

Background: The view that, unlike kinases, phosphatases are “nonspecific” pervades the field.
Results: PTP1B inhibited BRK by dephosphorylating Tyr-342, but activated SRC by antagonizing PAG-dependent inhibition by CSK.
Conclusion: Signaling is regulated by combinatorial effects of PTKs and PTPs, with both enzyme classes displaying exquisite specificity.
Significance: Defining phosphatase substrate specificity will reveal new, more effective strategies for therapeutic intervention in major human diseases.
Keywords: phosphorylation; phosphotyrosine; Src; tyrosine-protein kinase (tyrosine kinase); tyrosine-protein phosphatase (tyrosine phosphatase); PTP1B, SRC, BRK, CSK, and CBP/PAG
Abstract
Despite significant evidence to the contrary, the view that phosphatases are “nonspecific” still pervades the field. Systems biology approaches to defining how signal transduction pathways are integrated at the level of whole organisms also often downplay the contribution of phosphatases, defining them as “erasers” that serve merely to restore the system to its basal state. Here, we present a study that counteracts the idea of “nonspecific phosphatases.” We have characterized two structurally similar and functionally related kinases, BRK and SRC, which are regulated by combinations of activating autophosphorylation and inhibitory C-terminal sites of tyrosine phosphorylation. We demonstrated specificity at the level of the kinases in that SRMS phosphorylated the C terminus of BRK, but not SRC; in contrast, CSK is the kinase responsible for C-terminal phosphorylation of SRC, but not BRK. For the phosphatases, we observed that RNAi-mediated suppression of PTP1B resulted in opposing effects on the activity of BRK and SRC and have defined the mechanisms underlying this specificity. PTP1B inhibited BRK by directly dephosphorylating the Tyr-342 autophosphorylation site. In contrast, PTP1B potentiated SRC activity, but not by dephosphorylating SRC itself directly; instead, PTP1B regulated the interaction between CBP/PAG and CSK. SRC associated with, and phosphorylated, the transmembrane protein CBP/PAG at Tyr-317, resulting in CSK recruitment. We identified PAG as a substrate of PTP1B, and dephosphorylation abolished recruitment of the inhibitory kinase CSK. Overall, these findings illustrate how the combinatorial effects of PTKs and PTPs may be integrated to regulate signaling, with both classes of enzymes displaying exquisite specificity.
Introduction
From their studies of the regulation of glycogen phosphorylase in the early 1960s, Danforth et al. (1) published that “kinetic analysis suggests that changes in the phosphorylase b kinase rather than phosphorylase phosphatase activity are responsible for the increase and decrease in phosphorylase a”. This typified an initial view that the sophistication in the regulation of signaling was manifested at the level of the kinases, with the phosphatases serving a general housekeeping function associated with maintenance of the basal state. This view has been pervasive within the field and has fueled the idea that protein phosphatases are simply a barrier that must be overcome to study kinase function (2). This represents an obstacle to appreciating fully the importance of protein phosphatases in the regulation of cell signaling.
Reversible tyrosine phosphorylation, orchestrated by the activity of protein-tyrosine kinases (PTKs) and protein-tyrosine phosphatases (PTPs),4 must be tightly regulated to allow cells to sense and respond to perturbations in the environment. The family of PTPs in humans is encoded by ∼100 distinct genes (3–5); this level of structural diversity alone suggests functional diversity beyond simple housekeeping. The existence of receptor-like PTPs suggests a direct role in initiating a signaling response to extracellular ligands. The ability of PTPs to act positively as well as negatively, for example serving an oncogenic function in addition to being tumor suppressors (6), also suggests an active, rather than a passive, role for PTPs in the regulation of signal transduction. A striking level of specificity is illustrated by gene duplication in the nontransmembrane PTPs, for example giving rise to PTP1B and TCPTP, as well as SHP1 and SHP2; these pairs of enzymes are very closely related in sequence but display distinct, non-redundant functions in vivo (7). Regulatory sequences adjacent to PTP catalytic domains can contribute to substrate specificity, including the KIM domain in the MAP kinase phosphatases such as STEP and HePTP (8) and poly-Pro sequences in PTP-PEST, which direct its interaction with p130CAS (9). There are also examples of intrinsic specificity within the PTP catalytic domains themselves (10–12). Nevertheless, new systems biology approaches to defining how signal transduction pathways are integrated at the level of the whole organism often downplay the contribution of phosphatases. Recent analysis of the evolution of phosphotyrosine-based signals has described a three-part toolkit that involves a “writer” (kinase), “reader” (SH2 domain), and “eraser” (phosphatase) (13). Although the study indicated that protein phosphatases likely contributed to the evolution of phosphotyrosine signaling in ways that went beyond simply reversing kinase signals, the choice of “eraser” to describe the PTPs is unfortunate as it once again conjures up the old images of phosphatases as merely switching pathways off and cleaning up after kinases. In fact, recent publications continue to refer to PTPs as “nonspecific” (14). This study is derived from an original observation that RNAi-mediated suppression of PTP1B in MCF10A mammary epithelial cells resulted in opposing effects on the activity of two structurally similar and functionally related kinases, BRK and SRC. Our goal was to define the mechanistic basis for this effect and to illustrate how specificity in function of both kinases and phosphatases may be integrated to determine signaling outcome.
The non-receptor tyrosine kinase SRC is the prototypical member of the SRC family of tyrosine kinases (SFKs) and modulates a wide range of events, including proliferation, migration, invasion, and survival. SRC is regulated by the reversible phosphorylation of two critical tyrosine residues (15). Phosphorylation of a C-terminal tyrosine in SRC, Tyr-527, by a distinct kinase, CSK, promotes an inactive conformation in which the Tyr(P) residue is engaged in an intramolecular interaction with the SRC SH2 domain. Dephosphorylation of Tyr-527 by PTPs represents one mechanism by which these enzymes can function positively to promote tyrosine phosphorylation-dependent signaling (16). Following dephosphorylation of Tyr-527, SRC adopts an open, active conformation in which it autophosphorylates Tyr-416 in its activation loop. Dephosphorylation of this autophosphorylation site allows PTPs to switch SRC back off and return the system to its ground state. Several members of the PTP family have been reported to act on these sites in SRC (6).
Breast tumor kinase (BRK)/Protein Tyrosine Kinase 6 (PTK6) was first identified in a study of PTK overexpression in human metastatic breast tumors (17). BRK is an important oncogenic effector of EGF and IGF stimulation (18, 19), and by itself has potential to transform NIH3T3 cells (18). Although related to SRC, it is not a member of the SFK family; however, like SRC, BRK possesses SH3, SH2, and kinase catalytic domains, and its kinase activity is also negatively regulated by intramolecular interactions between its SH2 and SH3 domains and their cognate binding motifs (20, 21). In contrast to SRC, the lack of an N-terminal consensus sequence for myristoylation suggests that membrane localization is not required for BRK activation, thus some of its regulatory mechanisms may be different from those of SRC.
