

Background: Group 1 mGluRs induce dendritic spine remodeling, but the underlying molecular mechanisms remain uncharacterized.
Results: α-Actinin-4 regulates dendritic protrusion dynamics and morphogenesis and is required for the receptor-induced dynamic remodeling of dendritic protrusions.
Conclusion: α-Actinin-4 is a novel molecular effector of mGluR-dependent spine remodeling.
Significance: mGluR signaling via actinins could contribute to synaptic plasticity and spine dysmorphogenesis in neurodevelopmental disorders.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII), dendritic spine, metabotropic glutamate receptor (mGluR), neuron, synaptic plasticity, actin-binding proteins, actinin, dendritic protrusion dynamics
Abstract
Dendritic spines are dynamic, actin-rich protrusions in neurons that undergo remodeling during neuronal development and activity-dependent plasticity within the central nervous system. Although group 1 metabotropic glutamate receptors (mGluRs) are critical for spine remodeling under physiopathological conditions, the molecular components linking receptor activity to structural plasticity remain unknown. Here we identify a Ca2+-sensitive actin-binding protein, α-actinin-4, as a novel group 1 mGluR-interacting partner that orchestrates spine dynamics and morphogenesis in primary neurons. Functional silencing of α-actinin-4 abolished spine elongation and turnover stimulated by group 1 mGluRs despite intact surface receptor expression and downstream ERK1/2 signaling. This function of α-actinin-4 in spine dynamics was underscored by gain-of-function phenotypes in untreated neurons. Here α-actinin-4 induced spine head enlargement, a morphological change requiring the C-terminal domain of α-actinin-4 that binds to CaMKII, an interaction we showed to be regulated by group 1 mGluR activation. Our data provide mechanistic insights into spine remodeling by metabotropic signaling and identify α-actinin-4 as a critical effector of structural plasticity within neurons.
Introduction
Dendritic spines are sites of excitatory synapses and dynamic protrusions that undergo extensive remodeling during development (1), activity-dependent synaptic plasticity (2, 3), and behavioral learning (4). The motility of dendritic protrusions is elevated early in development, during neuronal maturation (5), when there is an initial overproduction of unstable filopodia, followed by a period of massive synaptic pruning and circuitry refinement (6, 7). Although the mechanisms by which activity shapes synaptic circuits during excitatory synapse formation and elimination remain unclear, changes in spine dynamics have been shown to depend on modifications of the actin cytoskeleton (8, 9). The group 1 metabotropic glutamate receptors (mGluRs)2 mGlu1 and mGlu5 are G protein-coupled receptors (10) critical for the formation and maintenance of brain circuitry (11–13) and activity-dependent synaptic plasticity (14, 15) and are implicated in neurodevelopmental disorders including fragile X syndrome, autism (16), and schizophrenia (17). Group 1 mGluRs participate in remodeling of dendritic protrusions; their stimulation has been shown to promote spine elongation (18–20), whereas coactivation with NMDA receptors is required for the shrinkage of a subset of mushroom spines (21, 22). Although Ca2+ mobilization underlies the effect(s) of receptor activation (19, 21), the specific molecular effectors engaged by group 1 mGluRs to drive structural remodeling of dendritic protrusions remain unclear.
α-Actinin-4 is a Ca2+-sensitive member of the evolutionarily conserved family of α-actinins (α-actinin-1 to α-actinin-4), a class of actin-binding proteins (23, 24). In addition to binding to actin filaments, actinins interact with membrane receptors (25–28), signaling and adhesion proteins, and phosphoinositides (29, 30), and their positioning at the interface between the plasma membrane and cortical actin meshwork may afford a functional link between receptor activation and modification of the actin cytoskeleton. By combining genetic, pharmacological, and imaging approaches, we find that group 1 mGluRs directly bind α-actinin-4, an interaction mediated by the Calponin homology region of α-actinin-4 and the cytoplasmic tail of group 1 mGluRs. We show that α-actinin-4 is enriched at excitatory synapses, where it colocalizes with the receptors, and that its expression is necessary to support group 1 mGluR-dependent regulation of dendritic protrusion dynamics. We further demonstrate that α-actinin-4 plays a critical role in promoting protrusion motility and morphogenesis, the latter being a function dependent on the α-actinin-4 carboxyl terminus that mediates Ca2+/Calmodulin-dependent protein kinase II (CaMKII) binding and show that this interaction is regulated by group 1 mGluRs. Our findings provide molecular insights into effectors governing dendritic spine dynamics and identify novel mechanisms by which metabotropic signaling regulates structural plasticity.
Experimental Procedures
Antibodies and Reagents
The following antibodies were used: goat polyclonal anti-GAPDH (GenScript); chicken polyclonal anti-MAP2 (EnCor Biotech); the rabbit monoclonal antibodies anti-Actn4, anti-Actn1, and anti-mGlu5 (Epitomics); the rabbit polyclonal antibodies anti-GFP (Santa Cruz Biotechnology), anti-pan-mGlu1, anti-pan-mGlu5 (Alomone Labs), anti-mGlu5 (GenScript), anti-PSD95 (Zymed Laboratories Inc.), anti-Actn4 (Enzo Life Sciences), anti-phospho-ERK1/2(Thr-202/Tyr-204), anti-ERK1/2, anti-phospho-CaMKII(Thr-286/Thr-287), and anti-CaMKII (Cell Signaling Technology); and the mouse monoclonal antibodies anti-MAP2 clone AP20 (Roche Applied Science), anti-actinin clone EA-53 (Sigma-Aldrich), anti-Actn4 (Abnova), anti-mGlu1a (BD Biosciences), anti-PSD95 clone K28/43 (University of California Davis/National Institutes of Health NeuroMab Facility), and anti-His tag (Aviva Systems Biology). The mouse monoclonal anti-SV2 antibody was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD/National Institutes of Health, and maintained at the University of Iowa Department of Biology. The anti-mGlu1b antibody has been described previously (31). Antibodies against different actinin isoforms were validated for specificity by Western blot analysis of lysates from HEK293 cells transfected with Actn1-GFP, Actn2-GFP, or Actn4-GFP. Rabbit polyclonal anti-Actn4 (Enzo Life Sciences) and rabbit monoclonal anti-Actn4 (Epitomics) antibodies were used for Western blot analysis and immunofluorescence. Mouse monoclonal anti-Actn4 antibody (Abnova) was used for immunofluorescence and immunoprecipitation. (S)-3,5-dihydroxyphenylglycine (DHPG), BAY36-7620 (BAY), and 2-methyl-6-(phenylethynyl)pyridine (MPEP) were obtained from Tocris/R&D Systems.
Plasmids and siRNAs
Human α-actinin-1 fused to GFP developed by the laboratory of Carol Otey was obtained from Addgene. Human α-actinin-2 and human full-length (1–911) and mutant (1–762) α-actinin-4 were subcloned into pEGFP-N1 (Clontech). Wild type full-length cDNA (1–911) encoding human α-actinin-4 and deletion mutants (residues 1–762, 1–516, 1–296, and 296–911) generated by polymerase chain reaction were subcloned in-frame to GST into pGEX4T-2 (GE Healthcare). The rat mGlu1 fragment (residues 841–886) was subcloned in pPROEX HTa. Additional plasmids used were pDsRed-1 (Clontech) and pmaxGFP (Lonza). Individual siRNAs (Accell, Dharmacon) used to knock down rat Actn4 were 5′-UUAUUAACUUACGGAUUUA-3′ (siRNA#1) and 5′-GCCUGGUCGUUAAUGGGUU-3′ (siRNA#2). Both siRNAs selectively down-regulate Actn4. The knockdown efficiency is ≈80% for siRNA#1 (Fig. 4) and ≈ 50% for siRNA#2 (data not shown). SiRNA#1 was used in all experiments involving knockdown, and siRNA#2 was used in the experiments illustrated in Fig. 5G.
FIGURE 4.
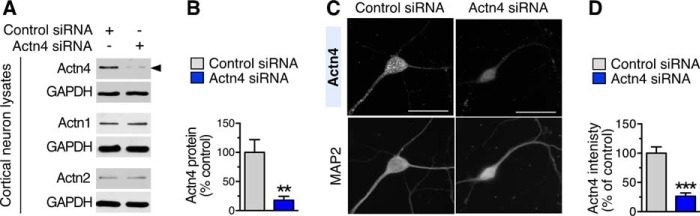
Efficiency and selectivity of Actn4 knockdown in neurons. A, representative immunoblots of lysates from DIV 12 cortical neurons treated with control or Actn4 siRNAs and probed with antibodies against Actn4, Actn1, and Actn2 and control GAPDH. Arrowhead points to Actn4. B, quantification of Actn4 protein abundance in control or Actn4 siRNA-treated neurons. Mean ± S.E.; n = 3 independent experiments; **, p < 0.01. C, representative images of DIV 12 cortical neurons treated with control or Actn4 siRNAs and labeled with anti-Actn4 and anti-MAP2 antibodies. Scale bars = 35 μm. D, quantification of Actn4 fluorescence intensity in dendritic segments from images like those in C. Mean ± S.E.; control siRNA, n = 13 neurons; Actn4 siRNA, n = 12 neurons; ***, p < 0.001.
