GS, Fu S, Piha-Paul S, Tsimberidou AM, Kurzrock R. P53 mutations in advanced cancers: clinical characteristics, outcomes, and correlation between progression-free survival and bevacizumab-containing therapy. Oncotarget. 2013;4:705–14. doi: 10.18632/oncotarget.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrelli F, Coinu A, Cabiddu M, Ghilardi M, Barni S. KRAS as prognostic biomarker in metastatic colorectal cancer patients treated with bevacizumab: a pooled analysis of 12 published trials. Medical oncology. 2013;30:650. doi: 10.1007/s12032-013-0650-4. [DOI] [PubMed] [Google Scholar]
- 6.Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, Wu D, Lee MK, Dintzis S, Adey A, Liu Y, Eaton KD, Martins R, Stricker K, Margolin KA, Hoffman N, Churpek JE, Tait JF, King MC, Walsh T. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. The Journal of molecular diagnostics : JMD. 2014;16:56–67. doi: 10.1016/j.jmoldx.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, Iwanik K, Maillet A, Buell J, White E, Zhao M, Balasubramanian S, Terzic S, Richards T, Banning V, Garcia L, Mahoney K, Zwirko Z, Donahue A, Beltran H, Mosquera JM, Rubin MA, Dogan S, Hedvat CV, Berger MF, Pusztai L, Lechner M, Boshoff C, Jarosz M, Vietz C, Parker A, Miller VA, Ross JS, Curran J, Cronin MT, Stephens PJ, Lipson D, Yelensky R. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nature biotechnology. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC, Meyerson M, Gabriel SB, Garraway LA. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer discovery. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pant S, Weiner R, Marton MJ. Navigating the rapids: the development of regulated next-generation sequencing-based clinical trial assays and companion diagnostics. Frontiers in oncology. 2014;4:78. doi: 10.3389/fonc.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zutter MM, Bloom KJ, Cheng L, Hagemann IS, Kaufman JH, Krasinskas AM, Lazar AJ, Leonard DG, Lindeman NI, Moyer AM, Nikiforova MN, Nowak JA, Pfeifer JD, Sepulveda AR, Willis JE, Yohe SL. The Cancer Genomics Resource List 2014. Archives of pathology & laboratory medicine. 2014 doi: 10.5858/arpa.2014-0330-CP. [DOI] [PubMed] [Google Scholar]
- 11.Cottrell CE, Al-Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ, Lockwood CM, Hagemann IS, O’Guin SM, Burcea LC, Sawyer CS, Oschwald DM, Stratman JL, Sher DA, Johnson MR, Brown JT, Cliften PF, George B, McIntosh LD, Shrivastava S, Nguyen TT, Payton JE, Watson MA, Crosby SD, Head RD, Mitra RD, Nagarajan R, Kulkarni S, Seibert K, Virgin HWt, Milbrandt J, Pfeifer JD. Validation of a next-generation sequencing assay for clinical molecular oncology. The Journal of molecular diagnostics : JMD. 2014;16:89–105. doi: 10.1016/j.jmoldx.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh RR, Patel KP, Routbort MJ, Aldape K, Lu X, Manekia J, Abraham R, Reddy NG, Barkoh BA, Veliyathu J, Medeiros LJ, Luthra R. Clinical massively parallel next-generation sequencing analysis of 409 cancer-related genes for mutations and copy number variations in solid tumours. British journal of cancer. 2014;111:2014–23. doi: 10.1038/bjc.2014.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simen BB, Yin L, Goswami CP, Davis KO, Bajaj R, Gong JZ, Peiper SC, Johnson ES, Wang ZX. Validation of a Next-Generation-Sequencing Cancer Panel for Use in the Clinical Laboratory. Archives of pathology & laboratory medicine. 2014 doi: 10.5858/arpa.2013-0710-OA. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Chen L, Sah S, Latham GJ, Patel R, Song Q, Koeppen H, Tam R, Schleifman E, Mashhedi H, Chalasani S, Fu L, Sumiyoshi T, Raja R, Forrest W, Hampton GM, Lackner MR, Hegde P, Jia S. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted next-generation sequencing. The oncologist. 2014;19:336–43. doi: 10.1634/theoncologist.2013-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsongalis GJ, Peterson JD, de Abreu FB, Tunkey CD, Gallagher TL, Strausbaugh LD, Wells WA, Amos CI. Routine use of the Ion Torrent AmpliSeq Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clinical chemistry and laboratory medicine : CCLM / FESCC. 2014;52:707–14. doi: 10.1515/cclm-2013-0883. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL. CONTRA: copy number analysis for targeted resequencing. Bioinformatics (Oxford, England) 2012;28:1307–13. doi: 10.1093/bioinformatics/bts146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Paramita P, Choi KP, Karuturi RK. ConReg-R: Extrapolative recalibration of the empirical distribution of p-values to improve false discovery rate estimates. Biology direct. 2011;6:27. doi: 10.1186/1745-6150-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nature biotechnology. 2014;32:246–51. doi: 10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
- 24.Egas-Bejar D, Anderson PM, Agarwal R, Corrales-Medina F, Devarajan E, Huh WW, Brown RE, Subbiah V. Theranostic Profiling for Actionable Aberrations in Advanced High Risk Osteosarcoma with Aggressive Biology Reveals High Molecular Diversity: The Human Fingerprint Hypothesis. Oncoscience. 2014;1:167–79. doi: 10.18632/oncoscience.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rafii A, Touboul C, Al Thani H, Suhre K, Malek JA. Where cancer genomics should go next: a clinician’s perspective. Human molecular genetics. 2014;23:R69–R75. doi: 10.1093/hmg/ddu234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.