We have characterized how kinases and phosphatases coordinate to regulate the activity of BRK and SRC. CSK, the established C-terminal kinase for the SRC kinases, does not phosphorylate BRK (20). In this study, we report that the non-receptor tyrosine kinase SRMS, but not CSK or CHK, phosphorylated the C terminus of BRK, but not SRC. For the phosphatases, we have demonstrated that whereas the activity of BRK was inhibited by direct dephosphorylation by PTP1B, the activity of SRC was enhanced by the same phosphatase, but not by dephosphorylating SRC itself directly; instead, PTP1B promoted SRC activity by regulating the interaction between CBP/PAG and the inhibitory kinase CSK. Therefore, these findings illustrate how the combinatorial effects of PTKs and PTPs may be integrated to regulate signaling, with both classes of enzymes displaying exquisite specificity.
Experimental Procedures
Cell Culture
MCF-10A cells were cultured in DMEM/F-12 medium supplemented with 5% donor horse serum, 20 ng/ml epidermal growth factor (EGF), 10 μg/ml insulin, 100 ng/ml hydrocortisone, 100 ng/ml cholera toxin, 100 units/ml penicillin, and 100 μg/ml streptomycin. T47D cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 10 μg/ml insulin, 1 mm sodium pyruvate, 2.5 μg/ml glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin. SYF (SRC/Yes/Fyn−/−/−) mouse fibroblasts were cultured in DMEM with 10% fetal bovine serum, 100 units/ml streptomycin sulfate, and 100 g/ml amphotericin B.
Plasmids, Transfection, and Infection
Mammalian expression plasmids used in this study were as follows: pMT2-PTP1B (WT, C215S, and D181A); pMT2-TCPTP (WT and DA); BRK (WT and K219M kinase dead, K219M/Y447F), and CSK in 3×FLAG vector from Sigma; SRC (WT and K296R kinase dead) in pUSE vector from Upstate; SRMS in pcDNA6-V5HisA from Invitrogen; pCX4bsr-Myc-CBP/PAG was a gift from Dr. M. Okada (Osaka University); PAG-FLAG (WT, Y317F, and 9YF Y317) was a gift from Dr. T. Berge (University of Oslo); pCDNA3-PTPN14 (WT and D1079A) was a gift from Dr. Y. Khew-Goodall (University of Adelaide).
Small interfering RNA sequences PTP1B sh1 (5′-CTTTGACCATAGTCGGATT-3′), PTP1B sh2 (5′-CTTAGTAAATCTATGGGAT-3′), CSK shRNA (CSK1562), and PAG shRNA (PAG617: 5′-TCTTTCGAGGCAGAAGTAG-3′) were kindly provided by Dr. S. Roche (University of Montpellier). A construct expressing a shRNA Luc directed against the unrelated luciferase was used as a negative control.
Lentiviral infection followed by puromycin selection were adopted to establish shRNA knockdown cell lines. Specifically, lentivirus expressing targeted shRNA was generated in 293T cells by co-transfecting shRNA vector, deltaR8.91, and VSVG at a ratio of 3:2:1. Twenty four hours after transfection, viral particles were harvested and passed through a 0.45-μm filter to remove cell debris. The cleared supernatants were then incubated with recipient cells in the presence of Polybrene (8 μg/ml). Twenty four hours after infection, cells were placed under puromycin selection (2 μg/ml), and the effectiveness of infection was further confirmed by immunoblotting with indicated antibody.
Protein Purification
His-tagged BRK (wild-type and K219M) (20) and SBP (streptavidin-binding protein)-tagged SRMS were expressed in Spodoptera frugiperda (Sf9) insect cells using the Bac-to-Bac baculovirus system (Invitrogen). The murine SRMS cDNA was subcloned into pFastBacA using EcoRI/NotI restriction sites. A vector encoding the SBP tag was a gift from Dr. Takashi Murayama. PCR was used to amplify the sequence and introduce the tag into the pFastBacA-SRMS plasmid.
For protein production, 30 ml of recombinant virus was used to infect 1.08 × 109 Sf9 cells in 600-ml total volume, and cells were harvested after 3 days. The pellets were washed with 0.9% NaCl(aq), and stored at −80 °C. BRK was purified with 500 μl of Ni-NTA resin (Qiagen). Between 2 and 4 of the harvested pellets were combined and lysed in 30–50 ml of detergent-lysis buffer (50 mm Tris/HCl, pH 8.0, 300 mm NaCl, 1% Nonidet P-40) supplemented with 0.1 mm vanadate, 5 mm β-mercaptoethanol, and a protease/inhibitor mixture (5 mg/liter aprotinin, 5 mg/liter leupeptin, 0.1 mm phenylmethylsulfonyl fluoride) for 1 h. The lysates were first cleared by centrifugation at 30,000 × g for 30 min and then incubated with resin for 1 h. The nickel resin was washed with 10 ml of Buffer A (50 mm Tris/HCl, pH 8.0, 500 mm NaCl, 20 mm imidazole), 5 ml of Buffer B (50 mm Tris/HCl, pH 8.0, 1 m NaCl, 20 mm imidazole), 5 ml of Buffer C (50 mm Tris/HCl, pH 8.0, 500 mm NaCl, 40 mm imidazole), and 5 ml of Buffer A. BRK was eluted from the resin by incubation with 500 μl of Elution Buffer (50 mm Tris/HCl, pH 8.0, 5% glycerol, 200 mm imidazole) for 10 min and then passing the buffers through a fritted column. SRMS was purified using a similar protocol using Strep-Tactin resin (Qiagen). Following lysis and resin binding as described above, these beads were washed twice with 25 ml of Buffer NP (50 mm Tris/HCl, pH 8.0, 300 mm NaCl). SRMS was eluted with Buffer NPD (50 mm Tris/HCl, pH 8.0, 300 mm NaCl, 2.5 mm desthiobiotin) following the same protocol as described above. Fractions from both purifications were either used immediately or dialyzed with 50 mm Tris/HCl, pH 8.0, 500 mm NaCl, 20% glycerol and stored at −20 °C.
Kinase-inactive (K219M) BRK was also produced in bacteria. The BRK cDNA was subcloned into the pT7-TEV-HMBP modified pET28a vector using EcoRI/NotI restriction sites and used as a template to generate the K219M mutant by site-directed mutagenesis. This modified vector was a gift from Dr. Miguel Garcia-Diaz. Following transformation into Arctic Express RIL Escherichia coli (Agilent), bacterial cells were grown in Luria Broth at 30 °C to an absorbance (600 nm) of 0.5. The growth mixture was then cooled to 12 °C and induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside. The cells were harvested after 24 h, washed with 0.9% NaCl(aq), and stored at −80 °C. For protein purification, two of the harvested pellets were combined and resuspended in 40 ml of lysis buffer lacking detergent. Following sonication (1 s on/1 s off for 1 min, followed by 1 min on ice, repeated three times), the lysed cells were centrifuged at 30,000 × g for 1 h. K219M protein was purified from the cleared lysate with 1 ml Ni-NTA resin, as described above. GST-tagged SRC D385N, and GST-tagged CSK proteins were gifts from Dr. Markus Seeliger.