FIGURE 5.

Actn4 supports dendritic protrusion dynamics and is required for protrusion remodeling by group 1 mGluRs. A, representative time-lapse images of dendritic segments before (Pre) and after (Post) DHPG application from neurons treated with control (top row) or Actn4 siRNAs (bottom row). Images are representative frames of ≈45-min-long imaging epochs initiated in the absence of agonist (Pre, 15 min) and continued after application of DHPG (Post). Existing protrusions that appear stable throughout epochs are marked by dots and motile protrusions by asterisks. Arrowheads point to protrusions that appear/disappear over time (turnover). Scale bar = 5 μm. B–D, quantification of time-lapse frames. B, percent change in length after DHPG treatment (Post, average of 20- to 25-min epoch) relative to basal (Pre) measured for individual protrusions over time. Control siRNA, n = 7 neurons, n = 42 protrusions; Actn4 siRNA, n = 5, n = 35; *, p < 0.05 paired t test of pre/post change for individual protrusions; ns, not significant. C, quantification of turnover ratio. Control siRNA, n = 6 neurons, n = 9 dendritic branches; Actn4 siRNA, n = 5, n = 5; *, p < 0.05; **, p < 0.01. D, quantification of motility. Control siRNA, n = 5 neurons; Actn4 siRNA, n = 5; ***p < 0.001. E, representative images of dendritic segments of fixed DIV 12 cortical neurons treated with control or Actn4 siRNA and incubated with vehicle (basal) or DHPG. Scale bar = 5 μm. F, quantification from images like those in E of mean protrusion length in matched cultures. Control siRNA basal, n = 362 protrusions; DHPG, n = 92; Actn4 siRNA (#1) basal, n = 124; DHPG, n = 293; *, p < 0.05; one-way analysis of variance. G, quantification of percent change from basal in spine length. Control siRNA basal, n = 48 neurons; DHPG, n = 35; Actn4 siRNA #1 basal, n = 28; DHPG, n = 25; Actn4 siRNA #2 basal, n = 24; DHPG, n = 10; ***, p < 0.001; one-way analysis of variance.
Immunoprecipitation and Pulldown Assays
All procedures involving animals were carried out according to protocols approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee and in accordance with the Guide for the Care and Use of Laboratory Animals by the United States Public Health Service. Dissected cerebrum from adult wild-type mice was homogenized on ice in a buffer of 10 mm Tris-HCl, 5 mm EDTA, and 320 mm sucrose (pH 7.4) with protease inhibitor mixture and sodium orthovanadate. The homogenate was centrifuged at 800 × g for 10 min, and the supernatant was spun at 10,000 × g for 15 min. The resulting pellet and supernatant were equilibrated to 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 1 mm EDTA with 1% Triton X-100 and 0.5% sodium deoxycholate. For immunoprecipitation, brain lysate was precleared by incubation with goat anti-rabbit IgG coupled to agarose beads (TrueBlot, eBioscience) for 1 h at 4 °C with constant rotation. Precleared lysate was incubated with primary antibody for 1 h on ice, and immunocomplexes were captured by incubation with anti-rabbit IgG-agarose beads for 16 h at 4 °C. Cortical neurons were rinsed with PBS and lysed in a buffer of 20 mm Tris-HCl (pH 7.4), 150 mm NaCl, and 1% Triton X-100 with protease inhibitors. For immunoprecipitation, lysates were precleared by incubation with protein G-coupled magnetic beads (Dynabeads, Life Technologies) for 10 min at 4 °C under constant rotation. Precleared lysates were incubated for 16 h at 4 °C with primary antibody bound onto magnetic beads according to the protocol of the manufacturer. Western blot analysis and detection with horseradish peroxidase-conjugated secondary antibodies was carried out according to standard protocols as described previously (31). For pulldown assays with cell lysates, preparation of GST fusion proteins and in vitro binding were carried out as described previously (31) with minor modifications. Briefly, 100 pmol of purified recombinant proteins were immobilized onto glutathione-agarose beads and incubated for ∼16 h at 4 °C with 2 mg of cell lysate, followed by wash with 1% Triton X-100 in PBS and elution with denaturing sample buffer. His-tagged proteins expressed in Escherichia coli BL21(D3) induced with 1 mm isopropylthio-galactoside for 1 h at 25 °C were purified by binding to nickel-NTA agarose (Thermo Scientific). For the in vitro binding assay, bound His-tagged proteins were washed extensively with a buffer of 50 mm NaH2PO4, 300 mm NaCl, and 20 mm imidazole (pH 8.0) and equilibrated in binding buffer of 50 mm Tris-Cl (pH 7.5), 200 mm NaCl, and 0.5% Triton X-100. GST-tagged fusion proteins (250 nm) were incubated for 2.5 h at 4 °C with bound His-tagged proteins in binding buffer. After an extensive wash with binding buffer, bound proteins were eluted with denaturing sample buffer.
Cell Culture, Transfection, RNAi, and Pharmacological Treatments
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% non-essential amino acids, 100 units/ml penicillin, and 100 μg/ml streptomycin. Transfection was carried out with Lipofectamine 2000 (Invitrogen) according to the specifications of the manufacturer. Cortical and hippocampal neurons were prepared from newborn rat pups and plated onto poly-l-lysine-coated coverslips, glass bottom dishes (MatTek Corp.), culture plates. Cultures were maintained in Neurobasal medium supplemented with 2% B27, 2 mm GlutaMax, 37 mm uridine and 27 mm 5-fluoro-2-deoxyuridine. Neurons were transfected by Nucleofection (Lonza) or calcium phosphate precipitation as described previously (31). For RNAi, siRNAs (1 μm; Accell, Dharmacon) were applied to DIV 7/8 neurons in culture medium and maintained for 4 days. For drug treatment, neurons plated on coverslips or culture dishes were rinsed twice with Krebs buffer (10 mm HEPES, 118 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 25 mm NaHCO3, 1.3 mm CaCl2, and 11 mm glucose (pH 7.4)), and DHPG was applied for 30 min at 37 °C. For live imaging experiments, neurons on glass bottom dishes were imaged at 37 °C in Neurobasal medium without phenol red supplemented with 2% B27 and bubbled with 95% O2 and 5% CO2. Cells were selected for imaging on the basis of morphology and GFP expression. Images were acquired every 15 s or 1 min under basal conditions (baseline, 10 min), followed by bath application of drugs in Neurobasal medium. Neurons displaying photodamage at the end of imaging session were not included in the quantification. Antagonists were applied alone for 15 min after baseline and then together with DHPG.
Biotinylation of Neuronal Surface Proteins
Cortical neurons (DIV 12) were incubated for 1 h in the presence of leupeptin (100 μm), and surface proteins were labeled as described previously (31). Briefly, cells were transferred onto ice, rinsed with cold phosphate buffer (pH 8.0), and incubated with 10 mm Sulfo-NHS-SS-Biotin (Pierce) for 20 min at 8 °C. Cells were washed twice with 50 mm glycine and once with 0.1% bovine serum albumin and lysed in a buffer of 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS with protease and phosphatase inhibitors. Biotin-labeled proteins were bound to NeutrAvidin beads (Pierce) for 2 h at 4 °C, washed with phosphate buffer with 1% Triton X-100, and eluted in denaturing sample buffer.
Immunolabeling and Microscopy
Cells were fixed with 4% paraformaldehyde for 15 min and incubated in blocking solution made of 5% normal donkey or goat serum for 1 h. Primary antibodies were applied in blocking solution overnight at 4 °C or for 1 h at room temperature. After four washes with PBS, appropriate fluorophore-conjugated secondary antibodies, including Alexa Fluor 488, Alexa Fluor 647, aminomethylcoumarin, or Cy3, were applied for 1 h at room temperature. After washing with PBS, coverslips were mounted on glass slides with Prolong (Invitrogen). Epifluorescence was imaged with 40× (numerical aperture 1.3) or 60× (numerical aperture 1.35) oil objectives mounted on an Olympus IX81 motorized inverted microscope equipped with a digital charge-coupled device ORCA-R2 camera (Hamamatsu). Confocal images were acquired with 40× or 60× oil objectives mounted on an Olympus Fluoview 500 confocal scanning microscope. For live imaging, images were acquired with a 60× oil objective mounted on a customized Olympus IX70 motorized inverted microscope equipped with a charge-coupled device ORCA-R2 camera.
Image Analysis
Analysis in fixed cells was performed blind with MetaMorph software (Molecular Devices). Dendritic branches ≥25 μm away from soma were analyzed. For confocal microscopy, stacks of images were acquired with 0.15-μm Z step. Two-dimensional average intensity projection images were used for spine quantification. Morphometric analysis was performed in the DsRed channel with the assumption that the fluorescent protein filled each spine homogeneously. Spine density was calculated as the number of protrusions (<10 μm) divided by branch length. Protrusion length was determined by measuring the distance between the tip and base. Spine head width was measured by drawing a line across the widest region of spines that possessed a distinguishable narrow neck and a wider head. Analysis of Actn4 fluorescence intensity was performed on images of neurons colabeled with MAP2 to visualize somatodendritic compartments. Images were thresholded using the same settings between different conditions. Dendritic segments were drawn manually in the MAP2 channel to define regions of interest for quantification of integrated pixel intensity relative to region of interest area. For live imaging, consecutive 1/min frames were used for quantification. Change in length was determined from the average length of individual protrusions before stimulation (the last 5 min of baseline normalized to the entire baseline) and at 20–25 min post-DHPG. Motility was calculated as the sum of the absolute value of change in spine length from frame to frame divided by number of frames. Turnover was determined as the sum of lost and gained protrusions divided by twice the total number of protrusions (18). New protrusions extending ≥0.55 μm from the dendritic shaft were considered “gained.” For display purposes, time-lapse videos were converted to 640 × 480 pixel resolution and captioned using Windows Movie Maker 6.
Electrophysiology
Whole-cell patch clamp recordings were carried out in DIV 11/13 hippocampal and cortical neurons voltage-clamped at −60 mV using patch-type pipette electrodes (∼3–4 MΩ) containing 131 mm Cs-gluconate, 8 mm NaCl, 1 mmCaCl2, 10 mm EGTA, 10 mm glucose, and 10 mm HEPES (pH 7.4); osmolarity, 285–292 mmol/kg. mEPSCs were recorded for 10–15 min at 32 °C in extracellular artificial cerebrospinal solution containing (in mm): 124 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 glucose. Picrotoxin (100 μm) and tetrodotoxin (0.5 μm) were added to this solution to block inhibitory transmission and action potentials, respectively. mEPSCs were filtered at 2.4 kHz, acquired at 5 kHz, and analyzed using a custom-made software written in Igor Pro 4.09A (Wavemetrics, Inc., Lake Oswego, OR). In all cases, the experimenter was blind to conditions during data acquisition and analysis.
Statistics
Data are presented as means ± S.E. Statistical significance was determined by Student's t test or analysis of variance, with p < 0.05 considered significant.
Results
The mGlu1 Cytoplasmic Tail Interacts with the α-Actinin-4 Calponin Homology Region
Modifications of the actin cytoskeleton by actin-binding proteins provide the principal driving force underlying rapid morphological changes of dendritic protrusions during development and in response to activity (8). We investigated whether group 1 mGluRs function via the actin-binding protein α-actinin-4 (Actn4) to regulate protrusion dynamics. We have reported previously that mGlu1 is present in a complex with Actn4, an association detected in heterologous cells and preserved in the brain (31). The GST-Actn4 fusion protein interacts in vitro with myc-mGlu1 expressed in HEK293 cells (Fig. 1, A and B). Therefore, we employed in vitro pulldown assays to identify the domains that mediate Actn4 interaction with mGlu1. To this end, we used Actn4 deletion mutants fused to GST and examined their capacity to pull down myc-mGlu1 expressed in HEK293 cells (Fig. 1, A and B). Actn4 is composed of two amino-terminal Calponin homology (CH) domains that bind F-actin, an extended rod-like region formed by Spectrin repeats, and a carboxy-terminal Calmodulin-like domain (CaM) that confers Ca2+ affinity (Fig. 1A). Deletion of the Actn4 amino-terminal region (residues 1–296) encompassing both CH domains drastically decreased binding, whereas residues 1–296 fused to GST strongly bound mGlu1 (Fig. 1B). Deletion of the CaM domain attenuated binding but not when deleted in conjunction with the last two Spectrin repeats, nor was the CaM domain sufficient to promote efficient interaction in the absence of the CH region (Fig. 1B). Together, these findings indicate that the Actn4 CH domain-containing region is largely responsible for binding to mGlu1 in vitro. The distal CaM domain may contribute to support the interaction, potentially by facilitating intra- and/or intermolecular folding of the binding site(s). We hypothesized that Actn4 could associate with mGlu1 by interacting with the carboxyl tail of the receptor that is exposed to the intracellular milieu. To test this possibility, we expressed, in HEK293 cells, chimeric constructs harboring the tail of mGlu1a (residues 841–1199) or mGlu1b (residues 841–906) fused to a truncated form of vesicular stomatitis virus glycoprotein G (VSVG) (Fig. 1, C and D) (32) and performed pulldown assays with GST-Actn4. GST-Actn4, but not GST alone, pulled down VSVG-mGlu1a-Tail (Fig. 1D) or VSVG-mGlu1b-Tail (Fig. 1C), suggesting that a conserved region (CR, residues 841–886) in the tail of both mGlu1 isoforms could mediate the interaction with Actn4. To determine whether Actn4 binds the CR region of mGlu1 tail directly, we performed in vitro binding assays with purified GST-Actn4, GST-Actn41–296 harboring the CH domain, and the His-tagged mGlu1 CR fragment (Fig. 1E) and found that His-CR precipitated GST-Actn4 (Fig. 1E, left panel) and GST-Actn41–296 (Fig. 1E, right panel) but not control GST. Therefore, different mGlu1 isoforms bind the Actn4 amino-terminal domain, an interaction mediated, at least in part, via a conserved region present in the intracellular tail of the receptor. The group 1 receptor mGlu5 possesses a high degree of homology with mGlu1 (33). In particular, the membrane-proximal tail region harboring the CR domain is highly conserved between mGlu1 and mGlu5, with 95.8% identity of the first 24 residues (64.3% identity and 83.3% similarity in first 42 overlapping residues). We previously reported the formation of a complex between Actn4 and mGlu1 in vivo in both the cortex and cerebellum (31). To examine whether Actn4 also interacts with mGlu5, we performed coimmunoprecipitation from cortical lysates and found that an anti-mGlu5 antibody, but not an unrelated IgG, coprecipitated Actn4 (Fig. 1F), attesting to an interaction between native proteins. Together, these findings indicate that Actn4 directly connects group 1 mGluRs to the actin cytoskeleton.
FIGURE 1.

Group 1 mGluRs interact with Actn4. A, schematic of wild type Actn4 and deletion mutants fused to GST used in a pulldown assay with mGlu1b. B, the Actn4 CH region is necessary and sufficient for mGlu1b binding. Shown are representative immunoblots (IB)of a pulldown assay between GST-Actn4 fusion proteins from A and lysates of HEK293 cells transfected with mGlu1b, probed with anti-mGlu1b, and stained with Ponceau. C and D, the mGlu1 cytoplasmic tail is sufficient for Actn4 binding. Shown is a schematic of chimeric constructs of VSVG protein fused to mGlu1b or mGlu1a carboxyl tails. Yellow highlights the membrane-proximal region of the tail that is conserved between receptor isoforms (CR region, see below). Also shown are representative immunoblots of input (20 μg) and pulldown (input lysate, 2 mg) between GST-Actn4 and VSVG-mGlu1a-Tail (D) and VSVG-mGlu1b-Tail (C) probed with anti-mGlu1a or anti-mGlu1b antibody, respectively. The estimated pulldown fraction in vitro is ∼1% of input receptors. E, Actn4 interacts directly with the CR region of the mGlu1 intracellular tail. Shown is a schematic of the CR construct and immunoblots illustrating in vitro binding of a purified His-tagged CR fragment and GST-tagged full-length Actn4 (GST-Actn4, left panel) or Actn4 CH region (GST-Actn41–296, right panel). Shown are bound and unbound (14% of input GST-tagged proteins) fractions probed with anti-GST and anti-His tag antibodies. The arrow points to full-length GST-Actn4 (left panel). Lower molecular mass bands were derived from partial protein fragmentation. The estimated relative bound fractions are 40% and 26% of input for GST-Actn4 and GST-Actn41–296, respectively. F, Actn4 coprecipitates with mGlu5 in the brain. Shown are representative immunoblots of input lysate and precipitated proteins (from 2.5 mg of input lysate) probed with anti-Actn4 (input, 10 μg) and anti-mGlu5 (input, 40 μg). The estimated immunoprecipitation (IP) is 5% and 3% of input for mGlu5 and Actn4, respectively.
Actn4 Is Enriched at Synaptic Sites and Colocalizes with Group 1 mGluRs
Actn4 is broadly expressed in the brain, as shown by Western blot with Actn4-specific antibodies (Fig. 2A) of hippocampus, cortex, and cerebellum lysates (Fig. 2B). To gain insight into Actn4 function(s) in the brain, we examined its expression and subcellular localization in primary neurons. We found that, in primary cortical neurons, Actn4 is already relatively abundant by DIV 7, a developmental time of synaptogenesis (34, 35), reaches a plateau by DIV 14, and remains elevated in mature neurons (Fig. 2, C and D). To closely examine Actn4 cellular localization, we performed immunolabeling of cortical (data not shown) and hippocampal neurons. Actn4 immunoreactivity is present in MAP2-positive dendrites and strikingly prominent at excitatory synapses, where it colocalizes with PSD95 apposed to the synaptic vesicle marker SV2 (Fig. 3A), as illustrated by line scan analysis of fluorescence (Fig. 3C), and with mGlu1b (Fig. 3, B and D) or mGlu5 clusters (data not shown). Overall, these findings indicate that Actn4 is enriched at excitatory synapses and colocalizes with group 1 mGluRs.