Kinase Assays
Two assays were used for SRMS. A continuous spectrophotometric assay of SRMS kinase activity was performed at 30 °C in 75-μl reaction volumes with 100 mm Tris/HCl buffer, pH 7.5, 10 mm MgCl2, 228.8 units/ml pyruvate kinase, 312 units/ml lactate dehydrogenase, 0.6 mg/ml NADH, 1 mm phosphoenolpyruvate, 1 mm ATP, and varying concentrations of kinase and poly(Glu,Tyr) peptide substrate (22). Initial rates were measured in duplicate, and kinetic parameters were calculated by fitting the data to the Michaelis-Menten equation using a non-linear regression analysis on GraphPad Prism software. SRMS was also assayed by measuring transfer of 32P-labeled phosphate from [γ-32P]ATP to various peptide substrates. For SRMS kinase activity assays against the poly(Glu,Tyr) peptide substrate, 500 nm purified protein was incubated with [γ-32P]ATP (1 μCi/reaction) in a total reaction volume of 50 μl that contained 150 mm Tris/HCl, pH 7.5, 100 mm MgCl2, 800 μm ATP, 20 μm poly(Glu,Tyr), at 30 °C, for 30 min. Reactions were terminated by spotting 35 μl of the mixture onto 1 × 1-inch square pieces of filter paper, which were washed three times in 600 ml of hot (65 °C) 5% trichloroacetic acid, dried, and analyzed in a scintillation counter. SRMS kinase activity toward other synthetic peptides was measured with [γ-32P]ATP and a phosphocellulose filter paper binding assay (23).
For measuring phosphorylation by autoradiography, purified BRK K219M or SRC D385N (2.5 μm) were incubated under the above phosphorylation conditions alone, or with either 1 μm SRMS or CSK, in a total volume of 50 μl. After 30 min, the samples were boiled in 25 μl Laemmli sample buffer and analyzed by SDS-PAGE and autoradiography.
Synthetic Peptides
The synthetic peptides used in this study were as follows: SRC substrate, Ala-Glu-Glu-Glu-Ile-Tyr-Gly-Glu-Phe-Glu-Ala-Lys-Lys-Lys-Lys-Gly; insulin receptor substrate, Lys-Lys-Glu-Glu-Glu-Glu-Tyr-Met-Met-Met-Met-Gly; protein kinase A substrate, Leu-Arg-Arg-Ala-Ser-Ala-Gly; insulin receptor substrate 1 analog, Lys-Lys-Ser-Arg-Gly-Asp-Tyr(P)-Met-Thr-Ala-Gln-Ile-Gly; SH2 control, Arg-Arg-Leu-Glu-Asp-Ala-Ile-Tyr-Ala-Ala-Gly-Gly-Gly-Gly-Gly-Glu-Pro-Pro-Gln-Phe-Glu-Glu-Ile-Gly; SH2 substrate, Arg-Arg-Leu-Glu-Asp-Ala-Ile-Tyr-Ala-Ala-Gly-Gly-Gly-Gly-Gly-Glu-Pro-Pro-Gln-Tyr(P)-Glu-Glu-Ile-Gly; SH3 control, Ala-Glu-Glu-Glu-Ile-Tyr-Gly-Glu-Phe-Gly-Gly-Arg-Gly-Ala-Ala-Ala-Ala-Ala-Ala-Ala-Val-Ala-Arg-Gly-Arg-Gly; and SH3 substrate, Ala-Glu-Glu-Glu-Ile-Tyr-Gly-Glu-Phe-Gly-Gly-Arg-Gly-Ala-Ala-Pro-Pro-Pro-Pro-Pro-Val-Pro-Arg-Gly-Arg-Gly. All peptides were purified by reverse phase HPLC prior to use; poly(Glu:Tyr) 4:1 was purchased from Sigma.
Phosphatase Assay
FLAG-BRK or SRC K296R kinase-dead mutant were co-transfected with CSK plasmids into 293T cells for 24 h, followed by immunoprecipitation with antibodies against either FLAG or SRC. Each immunoprecipitate was split into equal aliquots and incubated with purified PTP1B (range 0–800 ng) at 30 °C for 30 min. The in vitro dephosphorylation reaction was terminated with Laemmli sample buffer, and proteins were separated by SDS-PAGE. Substrate dephosphorylation was visualized by immunoblotting.
Mass Spectrometry
Mass spectrometry analysis was performed at the Stony Brook Proteomics Facility. Purified bacterially expressed BRK K219M and SRMS proteins, which were devoid of tyrosine phosphorylation, were incubated in 50-μl total volume containing 150 mm Tris/HCl, pH 7.5, 100 mm MgCl2, 800 μm ATP, for 30 min, at 30 °C. Following SDS-PAGE, the bands corresponding to BRK were excised and digested with trypsin. The mixture of peptide fragments were then separated by HPLC and analyzed by MS/MS on a Thermo Fisher Scientific LTQ XL ion trap mass spectrometer.
Antibodies and Immunoprecipitation
The following antibodies were used in the analysis: PTP1B (FG6; 50 kDa), TCPTP (CF4; 48 kDa), Myc (9E10); PAG (70–85 kDa due to multiple sites of tyrosine phosphorylation), and PTPN14 (135 kDa) (R&D Systems); phospho-Tyr-1068 and total EGFR (175 kDa), phospho-Tyr-416, Tyr-527, and total SRC (60 kDa), phospho-Thr-308 and total AKT (60 kDa), phospho- and total ERK (42 and 44 kDa), IRS-1 (180 kDa), β-catenin (92 kDa), SHP2 (72 kDa), and CSK (50 kDa) (Cell Signaling); 4G10, phospho-Tyr-342 and total BRK (50 kDa) (Millipore); phospho-Tyr-612 IRS-1 (Invitrogen; 180 kDa); β-tubulin (50 kDa), β-actin (45 kDa), and FLAG (Sigma); SRMS (Santa Cruz Biotechnology; 55 kDa).
Cell extracts were prepared in RIPA lysis buffer (50 mm Tris HCl, pH 8, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) and analyzed by SDS-PAGE and immunoblotting. Pre-cleared cell extracts were incubated with the indicated antibody for 4 h in a cold room with rotation, followed by a 1-h pulldown by 1:1 protein A/G-agarose beads. Immunoprecipitates were washed in lysis buffer three times before electrophoresis.
Preparation of Lipid Raft Fraction
T47D cells were lysed in homogenization buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA. 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm PMSF, 1 mm sodium orthovanadate, 50 mm NaF, 5 mm 2-mercaptoethanol) supplemented with 0.35% Triton X-100 and subjected to ultracentrifugation on a discontinuous sucrose density gradient (4 ml; 40% to 35% to 5%). The raft fraction was solubilized with ODG buffer (homogenization buffer plus 1.5% octyl-d-glucoside and 1% Nonidet P-40).