FIGURE 2.
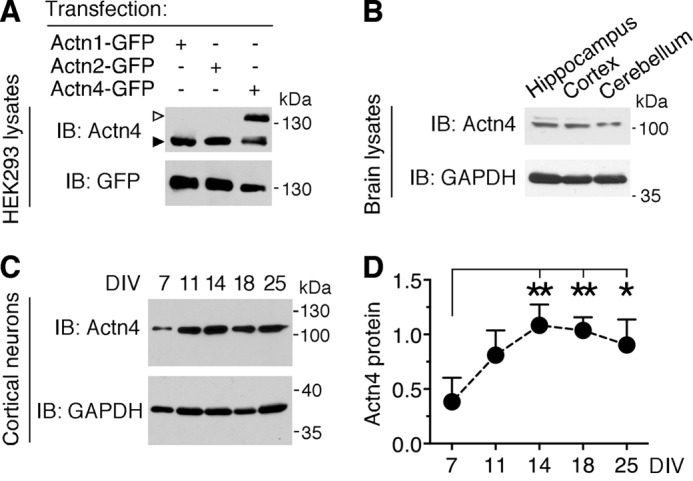
Actn4 is broadly expressed in the brain and in primary neurons at different stages of maturation. A, specificity of anti-Actn4 antibody. Shown are representative immunoblots (IB) of lysates from HEK293 cells transfected with Actn1-GFP, Actn2-GFP, or Actn4-GFP probed with anti-Actn4 and anti-GFP antibodies. Empty arrowhead, Actn4-GFP; black arrowhead, endogenous Actn4 present in HEK293 cells. B, representative immunoblots of lysates probed with anti-Actn4 and anti-GAPDH demonstrating Actn4 expression in adult mouse hippocampus, cortex, and cerebellum. C, developmental expression of Actn4 in primary cortical neurons. Shown are representative immunoblots of lysates from neurons at the indicated DIV probed with anti-Actn4 and anti-GAPDH. D, quantification of Actn4 expression relative to the loading control in cortical neurons at different ages in vitro. Mean ± S.E.; n = 3 independent experiments; *, p < 0.05, **, p < 0.01; one way analysis of variance.
FIGURE 3.
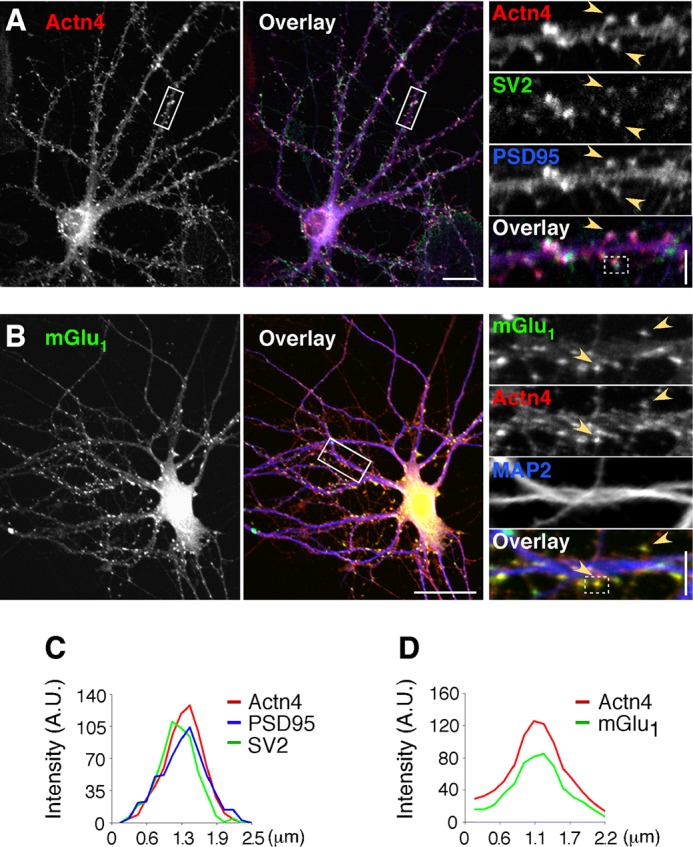
Actn4 is expressed at synapses and colocalizes with mGlu1. A, Actn4 localizes to synapses in hippocampal neurons. Shown are representative confocal images of cells labeled with anti-Actn4, anti-PSD95, and anti-SV2. Left panel, Actn4 alone. Center panel, overlay with PSD95 and SV2. Colocalization appears in white. Scale bar = 20 μm. Right panel, magnified boxed regions. Arrowheads point to areas of colocalization. Scale bar = 5 μm. B, Actn4 colocalizes with mGlu1. Shown are representative images of cells labeled with anti-mGlu1, anti-Actn4, and anti-MAP2. Left panel, mGlu1 alone. Center panel, overlay with Actn4 and MAP2. Scale bar = 35 μm. Right panel, arrowheads point to mGlu1 puncta colocalized with Actn4. Scale bar = 5 μm. C, line scan analysis of fluorescence intensity across a boxed region from the right panel in A. A.U., fluorescence intensity in arbitrary units. D, line scan of a boxed region from the right panel in B.
Actn4 Is Required for Group 1 mGluR-induced Remodeling of Dendritic Protrusions and Regulates Protrusion Dynamics
Activation of group 1 mGluRs has been shown previously to promote dendritic protrusion elongation in both primary neurons and brain slices (18–20). We hypothesized that, by virtue of the ability to bind to group 1 mGluRs, enrichment at synaptic sites, and early expression during neuronal maturation, Actn4 could play a role in group 1 mGluR-dependent protrusion remodeling. We first established conditions for selective and efficient Actn4 down-regulation in neurons using Actn4-specific siRNAs, as determined by Western blot analysis (Fig. 4, A and B) and quantitative analysis of fluorescence of labeled Actn4 (Fig. 4, C and D). Next we examined protrusion dynamics by time-lapse imaging in GFP-transfected cortical neurons (DIV 12–13) treated with Actn4-specific or control siRNAs. By measuring changes in protrusion length over an ∼45-min time period, we found that, in control siRNA-treated cells, application of the selective group 1 mGluR agonist DHPG (50 μm) induced a small but significant increase in the mean length of individual protrusions within 20–25 min (Fig. 5, A and B; supplemental Movie 1), an effect concordant with previous findings by others in primary hippocampal neurons (19, 20) and slice cultures (19) as well as neocortical pyramidal neurons in acute slice preparations (18). The effect of DHPG on protrusion length was blocked by coapplication of either the mGlu1-selective antagonist BAY 36-7620 (10 μm) or the mGlu5 antagonist MPEP (10 μm) (percent change of baseline; means ± S.E.; pre, 107.3 ± 5.639; DHPG with BAY 36-7620, 100.3 ± 3.730; n = 4 neurons; Pre, 104.1 ± 5.434; DHPG with MPEP, 94.03 ± 2.035; n = 5 neurons; p > 0.05; data not shown), suggesting that both mGlu1 and mGlu5 contribute to the regulation of protrusion dynamics. In contrast, DHPG did not significantly increase individual protrusion length over time in neurons treated with Actn4 siRNA (Fig. 5, A and B; supplemental Movie 2). To independently validate this observation, we measured net protrusion length in fixed preparations of neurons treated with one of two independent siRNAs targeting Actn4 or control siRNA and incubated with either vehicle (basal) or DHPG for 30 min (Fig. 5, E–G). In agreement with live imaging analysis, in control siRNA-treated neurons, overall protrusions length was increased by DHPG compared with basal but was not altered significantly in neurons treated with Actn4 siRNA (Fig. 5, F and G).