Results
Opposing Regulatory Effects of PTP1B on Kinase Activities of SRC and BRK
We used short hairpin RNAs to silence the PTPN1 gene, which encodes PTP1B, in MCF-10A mammary epithelial cell lines and compared the status of activation of the kinases SRC and BRK following EGF stimulation. Phosphorylation of Tyr-1068 in the EGF receptor illustrated the effectiveness of EGF treatment, and immunoblotting against PTP1B revealed the efficiency of silencing (Fig. 1). If PTP1B served a general housekeeping function, one would anticipate that its suppression would result in a general increase in overall tyrosine phosphorylation. In contrast, we observed that suppression of PTP1B did not impact EGF-induced phosphorylation of Tyr-1068 in the receptor and did not alter the kinetics of phosphorylation of Thr-308 of AKT; however, activation of ERK was attenuated. Most strikingly, we observed a differential effect of the loss of PTP1B on activation of SRC and BRK kinases. Although the activity of BRK was enhanced and sustained in PTP1B-deficient cells, activation of SRC was attenuated by suppression of PTP1B (Fig. 1).
FIGURE 1.

PTP1B exerted opposite regulatory effects on BRK and SRC. (Left) MCF-10A cells with either luciferase control or PTP1B shRNA were serum-starved for 16 h and stimulated with EGF (100 ng/ml) for the times indicated, lysed, and immunoblotted with the indicated antibodies. Knockdown efficiency was illustrated by blotting with anti-PTP1B antibody, and actin was probed as a loading control. (Right) The activation status of BRK and SRC following PTP1B suppression was quantified by ImageJ and normalized to total protein levels.
Using a complementary approach, we tested the effects of a small molecule inhibitor of PTP1B, MSI-1436 (24, 25), on signaling in T47D breast cancer cells. In cells treated with MSI-1436, we observed robust elevation of the tyrosine phosphorylation of IRS1, a known substrate of PTP1B (26). Consistent with the RNAi experiment, inhibition of PTP1B resulted in decreased SRC activation, by ∼60%, whereas the active form of BRK was increased under the same conditions (Fig. 2).
FIGURE 2.
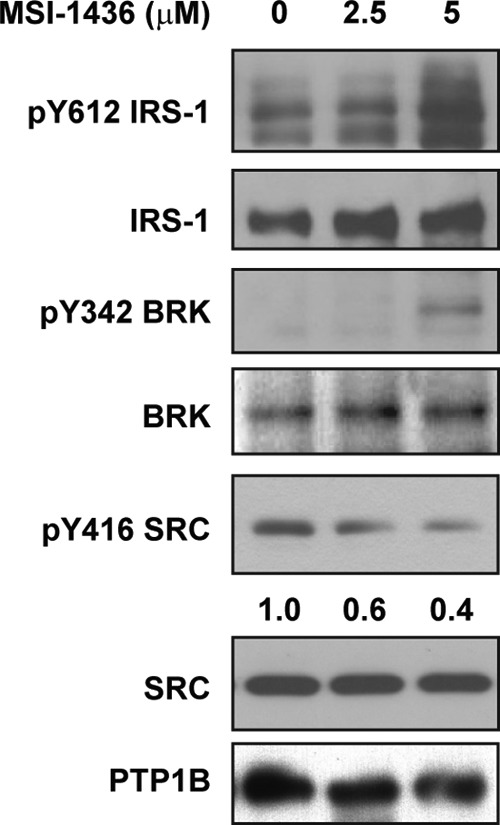
Small molecule inhibitor of PTP1B MSI-1436 exerted opposite regulatory effects on BRK and SRC. T47D breast cancer cell lines were treated with a specific PTP1B inhibitor, MSI-1436, for 4 h at the indicated concentration. Phosphorylation of IRS-1, BRK, and SRC was compared after treatment. The activation status of SRC was quantified by ImageJ and normalized to total protein levels.
By investigating whether the effects of PTP1B on kinase activation were direct, we observed a dose-response relationship between increased levels of PTP1B, either in vitro (Fig. 3A) or ectopically expressed in 293T cells (Fig. 3B), and we decreased phosphorylation of Tyr-342 from the activation loop of BRK. In contrast, there was minimal dephosphorylation of the inhibitory Tyr-527 site in SRC following incubation with increasing doses of purified PTP1B in vitro (Fig. 3C). Furthermore, the substrate-trapping PTP1B-D181A mutant, but not the wild-type form of the phosphatase, formed a stable complex with BRK (Fig. 3D), consistent with a direct enzyme-substrate interaction, as reported previously (27). Again in contrast, no complex was detected between the PTP1B substrate-trapping mutant and SRC in 293T cells, although Tyr-527 of SRC was clearly phosphorylated (Fig. 3D). Together, these results are consistent with two distinct molecular mechanisms, one direct the other indirect, by which PTP1B may regulate BRK and SRC, respectively.
FIGURE 3.

PTP1B dephosphorylated and inactivated BRK, but not SRC. (A) BRK was expressed in 293T cells, then isolated by immunoprecipitation with antibodies against the FLAG tag. Equal amounts of the immunoprecipitate were incubated with the indicated amounts of purified PTP1B at 30 °C for 30 min. The in vitro dephosphorylation reaction was stopped by addition of sample buffer, and dephosphorylation was assessed by immunoblotting with the indicated antibodies. (B) BRK was co-expressed with increasing amounts of PTP1B cDNA in 293T cells, and its activation status was determined by immunoblotting with a phospho-antibody against Tyr-342 of BRK. (C) The kinase-dead K296R mutant form of SRC was co-expressed in 293T cells with CSK and then immunoprecipitated. Dephosphorylation by PTP1B in vitro was assessed as described for C. (D) 293T cells were transiently transfected with PTP1B WT, D181A, BRK, and SRC expression plasmids, alone or in combination as indicated. PTP1B was immunoprecipitated, and its association with either BRK or SRC was examined by immunoblotting. Whole cell lysates were further probed for phospho- and total BRK or SRC, respectively.
Activity of SRC, but not BRK, Was Negatively Regulated by CSK
The phosphorylation of the C-terminal tyrosine residue of either SRC or BRK creates a docking site for their SH2 domains, and this intra-molecular interaction inhibits the kinases (20, 28). The enzyme responsible for phosphorylating SRC at its C terminus is the C-terminal SRC kinase (CSK) (29). As expected, suppression of CSK in the T47D human breast tumor cell line resulted in diminished phosphorylation of Tyr-527 in SRC, with concurrent elevation in phosphorylation of the activating autophosphorylation site Tyr-416 (Fig. 4A). Consistent with this, co-expression of CSK and a kinase-dead mutant of SRC resulted in enhanced phosphorylation of Tyr-527 (Fig. 4B). However, distinct results were observed for BRK. Expression of BRK resulted in the appearance of two bands in anti-phosphotyrosine immunoblots. The upper band was identified as SAM68 and the lower band as BRK, by immunoblotting. Due to the lack of a phospho-specific antibody that recognizes the C terminus of BRK, we precipitated the kinase-dead mutant protein (K219M) via a FLAG epitope tag and probed with antibody against phosphotyrosine. Although CSK was ectopically expressed in 293T cells, we did not detect any tyrosine phosphorylation of BRK K219M (Fig. 4C). Furthermore, following suppression of CSK in T47D cells, we did not observe activation of BRK (Fig. 4A). This suggests that a different kinase, distinct from CSK, may be responsible for phosphorylation of the C terminus of BRK.
FIGURE 4.

Activity of SRC, but not BRK, was negatively regulated by CSK. (A) (upper) T47D breast cancer cells, expressing either luciferase control or CSK shRNA, were lysed and immunoblotted with the indicated antibodies to determine the activation status of SRC. Knockdown efficiency was illustrated by blotting with anti-CSK antibody, and ERK1/2 was probed as loading control. (Lower) Immunoprecipitation followed by blotting analysis were applied to same lysates to check the activation of BRK upon CSK knockdown. (B) 293T cells were transiently transfected with SRC kinase-dead mutant and CSK expression plasmids, as indicated, and phosphorylation changes on SRC Tyr-527 and Tyr-416 were determined by phosphospecific antibodies. The intensity of the signal in the SRC Tyr(P)-527 blot was normalized to total SRC and quantitated by ImageJ. (C) Indicated constructs were transiently transfected in 293T cells. BRK was immunoprecipitated, and its activation status was examined by probing with specific Tyr-342 and Tyr(P) (4G10) antibodies.
SRMS: a Novel C-terminal Kinase for BRK
Like BRK, SRMS is a member of the FRK family of kinases, and the genes encoding BRK and SRMS are separated by only 1.1 kb on human chromosome 20q13.3 (30). Although poorly characterized, the kinase SRMS shares several key features with CSK, including the fact that it is not myristoylated at the N terminus, it has the same domain architecture, and it lacks a negative regulatory tyrosine at the C terminus (31). To characterize the kinase activity of SRMS, we expressed and purified the full-length protein. Of those substrates tested, we observed activity in vitro only against the random co-polymer, poly(Glu,Tyr) (4:1) (Fig. 5A). Among the tyrosine kinase inhibitors tested, SRMS was only sensitive to dasatinib (data not shown). Both BRK and SRC display high activity toward peptide substrates that incorporate SH3 or SH2 ligands (21, 32). Although SRMS possesses SH3 and SH2 domains in a similar arrangement to BRK and SRC, it did not phosphorylate such peptides to any significant extent (Fig. 5A). These results demonstrated that although SRMS is active as a tyrosine kinase, its substrate specificity is significantly different from BRK and the SFKs. The preferential activity toward poly(Glu,Tyr) is reminiscent of CSK (33), which prompted us to investigate a possible role for SRMS in regulating BRK or SFKs.
FIGURE 5.

SRMS, but not CSK, phosphorylated the C-terminal tyrosine of BRK. (A) The activity of purified WT SRMS protein was measured toward various peptides using the phosphocellulose paper binding assay. The inset shows an expanded range. (B) Purified SRMS and kinase-inactive BRK K219M proteins were incubated for 30 min at 30 °C in the presence of γ-32P-ATP. The reactions were analyzed by SDS-PAGE, and proteins were detected by autoradiography (top panel) or Coomassie Blue staining (lower panel). (C) Kinase-inactive BRK K219M was incubated either with or without SRMS under phosphorylation conditions. Following kinase reactions, proteins were separated by SDS-PAGE and stained with Coomassie Blue. Bands corresponding to BRK were excised, digested with trypsin, and analyzed by LC-MS. The LC peaks corresponding to the peptide fragments containing Tyr-447 in SRMS-treated BRK fractions were further analyzed by MS/MS. (D) The indicated constructs were expressed in SRC/Yes/Fyn−/−/− (SYF) fibroblasts. Proteins were immunoprecipitated from whole cell lysates with anti-FLAG and analyzed by Western blotting with the indicated antibodies. (E) In vitro reactions were performed with SRMS, CSK, and the kinase-inactive forms of SRC and BRK in the presence of γ-32P-ATP. The reactions were analyzed by SDS-PAGE, and proteins were detected by autoradiography or Coomassie Blue staining. (F) FLAG-BRK K219M, SRMS, and increasing amounts of PTP1B cDNA were co-transfected in 293T cells, as indicated. BRK K219M was immunoprecipitated by anti-FLAG, followed by Tyr(P) (4G10) blotting.
SRMS phosphorylated the kinase-inactive mutant form of BRK (K219M) in vitro (Fig. 5B). Using LC/MS/MS, we identified Tyr-447 as the primary site (phosphorylation: 73.8%) of SRMS-mediated phosphorylation on BRK (Fig. 5C). Tyr-13 and Tyr-342 (in the SH2 domain and activation loop, respectively) were also phosphorylated, but only to low levels (phosphorylation, 1.4 and 3.9%, respectively). To test for phosphorylation of BRK in mammalian cells, we co-expressed SRMS and the BRK K219M mutant in Src/Yes/Fyn triple knock-out fibroblasts (SYF cells). Co-expression with SRMS led to phosphorylation of BRK, with no signal seen in the absence of SRMS (Fig. 5D). SRMS-induced phosphorylation of BRK in SYF cells was essentially abrogated by mutation of Tyr-447 to Phe, which together with the mass spectrometry data identified BRK Tyr-447, the C-terminal inhibitory site, as a cellular substrate for SRMS (Fig. 5D).
Finally, when we compared the activity of SRMS toward kinase-inactive mutants of BRK and SRC in vitro, we observed that unlike CSK, which showed a strong preference for SRC over BRK, SRMS preferred BRK over SRC (Fig. 5E). By investigating the dephosphorylation of SRMS-phosphorylated BRK, we observed that co-expression of PTP1B with SRMS and BRK K219M in 293T cells did not alter the tyrosine phosphorylation on BRK (Fig. 5F). This suggests that whereas PTP1B was able to recognize Tyr-342 in the BRK activation loop as a substrate (Fig. 3), it was a relatively weak phosphatase for the C terminus of BRK, illustrating site specificity in the recognition of BRK as a substrate by PTP1B.
Phosphorylation of PAG by SRC, but Not BRK, Promoted CSK Recruitment
The transmembrane phosphoprotein designated CSK Binding Protein/Phosphoprotein Associated with Glycosphingolipid-enriched Microdomains (CBP/PAG) is a substrate for SRC family kinases and is essential for membrane recruitment of CSK (34, 35). Considering the differential response of SRC and BRK to CSK, we examined the ability of each kinase to recruit CSK to the membrane. MYC-tagged PAG was transiently expressed alone, or together with either BRK or SRC, in 293T cells, followed by immunoprecipitation with antibody against MYC. We observed a retardation of the electrophoretic mobility of the PAG protein when co-expressed with SRC (Fig. 6A), presumably due to hyperphosphorylation. In contrast, band shift detected when PAG and BRK were co-expressed was less pronounced. Nevertheless, both SRC and BRK formed complexes with PAG, to a comparable extent (Fig. 6A).
FIGURE 6.