We further analyzed dynamic events during the imaging epoch to measure protrusion formation and retraction (turnover) and overall motility. In neurons treated with control siRNA, the number of protrusions that emerged from dendrites and rapidly disappeared transiently increased during the initial 10 min of DHPG application (Fig. 5A; supplemental Movie 1), as indicated by an increased turnover ratio (Fig. 5C), although the effect of DHPG on turnover was not accompanied by a net change in protrusion density by 30 min (data not shown). In contrast, knockdown of Actn4 decreased significantly basal turnover and blocked enhancement by DHPG (Fig. 5, A and C; supplemental Movie 2). Overall, under basal conditions, control neurons displayed highly motile protrusions undergoing repeated rounds of elongation and retraction (Fig. 5, A and D; supplemental Movie 1), reflecting rapid, dynamic morphological changes occurring at times of synapse formation (36, 37). Application of DHPG did not alter net protrusion motility in control neurons (Fig. 5D). Similarly, incubation with BAY 36-7620 or MPEP did not affect motility (micrometers/minute; means ± S.E.; pre, 0.337 ± 0.034; BAY 36-7620, 0.298 ± 0.038; n = 4 neurons; pre 0.316 ± 0.052; MPEP, 0.256 ± 0.030; n = 5 neurons; p > 0.05; data not shown). Remarkably, motility was reduced drastically in neurons treated with Actn4 siRNA (Fig. 5, A and D; supplemental Movie 2) and was not affected by stimulation with DHPG. Analysis of the impact of Actn4 siRNAs on protrusion properties in fixed, GFP-transfected cortical neurons (DIV 12–13) showed that Actn4 down-regulation did not alter protrusion density (protrusions per micrometer; means ± S.E. from three experiments; control siRNA, 0.37 ± 0.02; n = 38 neurons; Actn4 siRNA, 0.38 ± 0.03; n = 27; p = 0.71). These findings indicate that Actn4 plays a critical function in promoting protrusion motility and participates in supporting protrusion elongation and turnover induced by group 1 mGluRs.
Actinins have been shown previously to play a role in the regulation of membrane protein surface expression, including L-type Ca2+ channels (25) and the AMPA receptor GluA1 subunit (38). To test whether Actn4 knockdown might impair group 1 mGluR agonist-dependent activity, potentially by perturbing receptor trafficking, we undertook two different approaches. First, we examined receptor expression at the plasma membrane by measuring the abundance of biotin-labeled mGlu1 and mGlu5 at the cell surface. Immunoblot analysis of biotinylated and total mGlu1 and mGlu5 in lysates from control and Actn4 siRNA-treated neurons did not reveal significant differences in receptor surface expression (Fig. 6, A and B). Next we examined group 1 mGluR-dependent activation of ERK-MAPK (39), a pathway involved in de novo spine formation (40). Application of DHPG (5 min) increased ERK1/2 phosphorylation with similar efficiency in control and Actn4 siRNA-treated neurons (Fig. 6, C and D), as determined by immunoblot analysis with anti-phospho-ERK1/2 (Thr-202/Tyr-204) and anti-ERK1/2 antibodies. These findings strongly suggest that impairment in DHPG-induced protrusion remodeling does not arise from group 1 mGluR misexpression upon Actn4 knockdown. Finally, to examine whether Actn4 down-regulation might affect the formation/stabilization of functional synapses, we recorded AMPA receptor-mediated mEPSCs as a measure of basal glutamatergic transmission. No significant differences in frequency or amplitude of mEPSCs between control and Actn4 siRNA-treated neurons were observed (Fig. 6E), suggesting that knockdown of Actn4 does not alter basal synaptic transmission and synapse formation in vitro.
FIGURE 6.

Actn4 knockdown does not impair group 1 mGluR expression, signaling, or basal excitatory transmission. A, representative immunoblots (IB) of mGlu1 or mGlu5 from biotin-labeled (surface; mGlu1, 850 μg; mGlu5, 450 μg input lysate) or total lysates (30 μg) of cortical neurons treated with control or Actn4 siRNA. Arrows point to bands corresponding to the receptor dimer and monomer. B, quantification from images like those in A of the mGlu1 (left panel) and mGlu5 (right panel) surface/total ratio in control or Actn4 siRNA-treated neurons. Means ± S.E.; n = 4 experiments; p > 0.05. ns, not significant. C, Actn4 knockdown does not impair DHPG-induced ERK-MAPK activation. Shown are representative immunoblots of DIV 12 cortical neuron extracts probed with anti-phospho-ERK1/2(Thr-202/Tyr-204) and anti-ERK1/2. D, quantification of ERK2 phosphorylation expressed as the ratio to total ERK2 from images like those shown in C. n = 3 independent experiments; *, p < 0.05, **, p < 0.01. E, representative mEPSC traces (top panel) and summary data (bottom panel) showing that Actn4 knockdown does not affect basal AMPA receptor-mediated transmission in hippocampal neurons (DIV 12/13) treated with control or Actn4 siRNA. n = 3 independent experiments; control siRNA, n = 18 cells; Actn4 siRNA, n = 23 cells. Calibration bar = 30 pA, 300 ms.
Actn4 Interacts with CaMKII to Regulate Dendritic Protrusion Morphogenesis
Actn4 has been shown previously to bind α and β subunits of CaMKII (41). CaMKIIβ governs actin cytoskeleton dynamics in dendritic spines (42) and regulates protrusion motility and spine morphogenesis (43, 44). We reasoned that Actn4 might cooperate with CaMKIIβ in controlling protrusion dynamics and that this functional association could be regulated by group 1 mGluRs because CaMKII interaction with actinins is antagonized by Ca2+/Calmodulin (45) or Ca2+ (28). To explore this hypothesis, we first examined whether activation of group 1 mGluRs could induce CaMKIIβ autophosphorylation at Thr-287, triggered by Ca2+/Calmodulin binding. Group 1 mGluRs have been shown previously to induce autophosphorylation of CaMKIIα in the striatum (46) and hippocampus (47), but their impact on CaMKIIβ is not known. Cortical neurons (DIV 13–17) were treated with DHPG (50 μm) or vehicle for different times, and CaMKIIβ autophosphorylation was assessed by Western blot with anti-phospho-CaMKII antibodies that recognize Thr-287 in CaMKIIβ. Application of DHPG rapidly increased CaMKIIβ phosphorylation, which reached significance at 5 min and was still elevated after 15 min but decreased to near basal levels by 30 min (Fig. 7, A and B). Next we tested whether group 1 mGluR activation affects CaMKII interaction with Actn4. To this end, we immunoprecipitated Actn4 from cortical neurons treated with DHPG (50 μm, 5 min) or control medium and assessed CaMKII binding by Western blot analysis. As shown in Fig. 7C, 5-min stimulation with DHPG dramatically decreased the abundance of CaMKII (α and β subunits) that coprecipitated with Actn4 (Fig. 7, C and D). Together, these findings indicate that group 1 mGluR stimulation can rapidly induce CaMKIIβ activation and dissociation of the CaMKII-Actn4 complex and further suggest that Actn4 may act in concert with CaMKII to regulate spine morphogenesis.
FIGURE 7.
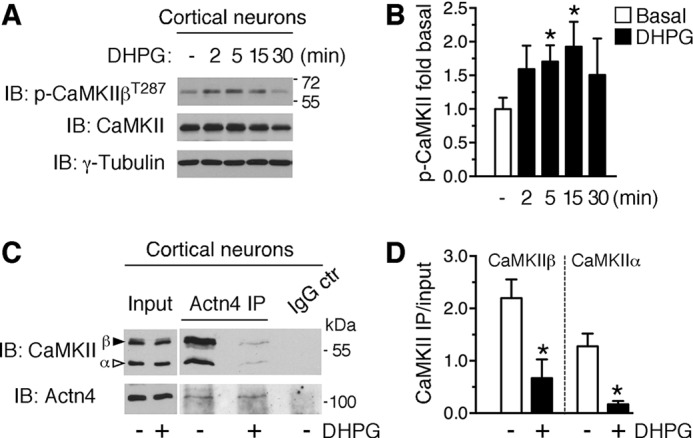
Group 1 mGluRs regulate CaMKIIβ Thr-287 autophosphorylation and CaMKII binding to actinin-4. A, DHPG induces CaMKIIβ autophosphorylation at Thr-287 in cortical neurons. Shown are representative immunoblots (IB) of extracts from cortical neurons treated with DHPG for the indicated times and probed with anti-phospho-CaMKII, anti-CaMKII, and anti-γ-tubulin. B, quantification of CaMKIIβ phosphorylation as the ratio to γ-tubulin from images like those in A, means ± S.E.; n = 4–5/group; *, p < 0.05. C, CaMKII coprecipitation with Actn4 from cortical neurons (DIV 7–13) treated with DHPG (5 min) or control medium. Shown are representative immunoblots illustrating total lysate (30 μg) and precipitated protein (1 mg input lysate) probed with anti-CaMKII and anti-Actn4. IP, immunoprecipitation. D, quantification from images like those in C of the amount of CaMKIIβ or CaMKIIα protein coprecipitated by Actn4 from neurons treated with control medium (basal) or DHPG. Mean ± S.E.; n = 3 independent experiments; *, p < 0.05. Efficiency of Actn4 immunoprecipitation per treatment: immunoprecipitation versus input ratio; basal, 1.15 ± 0.15; DHPG, 1.03 ± 0.26; n = 3; p = 0.727.