Phosphorylation of PAG by SRC, but not BRK, promoted CSK recruitment. (A–B) The indicated constructs were transiently transfected in 293T cells. Tagged PAG was immunoprecipitated with an antibody against MYC, and its interactions with (A) SRC, BRK, and (B) CSK were demonstrated by immunoblotting. (C) 293T cells co-transfected with SRC and MYC-PAG were treated in the presence or absence of SRC kinase inhibitor SU6656 (5 μm) for 1 h. PAG was immunoprecipitated by antibody against MYC, and the binding of CSK was compared by immunoblotting. (D) 293T cells were transiently transfected with wild-type or kinase-dead mutant BRK and MYC-PAG expression plasmids as indicated. PAG was immunoprecipitated with an antibody against MYC, and the immunoprecipitates were further examined for CSK binding. (E–F) 293T cells were transiently transfected with indicated plasmids, and FLAG-tagged PAG, PAG Y317F (E), or PAG 9YF 317Y (F) was immunoprecipitated by anti-FLAG. Pulldown efficiency, total tyrosine phosphorylation of PAG, and its interaction with CSK were determined with indicated antibodies. (G) (left) T47D breast cancer cell line with either luciferase control or PAG shRNA was lysed and immunoblotted with anti-Tyr(P) (4G10). (Right) The same lysates were also probed with the indicated antibodies to determine the activation status of SRC. Knockdown efficiency was illustrated by blotting with anti-PAG antibody, and ERK1/2 was probed as loading control.
We observed robust binding between PAG and CSK in the presence of SRC, which was abrogated when the kinase-dead mutant of SRC was co-expressed, suggesting the importance of tyrosine phosphorylation for the association (Fig. 6B). Furthermore, wild-type SRC, but not the kinase-dead form, co-immunoprecipitated with PAG, suggesting that SRC may create its own docking site on the membrane protein (Fig. 6B). Similarly, the association of CSK and PAG was disrupted when SRC activity was attenuated by the small molecule inhibitor SU6656 (Fig. 6C) (36), highlighting the importance of SRC kinase activity in CSK recruitment. In contrast, although wild-type BRK (but not K219M) could phosphorylate its own docking site on PAG, it did not promote the recruitment of CSK to PAG (Fig. 6D).
It has been reported that phosphorylation of Tyr-317 on PAG is essential for CSK recruitment (34, 35). Consistent with this, we observed that the Y317F PAG mutant failed to co-precipitate CSK in the presence of SRC (Fig. 6E). Therefore, we compared the ability of BRK and SRC to phosphorylate this particular site. To address this, a mutant PAG construct containing phenylalanine substitutions at all tyrosine residues except Tyr-317 was generated. As shown in Fig. 6F, SRC, but not BRK, was able to phosphorylate PAG at Tyr-317, and this phosphorylation was sufficient to induce association with CSK. Together, these data indicate that SRC-mediated tyrosine phosphorylation of PAG at Tyr-317 is necessary and sufficient for CSK recruitment.
We also generated a stable T47D cell line in which PAG was suppressed, and we addressed the impact of the loss of PAG on the kinase activity of SRC and BRK. As shown in Fig. 6G, although loss of PAG had a minimal effect on phosphorylation of the active site of BRK, SRC activity was increased. These results demonstrate that PAG was a negative regulator of SRC, but not BRK.
PAG Was a Substrate of PTP1B and Was Essential for PTP1B-mediated SRC Activation
Our data suggest that the mechanism by which PTP1B affects SRC activation may be indirect; therefore, we examined the contribution of PAG to that regulatory mechanism. In light of the positive role of PTP1B in SRC activation (Figs. 1 and 2), we hypothesized that the phosphatase might dephosphorylate Tyr-317 of PAG, thereby blocking CSK recruitment. As shown in Fig. 7A, expression of wild type PTP1B, but not the D181A substrate-trapping mutant, decreased the shift in electrophoretic mobility of PAG that was induced by SRC expression. Furthermore, expression of wild type PTP1B, but not the phosphatase-dead C215S mutant, led to dephosphorylation of PAG (Fig. 7B). In contrast, RNAi-mediated suppression of PTP1B increased the total tyrosine phosphorylation of PAG (Fig. 7C). A robust interaction was detected between PAG and the substrate-trapping mutant form of PTP1B in the presence of SRC, and this interaction was not observed with wild type PTP1B (Fig. 7D), suggesting that PAG was a direct substrate of PTP1B.
FIGURE 7.

Dephosphorylation of PAG by PTP1B-mediated SRC activation. (A) The indicated expression plasmids were transiently transfected in 293T cells, and lysates were collected for immunoblotting. Expression levels of the transfected plasmids and the electrophoretic mobility of the PAG protein band are shown. (B) Myc-tagged PAG was co-expressed with either WT PTP1B, C215S PTP1B, WT PTPN14, or WT TCPTP 45kDa in 293T cells, as indicated, and PAG was immunoprecipitated by antibody against Myc. Pulldown efficiency and total tyrosine phosphorylation of PAG were determined by the indicated antibodies. Expression levels of PTP1B, PTPN14. and TCPTP 45kDa were also examined. (C) Lipid raft enrichment was performed on lysates from MCF-10A cell with either luciferase control or PTP1B shRNAs. The fraction was fully dissolved with addition of 2% ODG, followed by anti-PAG antibody immunoprecipitation. The total tyrosine phosphorylation of PAG was examined by 4G10. Expression level of PAG and knockdown efficiency of PTP1B was also determined. (D) PAG and SRC were co-transfected with either WT or D181A mutant of PTP1B as indicated. PTP1B was immunoprecipitated, and its association with PAG was examined. Equal expression levels of PAG, SRC, and PTP1B in both samples were further demonstrated. (E) Breast cancer cell line T47D was homogenized in a buffer containing 0.35% Triton X-100, and the raft fractions were separated on a discontinuous sucrose gradient. The fractions (3 ml) were collected from the top of the gradient and subjected to immunoblotting with antibodies against indicated proteins. (F) The indicated plasmids were co-expressed, and cell lysates were processed as described in B. (G) The indicated plasmids were co-expressed, and cell lysates were processed as described in A. (H–I) Expression plasmids were transiently transfected, alone or in combination, in 293T cell as indicated. PAG was immunoprecipitated by antibody against Myc, and the immunoprecipitates were further examined for CSK binding. Expression levels of PTP1B and SRC were also examined. (J) Plasmids were transiently transfected in 293T cells as indicated. SRC activation was compared in the absence and presence of PTP1B.
Considering the exclusive localization of PAG in lipid raft-enriched membrane domains (37), as well as the fact that the activation/inactivation of SRC takes place in the same compartment (38), we investigated whether PTP1B could be detected in lipid rafts. We performed ultracentrifugation on sucrose gradients to isolate both lipid raft and cytosol fractions. Immunoblotting confirmed that PAG was exclusively enriched in early fractions (Fig. 7E), where lipid rafts were recovered. Consistent with the role of myristoylation in membrane anchorage (38), we also observed a pool of SRC that was associated with the lipid raft fractions; in contrast, we did not detect BRK in these fractions (Fig. 7E). The distribution pattern of PTP1B was similar to that of SRC, illustrating that a pool of all three proteins, PTP1B, SRC, and PAG, was recovered within lipid rafts, consistent with a role in SRC activation.
We did not detect SHP2 in lipid rafts, although it has been reported to dephosphorylate PAG (39); however, we did detect PTPN14 and TCPTP (Fig. 7E). Therefore, we tested whether these two phosphatases could also dephosphorylate PAG. As shown in Fig. 7F, PTPN14 was not able to dephosphorylate PAG. Although TCPTP, the closest relative of PTP1B, displayed limited activity toward PAG, this was less than the activity seen with PTP1B (67% versus 26%, at comparable levels of expression). In addition, expression of PTP1B abrogated the SRC-induced shift in the electrophoretic mobility of the PAG band, whereas the effects of PTPN14 and TCPTP were less pronounced (Fig. 7G). Furthermore, although expression of PTP1B antagonized CSK-PAG complex formation (Fig. 7H), the effects of PTPN14 and TCPTP were again less pronounced (Fig. 7I). Overall, these data highlight specificity in the effects of PTP1B in the dephosphorylation of PAG.
Finally, we tested the importance of dephosphorylation of PAG by PTP1B on CSK recruitment and SRC activation. In cells where both SRC and PAG were expressed, we observed a retardation in the electrophoretic mobility of PAG due to SRC-mediated phosphorylation (Fig. 7I). Under these conditions, we were able to co-precipitate endogenous CSK in a complex with PAG. When PTP1B was also expressed, tyrosine phosphorylation of PAG was attenuated but not suppressed completely, and CSK recruitment was substantially diminished (Fig. 7I).
It has been postulated that SRC could be activated not only by dephosphorylation of Tyr-527, but also by engagement of its SH2 domain by specific phosphorylated tyrosine motifs (40). Considering the potential for SH2 domain-mediated interaction between SRC and PAG (15), we co-expressed PTP1B with SRC and PAG 9YF Tyr-317 to focus on this CSK-binding site (Fig. 7J). The results demonstrated increased tyrosine phosphorylation of Tyr-317 in the PAG mutant upon SRC expression, and this elevated signal was substantially diminished by PTP1B (Fig. 7J). Although phosphorylation of SRC on Tyr-527 was not altered in the presence of PTP1B, we observed increased activation of SRC, by ∼2-fold, consistent with the importance of PTP1B-mediated dephosphorylation of Tyr-317 in PAG and the concomitant decrease in CSK recruitment, in promoting SRC activity.
Discussion
This study illustrates that distinct mechanisms have evolved to regulate the activities of two structurally similar and functionally related kinases, BRK and SRC. Our data show that this is achieved through the combinatorial effects of PTKs and PTPs, with both classes of enzymes displaying exquisite specificity.
We have demonstrated that the C termini of SRC and BRK are phosphorylated by distinct tyrosine kinases. CSK has been identified as the physiologically relevant kinase for the C terminus of SRC (29); in this study we have shown that it failed to phosphorylate BRK (Fig. 4C). In contrast, we report here that the non-receptor tyrosine kinase SRMS selectively phosphorylated the C terminus of BRK, both in vitro and in cells (Fig. 5B-D), but did not phosphorylate SRC (Fig. 5E). In this first enzymatic characterization of SRMS, we have demonstrated that, like CSK, it did not phosphorylate various synthetic peptide substrates, and had only moderate activity against the polymeric substrate poly(Glu,Tyr). Importantly, SRMS displayed specificity toward BRK over SRC and toward Tyr-447 (in the C terminus) over Tyr-342 (in the activation loop) and other tyrosine residues on BRK (Fig. 5). CSK is a negative regulator of kinase activity and has tumor suppressor function (15), but the cellular functions of SRMS are still unclear. It was reported that SRMS is overexpressed in breast tumors, whereas its expression level in normal mammary epithelial cells is very low (41). It was also reported that SRMS phosphorylates DOK1, which has been implicated in mitogenic signaling pathways in chronic myelogenous leukemia (41). Previous studies have indicated BRK is subject to similar regulatory mechanisms as SRC (20), and we are currently characterizing the SRMS-BRK interaction in different cell contexts and subcellular localizations.
Although there is a high degree of sequence similarity between BRK and SRC family kinases, the lack of an N-terminal myristoylation motif in BRK leads to a distinct pattern of localization within cells. Our work shows that this is coupled to differing regulatory mechanisms. We confirmed that a pool of SRC, but not BRK, was found in the cell membrane compartment, particularly in lipid rafts (Fig. 7E). This location exposes SRC to Phosphoprotein Associated with Glycosphingolipid-enriched microdomains (PAG), which is also known as CSK-Binding Protein (CBP). PAG is a ubiquitously expressed protein that is conserved in almost all vertebrate species (42). It is a membrane protein with a putative palmitoylation site immediately C-terminal to its helical transmembrane domain, which targets it to lipid rafts (35). PAG/CBP contains 10 tyrosyl residues in its intracellular segment, of which 9 are substrates of SRC family kinases (43). Consistent with this, we observed that co-expression of SRC and PAG caused a profound retardation in the electrophoretic mobility of PAG (Fig. 6A). Furthermore, we demonstrated that phosphorylation of Tyr-317 in PAG in particular was necessary and sufficient to promote recruitment and activation of CSK (Fig. 6E-F), which in turn inhibits SRC (34, 35). Not only does this provide a mechanism for feedback control of SRC activity, but also it represents an indirect mechanism by which PTPs may influence the activation status of SRC.
It is striking to note that the PTPs displayed comparable sophistication and specificity to the kinases in their regulation of SRC-like kinases. Despite the close relationship of BRK and SRC in the kinome, we show here that PTP1B differentially regulated their kinase activities. PTP1B could distinguish between the two kinases; although we did not detect dephosphorylation of either Tyr-416 or Tyr-527 in SRC, PTP1B was able to recognize BRK as a substrate. Furthermore, PTP1B displayed the ability to distinguish between individual phosphorylation sites, as in its ability to dephosphorylate the Tyr-342 autophosphorylation site but not the C-terminal Tyr-447 site of BRK. Instead of a direct effect on SRC, we propose a regulatory model in which PTP1B promotes SRC activation via dephosphorylation of PAG, with concomitant disruption of its association with CSK (Fig. 8). Although other PTPs have been reported to dephosphorylate PAG (42), it is interesting to note that in our study both TCPTP, the closest relative of PTP1B in the PTP family, and PTPN14 were detected in the lipid raft fraction but were less efficient than PTP1B in dephosphorylation of PAG (Fig. 7F). It is interesting to note that PTP1B has also been reported to promote RAS signaling through an indirect mechanism. It does so by dephosphorylation of the p62DOK scaffold protein, which in turn leads to decreased levels of the Ras GTPase-activating protein p120RasGAP, an inhibitor of RAS signaling (44). These observations provide further illustration of the potential for PTP1B to exert specific effects on the regulation of cell signaling.
FIGURE 8.