To gain insight into the impact of Actn4 on spine morphogenesis, we performed gain-of-function experiments by transfecting Actn4-GFP together with DsRed or DsRed alone in DIV 8 cortical neurons and examined dendritic protrusions at DIV 13. In control neurons, we observed both filopodia and spines that mostly displayed immature features (Fig. 8A, top panel) with a small head and long neck (48). Like endogenous Actn4, Actn4-GFP concentrated in spine heads (Fig. 8A, center panels) that appeared strikingly larger than in control neurons (Fig. 8, A and C). Moreover, Actn4-GFP overexpression modestly increased spine length and density (Fig. 8C) compared with the control. Actn4 interaction with CaMKIIα/β is mediated via its carboxyl-terminal region (41), termed CaM (Fig. 8B), which also harbors EF hands mediating Ca2+ binding (30). We hypothesized that the CaM domain may contribute to Actn4 function in spine morphogenesis by supporting interaction with CaMKII. To examine this possibility, we generated a GFP-tagged Actn4 truncation mutant, Actn41–762-GFP, and expressed it together with DsRed in DIV 8 neurons (Fig. 8A, bottom panels). In contrast to wild-type Actn4, overexpression of mutant Actn41–762-GFP did not significantly increase spine head size (Fig. 8, A and C) but markedly increased protrusion length and density (Fig. 8, A and C), together resulting in an overabundance of long protrusions with an immature, filopodium-like morphology. An overabundance of immature protrusions suggests delays and/or abnormalities in spine morphogenesis induced by Actn4 mutants lacking the CaM domain. To examine whether protrusion structural abnormalities may result in functional synapses, we measured mEPSCs in cortical neurons expressing either Actn4-GFP or Actn41-762-GFP. Although expression of Actn4-GFP did not significantly affect mEPSC amplitude or frequency (control versus Actn4-GFP; amplitude, p = 0.792; frequency, p = 0.569; n = 19 and 21 cells, respectively; data not shown), expression of the Actn41-762-GFP mutant significantly reduced the frequency but not the amplitude of mEPSCs (Fig. 8D), consistent with an overabundance of immature protrusions in these neurons (49). Collectively, these findings provide evidence for a role of Actn4 in the regulation of spine morphogenesis and suggest that this function involves interaction with CaMKII.
FIGURE 8.

Actn4 regulates spine morphogenesis. A, representative Z-projections of confocal image stacks displaying dendritic segments from cortical neurons transfected with the indicated plasmids. Scale bar = 5 μm. B, schematic of native Actn4 homodimers. ABD, actin-binding domain. C, quantification from images like those in A of spine head width (left panel), protrusion length (center panel), and density (right panel). DsRed, n = 29 cells; Actn4-GFP, n = 23; Actn41-762-GFP, n = 21; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. D, representative traces (left panel) and summary data (right panel) showing a reduction in the frequency of mEPSC recorded from cortical neurons transfected with Actn41-762-GFP compared with control neurons transfected with GFP (p = 0.00728, n = 10 cells/group). Calibration bar = 30 pA, 500 ms. E, working model of potential mechanisms underlying group 1 mGluR-dependent spine remodeling via the Actn4/CaMKII/F-actin interaction network. Actn4 enables the maturation of dendritic protrusions in conjunction with CaMKII by supporting CaMKII recruitment to F-actin. Activation of group 1 mGluR uncouples CaMKII from Actn4, potentially releasing it from F-actin, thereby enhancing spine dynamics by enabling remodeling of the actin cytoskeleton.
Discussion
Our studies identify Actn4 as a novel group 1 mGluR interacting partner and a critical cell-autonomous effector of protrusion dynamics that is necessary for group 1 mGluR-dependent spine remodeling. We find that group 1 mGluR activation enhances protrusion dynamics by inducing elongation and turnover. These effects are abrogated by Actn4 silencing in the absence of perturbations in receptor expression at the neuronal surface or signaling capacity. In addition, we discover a role for Actn4 as a central player in processes underlying protrusion dynamics. We show that Actn4 down-regulation reduces protrusion motility and turnover and further demonstrate that Actn4 participates in spine morphogenesis. Actn4 overexpression increases protrusion length and induces profound enlargement of spine heads. Mechanistically, our results provide evidence that Actn4 morphogenic function(s) are linked to its capacity to bind CaMKII and that this interaction is regulated by group 1 mGluRs.
Group 1 mGluRs play a critical function in circuitry tuning and remodeling of dendritic spines and synapses during development. In the barrel cortex, mGlu5 activation participates in sensory map formation and spinogenesis (11, 13), whereas mGlu1 is required for elimination of redundant synapses during postnatal development of the cerebellar cortex (12, 50). Abnormal group 1 mGluR activity is implicated in the pathology of fragile X syndrome, the most prevalent cause of inherited intellectual disability and autism (51), a condition characterized by spine dysmorphogenesis (52). Notably, genetic or pharmacological reduction of mGlu5 activity has been shown to ameliorate this pathological feature (53, 54). Despite emerging evidence that metabotropic glutamatergic signaling is critical to activity-dependent refinement of spine properties under physiopathological conditions, at present little is known about the effector mechanisms underlying group 1 mGluR capacity to regulate spine remodeling. Here we provide evidence indicating that Actn4 is a central player in this process.
Multiple actinin isoforms are expressed in the brain, including Actn1 (highly homologous to Actn4) and Actn2 (55). Mass spectrometry studies indicate that actinins are components of the postsynaptic density (PSD) (56, 57), underscoring their presence at postsynaptic sites. Consistent with biochemical findings, Actn2, for which ultrastructural information is available, has been shown to localize in apposition to the PSD and the spine apparatus within dendritic spines (58). Group 1 mGluRs localize to perisynaptic sites of mature spine synapses, whereby both mGlu1a/b and mGlu5 have been shown to localize at the periphery of the PSD at excitatory synapses (59, 60). Colocalization of group 1 mGluRs and actinin-4 within dendritic spines lends support for a potential coordinate action in promoting spine remodeling.
Native actinins form homodimers that bind F-actin within the cortical actin meshwork and can be transiently tethered to plasma membrane microdomains via interaction with phosphoinositides (29). Therefore, Actn4 may facilitate the functional interaction of membrane mGluRs with cortical F-actin and signaling proteins that contribute to the modification of the actin cytoskeleton. Chiefly among them is CaMKII, a central regulator of functional and structural plasticity in the central nervous system. Actinins bind CaMKII, an interaction mediated by the kinase regulatory segment and the CaM domain of actinins (45). CaMKIIβ bundles F-actin (43, 61), governs actin cytoskeleton dynamics in dendritic spines (42), and has been shown to regulate dendritic protrusion motility and morphogenesis (43, 44). We show that the capacity of Actn4 to induce spine enlargement is abrogated in the absence of an intact carboxyl terminus that harbors the CaM domain, strongly suggesting a potential requirement for interaction with CaMKII. In fact, overexpression of mutant Actn4 lacking the CaM domain induces the formation of overabundant, morphologically immature dendritic protrusions that is functionally accompanied by reduced basal synaptic transmission. These observations are further buttressed by the findings that native Actn4 binds CaMKII α and β subunits in neurons and, importantly, that group 1 mGluR activation leads to CaMKIIβ phosphorylation and weakening of the Actn4/CaMKII interaction. We propose a model in which group 1 mGluRs activate Ca2+/Calmodulin, which binds and activates CaMKII, concomitantly displacing Actn4 by competing for binding to the regulatory segment (Fig. 8E). Interaction of CaMKIIβ with Actn4 may cooperate in bundling of F-actin within spines (62) and regulate the cytoskeleton suprastructure to promote spine head enlargement. In immature motile protrusions, dissociation of this complex by group 1 mGluR activation may transiently decrease the proportion of CaMKIIβ bound to F-actin and enable protrusive motility and elongation. Actinins participate in the targeting of CaMKIIα to F-actin (63), and CaMKIIβ knockdown reduces dendritic protrusion motility (43), similar to Actn4 down-regulation. Importantly, CaMKII-mediated effects on spine morphogenesis depend on its interaction with F-actin and not on its enzymatic activity (42).
The motility of dendritic protrusions in developing neurons promotes synaptogenesis by facilitating cell-cell interactions (5, 64, 65) and regulates spine morphogenesis through changes in spine head morphology (62, 66). Although Actn4 is critical for protrusion motility, we found that synaptic transmission is not significantly affected by its down-regulation, indicating that synapses were formed normally. Other actinin isoforms could compensate for Actn4 functions in spinogenesis. Indeed, expression of Actn2, which competes with Ca2+/Calmodulin for binding to the NR1 subunit of NMDA receptors (67, 68), has been shown to promote increased density of filopodia and immature thin spines (55). Together, these observations suggest that Actn4 is not essential for the initial establishment of connections between axons and dendrites (69), which involves pre-/post-synaptic recognition, adhesion (70), and trans-synaptic signaling (71), but, instead, contributes to later stages of spine maturation by regulating morphogenesis. This is illustrated by dominant phenotypic abnormalities induced by overexpression of Actn4 mutants lacking the CaM domain, whereby dendritic protrusions are formed but show impaired structural/functional maturation of spine heads. Actinins may therefore cooperate in supporting the formation/stabilization of dendritic protrusions while retaining isoform-specific properties that differentially affect spine morphogenesis.
The extended central rod-like region of actinins is composed by Spectrin repeats that form a platform for protein-protein interactions, a characteristic that confers actinins the features of a central hub for effector proteins (72). Actinins interact via the rod region with proteins enriched at excitatory synapses, including synaptopodin (73, 74), an F-actin binding protein (75) tightly connected to the spine apparatus (76) in large mushroom spine heads. Therefore, in mature spines, actinins are positioned to participate in the group 1 mGluR-dependent modification of the actin cytoskeleton that may underlie spine shrinkage accompanying long-term depression. Recently, actinins have been identified as nodal points in the protein interactome of autism genes (77), including Shank (78, 79), and Actn4 mRNA has been found to be a target for regulation by fragile X mental retardation protein (FMRP) (80), encoded by the Fmr1 gene, which is silenced in fragile X syndrome. Whether abnormalities in actinin expression and/or function(s) are involved in spine dysmorphogenesis, which accompanies these pathological conditions, remains to be established.
Supplementary Material
Acknowledgments
We thank Catherine Castillo for technical assistance, Kevin Fisher for assistance with multimedia, Dr. Ranju Kumari for reagents, and Dr. Yuki Hashimotodani for assistance with mEPSC recordings.
This work was supported, in whole or in part, by National Institutes of Health Grants MH082870 (to A. F.), NS072238 (to F. F. B.), MH081935 (to P. E. C), and NICHD/National Institutes of Health Grant P30 HD071593. This work was also supported by a NARSAD Young Investigator grant from the Brain and Behavior Research Foundation (to A. E. C.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movies 1 and 2.
- mGluR
- metabotropic glutamate receptor
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- DHPG
- (S)-3,5-dihydroxyphenylglycine
- MPEP
- 2-methyl-6-(phenylethynyl)pyridine
- DIV
- days in vitro
- mEPSC
- miniature excitatory postsynaptic current
- CH
- Calponin homology
- VSVG
- vesicular stomatitis virus glycoprotein G
- CR
- conserved region
- PSD
- postsynaptic density
- CaM
- Calmodulin-like domain.
References
- 1. Bhatt D. H., Zhang S., Gan W. B. (2009) Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282 [DOI] [PubMed] [Google Scholar]
- 2. Nägerl U. V., Eberhorn N., Cambridge S. B., Bonhoeffer T. (2004) Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767 [DOI] [PubMed] [Google Scholar]
- 3. Yang Y., Wang X. B., Frerking M., Zhou Q. (2008) Spine expansion and stabilization associated with long-term potentiation. J. Neurosci. 28, 5740–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caroni P., Donato F., Muller D. (2012) Structural plasticity upon learning: regulation and functions. Nat. Rev. Neurosci. 13, 478–490 [DOI] [PubMed] [Google Scholar]
- 5. Bonhoeffer T., Yuste R. (2002) Spine motility: phenomenology, mechanisms, and function. Neuron 35, 1019–1027 [DOI] [PubMed] [Google Scholar]
- 6. Fiala J. C., Feinberg M., Popov V., Harris K. M. (1998) Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci. 18, 8900–8911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yuste R., Bonhoeffer T. (2004) Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 5, 24–34 [DOI] [PubMed] [Google Scholar]
- 8. Cingolani L. A., Goda Y. (2008) Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 9, 344–356 [DOI] [PubMed] [Google Scholar]
- 9. Ethell I. M., Pasquale E. B. (2005) Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 75, 161–205 [DOI] [PubMed] [Google Scholar]
- 10. Niswender C. M., Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hannan A. J., Blakemore C., Katsnelson A., Vitalis T., Huber K. M., Bear M., Roder J., Kim D., Shin H. S., Kind P. C. (2001) PLC-β1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat. Neurosci. 4, 282–288 [DOI] [PubMed] [Google Scholar]
- 12. Kano M., Hashimoto K., Kurihara H., Watanabe M., Inoue Y., Aiba A., Tonegawa S. (1997) Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18, 71–79 [DOI] [PubMed] [Google Scholar]
- 13. Wijetunge L. S., Till S. M., Gillingwater T. H., Ingham C. A., Kind P. C. (2008) mGluR5 regulates glutamate-dependent development of the mouse somatosensory cortex. J. Neurosci. 28, 13028–13037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bellone C., Lüscher C., Mameli M. (2008) Mechanisms of synaptic depression triggered by metabotropic glutamate receptors. Cell. Mol. Life Sci. 65, 2913–2923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lüscher C., Huber K. M. (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhakar A. L., Dölen G., Bear M. F. (2012) The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herman E. J., Bubser M., Conn P. J., Jones C. K. (2012) Metabotropic glutamate receptors for new treatments in schizophrenia. Handb. Exp. Pharmacol. 213, 297–365 [DOI] [PubMed] [Google Scholar]
- 18. Cruz-Martín A., Crespo M., Portera-Cailliau C. (2012) Glutamate induces the elongation of early dendritic protrusions via mGluRs in wild type mice, but not in fragile X mice. PLoS ONE 7, e32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanderklish P. W., Edelman G. M. (2002) Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 1639–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Z., Hu J., Passafaro M., Xie W., Jia Z. (2011) GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J. Neurosci. 31, 819–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oh W. C., Hill T. C., Zito K. (2013) Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc. Natl. Acad. Sci. U.S.A. 110, E305–E312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramiro-Cortés Y., Israely I. (2013) Long lasting protein synthesis- and activity-dependent spine shrinkage and elimination after synaptic depression. PLoS ONE 8, e71155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foley K. S., Young P. W. (2014) The non-muscle functions of actinins: an update. Biochem. J. 459, 1–13 [DOI] [PubMed] [Google Scholar]
- 24. Otey C. A., Carpen O. (2004) α-Actinin revisited: a fresh look at an old player. Cell Motil. Cytoskeleton 58, 104–111 [DOI] [PubMed] [Google Scholar]
- 25. Hall D. D., Dai S., Tseng P. Y., Malik Z., Nguyen M., Matt L., Schnizler K., Shephard A., Mohapatra D. P., Tsuruta F., Dolmetsch R. E., Christel C. J., Lee A., Burette A., Weinberg R. J., Hell J. W. (2013) Competition between alpha-actinin and Ca2+-calmodulin controls surface retention of the L-type Ca2+ channel Ca(V)1.2. Neuron 78, 483–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krupp J. J., Vissel B., Thomas C. G., Heinemann S. F., Westbrook G. L. (1999) Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J. Neurosci. 19, 1165–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu L., Timofeyev V., Li N., Rafizadeh S., Singapuri A., Harris T. R., Chiamvimonvat N. (2009) α-Actinin2 cytoskeletal protein is required for the functional membrane localization of a Ca2+-activated K+ channel (SK2 channel). Proc. Natl. Acad. Sci. U.S.A. 106, 18402–18407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Robison A. J., Bass M. A., Jiao Y., MacMillan L. B., Carmody L. C., Bartlett R. K., Colbran R. J. (2005) Multivalent interactions of calcium/calmodulin-dependent protein kinase II with the postsynaptic density proteins NR2B, densin-180, and α-actinin-2. J. Biol. Chem. 280, 35329–35336 [DOI] [PubMed] [Google Scholar]
- 29. Fraley T. S., Tran T. C., Corgan A. M., Nash C. A., Hao J., Critchley D. R., Greenwood J. A. (2003) Phosphoinositide binding inhibits α-actinin bundling activity. J. Biol. Chem. 278, 24039–24045 [DOI] [PubMed] [Google Scholar]
- 30. Sjöblom B., Salmazo A., Djinović-Carugo K. (2008) α-Actinin structure and regulation. Cell. Mol. Life Sci. 65, 2688–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Francesconi A., Kumari R., Zukin R. S. (2009) Proteomic analysis reveals novel binding partners of metabotropic glutamate receptor 1. J. Neurochem. 108, 1515–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Francesconi A., Duvoisin R. M. (2002) Alternative splicing unmasks dendritic and axonal targeting signals in metabotropic glutamate receptor 1. J. Neurosci. 22, 2196–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abe T., Sugihara H., Nawa H., Shigemoto R., Mizuno N., Nakanishi S. (1992) Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J. Biol. Chem. 267, 13361–13368 [PubMed] [Google Scholar]
- 34. Basarsky T. A., Parpura V., Haydon P. G. (1994) Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J. Neurosci. 14, 6402–6411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin Y. C., Huang Z. H., Jan I. S., Yeh C. C., Wu H. J., Chou Y. C., Chang Y. C. (2002) Development of excitatory synapses in cultured neurons dissociated from the cortices of rat embryos and rat pups at birth. J. Neurosci. Res. 67, 484–493 [DOI] [PubMed] [Google Scholar]
- 36. Dunaevsky A., Tashiro A., Majewska A., Mason C., Yuste R. (1999) Developmental regulation of spine motility in the mammalian central nervous system. Proc. Natl. Acad. Sci. U.S.A. 96, 13438–13443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korkotian E., Segal M. (2001) Regulation of dendritic spine motility in cultured hippocampal neurons. J. Neurosci. 21, 6115–6124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schulz T. W., Nakagawa T., Licznerski P., Pawlak V., Kolleker A., Rozov A., Kim J., Dittgen T., Köhr G., Sheng M., Seeburg P. H., Osten P. (2004) Actin/α-actinin-dependent transport of AMPA receptors in dendritic spines: role of the PDZ-LIM protein RIL. J. Neurosci. 24, 8584–8594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gallagher S. M., Daly C. A., Bear M. F., Huber K. M. (2004) Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J. Neurosci. 24, 4859–4864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kwon H. B., Sabatini B. L. (2011) Glutamate induces de novo growth of functional spines in developing cortex. Nature 474, 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walikonis R. S., Oguni A., Khorosheva E. M., Jeng C. J., Asuncion F. J., Kennedy M. B. (2001) Densin-180 forms a ternary complex with the α-subunit of Ca2+/calmodulin-dependent protein kinase II and α-actinin. J. Neurosci. 21, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Okamoto K., Narayanan R., Lee S. H., Murata K., Hayashi Y. (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. U.S.A. 104, 6418–6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fink C. C., Bayer K. U., Myers J. W., Ferrell J. E., Jr., Schulman H., Meyer T. (2003) Selective regulation of neurite extension and synapse formation by the β but not the α isoform of CaMKII. Neuron 39, 283–297 [DOI] [PubMed] [Google Scholar]
- 44. Okamoto K., Bosch M., Hayashi Y. (2009) The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag? Physiology 24, 357–366 [DOI] [PubMed] [Google Scholar]
- 45. Hell J. W. (2014) CaMKII: claiming center stage in postsynaptic function and organization. Neuron 81, 249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin D. Z., Guo M. L., Xue B., Fibuch E. E., Choe E. S., Mao L. M., Wang J. Q. (2013) Phosphorylation and feedback regulation of metabotropic glutamate receptor 1 by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 33, 3402–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mockett B. G., Guévremont D., Wutte M., Hulme S. R., Williams J. M., Abraham W. C. (2011) Calcium/calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J. Neurosci. 31, 7380–7391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papa M., Bundman M. C., Greenberger V., Segal M. (1995) Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J. Neurosci. 15, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsuzaki M., Ellis-Davies G. C., Nemoto T., Miyashita Y., Iino M., Kasai H. (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Uesaka N., Uchigashima M., Mikuni T., Nakazawa T., Nakao H., Hirai H., Aiba A., Watanabe M., Kano M. (2014) Retrograde semaphorin signaling regulates synapse elimination in the developing mouse brain. Science 344, 1020–1023 [DOI] [PubMed] [Google Scholar]
- 51. Bear M. F., Huber K. M., Warren S. T. (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377 [DOI] [PubMed] [Google Scholar]
- 52. Irwin S. A., Patel B., Idupulapati M., Harris J. B., Crisostomo R. A., Larsen B. P., Kooy F., Willems P. J., Cras P., Kozlowski P. B., Swain R. A., Weiler I. J., Greenough W. T. (2001) Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167 [DOI] [PubMed] [Google Scholar]
- 53. Dölen G., Osterweil E., Rao B. S., Smith G. B., Auerbach B. D., Chattarji S., Bear M. F. (2007) Correction of fragile X syndrome in mice. Neuron 56, 955–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Michalon A., Sidorov M., Ballard T. M., Ozmen L., Spooren W., Wettstein J. G., Jaeschke G., Bear M. F., Lindemann L. (2012) Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakagawa T., Engler J. A., Sheng M. (2004) The dynamic turnover and functional roles of alpha-actinin in dendritic spines. Neuropharmacology 47, 734–745 [DOI] [PubMed] [Google Scholar]
- 56. Peng J., Kim M. J., Cheng D., Duong D. M., Gygi S. P., Sheng M. (2004) Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 279, 21003–21011 [DOI] [PubMed] [Google Scholar]
- 57. Yun-Hong Y., Chih-Fan C., Chia-Wei C., Yen-Chung C. (2011) A study of the spatial protein organization of the postsynaptic density isolated from porcine cerebral cortex and cerebellum. Mol. Cell. Proteomics 10.1074/mcp.M110.007138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wyszynski M., Kharazia V., Shanghvi R., Rao A., Beggs A. H., Craig A. M., Weinberg R., Sheng M. (1998) Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J. Neurosci. 18, 1383–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lujan R., Nusser Z., Roberts J. D., Shigemoto R., Somogyi P. (1996) Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci. 8, 1488–1500 [DOI] [PubMed] [Google Scholar]
- 60. Hunt D. L., Puente N., Grandes P., Castillo P. E. (2013) Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat. Neurosci. 16, 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Leary H., Lasda E., Bayer K. U. (2006) CaMKIIβ association with the actin cytoskeleton is regulated by alternative splicing. Mol. Biol. Cell 17, 4656–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Honkura N., Matsuzaki M., Noguchi J., Ellis-Davies G. C., Kasai H. (2008) The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57, 719–729 [DOI] [PubMed] [Google Scholar]
- 63. Jalan-Sakrikar N., Bartlett R. K., Baucum A. J., 2nd, Colbran R. J. (2012) Substrate-selective and calcium-independent activation of CaMKII by α-actinin. J. Biol. Chem. 287, 15275–15283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kayser M. S., Nolt M. J., Dalva M. B. (2008) EphB receptors couple dendritic filopodia motility to synapse formation. Neuron 59, 56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ziv N. E., Smith S. J. (1996) Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron 17, 91–102 [DOI] [PubMed] [Google Scholar]
- 66. Brünig I., Kaech S., Brinkhaus H., Oertner T. G., Matus A. (2004) Influx of extracellular calcium regulates actin-dependent morphological plasticity in dendritic spines. Neuropharmacology 47, 669–676 [DOI] [PubMed] [Google Scholar]
- 67. Wyszynski M., Lin J., Rao A., Nigh E., Beggs A. H., Craig A. M., Sheng M. (1997) Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature 385, 439–442 [DOI] [PubMed] [Google Scholar]
- 68. Merrill M. A., Malik Z., Akyol Z., Bartos J. A., Leonard A. S., Hudmon A., Shea M. A., Hell J. W. (2007) Displacement of α-actinin from the NMDA receptor NR1 C0 domain By Ca2+/calmodulin promotes CaMKII binding. Biochemistry 46, 8485–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shen K., Scheiffele P. (2010) Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 33, 473–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ko J., Kim S., Chung H. S., Kim K., Han K., Kim H., Jun H., Kaang B. K., Kim E. (2006) SALM synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron 50, 233–245 [DOI] [PubMed] [Google Scholar]
- 71. Dalva M. B., McClelland A. C., Kayser M. S. (2007) Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Djinovic-Carugo K., Gautel M., Ylänne J., Young P. (2002) The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett. 513, 119–123 [DOI] [PubMed] [Google Scholar]
- 73. Kremerskothen J., Plaas C., Kindler S., Frotscher M., Barnekow A. (2005) Synaptopodin, a molecule involved in the formation of the dendritic spine apparatus, is a dual actin/α-actinin binding protein. J. Neurochem. 92, 597–606 [DOI] [PubMed] [Google Scholar]
- 74. Zhang X. L., Pöschel B., Faul C., Upreti C., Stanton P. K., Mundel P. (2013) Essential role for synaptopodin in dendritic spine plasticity of the developing hippocampus. J. Neurosci. 33, 12510–12518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Deller T., Merten T., Roth S. U., Mundel P., Frotscher M. (2000) Actin-associated protein synaptopodin in the rat hippocampal formation: localization in the spine neck and close association with the spine apparatus of principal neurons. J. Comp. Neurol. 418, 164–181 [DOI] [PubMed] [Google Scholar]
- 76. Deller T., Mundel P., Frotscher M. (2000) Potential role of synaptopodin in spine motility by coupling actin to the spine apparatus. Hippocampus 10, 569–581 [DOI] [PubMed] [Google Scholar]
- 77. Sakai Y., Shaw C. A., Dawson B. C., Dugas D. V., Al-Mohtaseb Z., Hill D. E., Zoghbi H. Y. (2011) Protein interactome reveals converging molecular pathways among autism disorders. Sci. Transl. Med. 3, 86ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Han K., Holder J. L., Jr., Schaaf C. P., Lu H., Chen H., Kang H., Tang J., Wu Z., Hao S., Cheung S. W., Yu P., Sun H., Breman A. M., Patel A., Lu H. C., Zoghbi H. Y. (2013) SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503, 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Won H., Lee H. R., Gee H. Y., Mah W., Kim J. I., Lee J., Ha S., Chung C., Jung E. S., Cho Y. S., Park S. G., Lee J. S., Lee K., Kim D., Bae Y. C., Kaang B. K., Lee M. G., Kim E. (2012) Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265 [DOI] [PubMed] [Google Scholar]
- 80. Ascano M., Jr., Mukherjee N., Bandaru P., Miller J. B., Nusbaum J. D., Corcoran D. L., Langlois C., Munschauer M., Dewell S., Hafner M., Williams Z., Ohler U., Tuschl T. (2012) FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.