Model to illustrate the regulation of BRK and SRC. (A) PTP1B played a negative role in controlling BRK activation by directly dephosphorylating the autophosphorylation site of the kinase. (B) By dephosphorylating PAG at Tyr-317 to exclude CSK-mediated SRC inactivation, PTP1B indirectly sustained and potentiated the activity of SRC.
PTP1B is a validated therapeutic target for diabetes and obesity due to its regulatory role in attenuating insulin and leptin signaling, respectively. In addition, PTP1B has a positive role in HER2-related breast cancers, reinforcing its potential as an anti-tumor target (25, 45, 46). This study also emphasizes its potential as a positive regulator of SRC. In the current quest for personalized medicine, in which therapeutic strategies are tailored to the changes in signaling that characterize a disease state, it is important to define these changes completely from the perspective of both kinases and phosphatases. An understanding of the signaling function of protein phosphatases, including definition of their substrate specificities, will allow us to exploit a greater spectrum of the changes in signaling in disease and generate new and more effective strategies for therapeutic intervention in major human diseases.
Acknowledgments
We thank Dr. M. Okada (Osaka University), Dr. T. Berge (University of Oslo), Dr. Y. Khew-Goodall (University of Adelaide), and Dr. S. Roche (University of Montpellier) for providing plasmid constructs; we also thank Dr. T. Koller and the Stony Brook Proteomics Center for mass spectrometry, and Dr. M. Seeliger (Stony Brook University) for purified SRC and CSK proteins.
This research by NIH Grants CA53840 (N. K. T.) and CA58530 (W. T. M) and the CSHL Cancer Centre Support Grant CA45508 (N. K. T.). N. K. T. is also grateful for the support from the following foundations: The Gladowsky Breast Cancer Foundation, The Don Monti Memorial Research Foundation, Hansen Memorial Foundation, West Islip Breast Cancer Coalition for Long Island, Glen Cove CARES, Find a Cure Today (FACT), Constance Silveri, Robertson Research Fund and the Masthead Cove Yacht Club Carol Marcincuk Fund.
This article was selected as a Paper of the Week.
- PTP
- protein-tyrosine phosphatase
- SRC
- proto-oncogene C-Src
- BRK
- breast tumor kinase
- CSK
- C-terminal SRC kinase
- SRMS
- Src-related kinase lacking C-terminal regulatory tyrosine and N-terminal myristylation sites
- PAG/CBP
- phosphoprotein associated with lycosphingolipid-enriched microdomains/CSK-binding protein
- EGF
- epidermal growth factor
- IGF
- insulin-like growth factor
- TCPTP
- T-cell protein tyrosine phosphatase
- p130CAS
- Crk-associated substrate p130CAS
- VSVG
- vesicular stomatitis virus glycoprotein
- ODG
- octyl-d-glucoside
- SH
- Src homology
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Danforth W. H., Helmreich E., Cori C. F. (1962) The effect of contraction and of epinephrine on the phosphorylase activity of frog sartorius muscle. Proc. Natl. Acad. Sci. U.S.A. 48, 1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hayashi K., Yonemura S., Matsui T., Tsukita S. (1999) Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxyl-terminal threonine phosphorylated in cultured cells and tissues. J. Cell Sci. 112, 1149–1158 [DOI] [PubMed] [Google Scholar]
- 3. Andersen J. N., Jansen P. G., Echwald S. M., Mortensen O. H., Fukada T., Del Vecchio R., Tonks N. K., Møller N. P. (2004) A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 18, 8–30 [DOI] [PubMed] [Google Scholar]
- 4. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 5. Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. (2004) Protein tyrosine phosphatases in the human genome. Cell 117, 699–711 [DOI] [PubMed] [Google Scholar]
- 6. Tonks N. K. (2013) Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J. 280, 346–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andersen J. N., Mortensen O. H., Peters G. H., Drake P. G., Iversen L. F., Olsen O. H., Jansen P. G., Andersen H. S., Tonks N. K., Møller N. P. (2001) Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 21, 7117–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pulido R., Zúñiga A., Ullrich A. (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 17, 7337–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garton A. J., Burnham M. R., Bouton A. H., Tonks N. K. (1997) Association of PTP-PEST with the SH3 domain of p130cas; a novel mechanism of protein tyrosine phosphatase substrate recognition. Oncogene 15, 877–885 [DOI] [PubMed] [Google Scholar]
- 10. Salmeen A., Andersen J. N., Myers M. P., Tonks N. K., Barford D. (2000) Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol. Cell 6, 1401–1412 [DOI] [PubMed] [Google Scholar]
- 11. Myers M. P., Andersen J. N., Cheng A., Tremblay M. L., Horvath C. M., Parisien J. P., Salmeen A., Barford D., Tonks N. K. (2001) TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 276, 47771–47774 [DOI] [PubMed] [Google Scholar]
- 12. Ren L., Chen X., Luechapanichkul R., Selner N. G., Meyer T. M., Wavreille A. S., Chan R., Iorio C., Zhou X., Neel B. G., Pei D. (2011) Substrate specificity of protein tyrosine phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry 50, 2339–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lim W. A., Pawson T. (2010) Phosphotyrosine signaling: evolving a new cellular communication system. Cell 142, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y., Ning H., Ren F., Zhang Y., Rong Y., Wang Y., Su F., Cai C., Jin Z., Li Z., Gong X., Zhai Y., Wang D., Jia B., Qiu Y., et al. (2014) GdX/UBL4A specifically stabilizes the TC45/STAT3 association and promotes dephosphorylation of STAT3 to repress tumorigenesis. Mol. Cell 53, 752–765 [DOI] [PubMed] [Google Scholar]
- 15. Okada M. (2012) Regulation of the SRC family kinases by Csk. Int. J. Biol. Sci. 8, 1385–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng X. M., Wang Y., Pallen C. J. (1992) Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 359, 336–339 [DOI] [PubMed] [Google Scholar]
- 17. Mitchell P. J., Barker K. T., Martindale J. E., Kamalati T., Lowe P. N., Page M. J., Gusterson B. A., Crompton M. R. (1994) Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 9, 2383–2390 [PubMed] [Google Scholar]
- 18. Kamalati T., Jolin H. E., Mitchell P. J., Barker K. T., Jackson L. E., Dean C. J., Page M. J., Gusterson B. A., Crompton M. R. (1996) Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J. Biol. Chem. 271, 30956–30963 [DOI] [PubMed] [Google Scholar]
- 19. Qiu H., Zappacosta F., Su W., Annan R. S., Miller W. T. (2005) Interaction between Brk kinase and insulin receptor substrate-4. Oncogene 24, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 20. Qiu H., Miller W. T. (2002) Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 277, 34634–34641 [DOI] [PubMed] [Google Scholar]
- 21. Qiu H., Miller W. T. (2004) Role of the Brk SH3 domain in substrate recognition. Oncogene 23, 2216–2223 [DOI] [PubMed] [Google Scholar]
- 22. Barker S. C., Kassel D. B., Weigl D., Huang X., Luther M. A., Knight W. B. (1995) Characterization of pp60c-src tyrosine kinase activities using a continuous assay: autoactivation of the enzyme is an intermolecular autophosphorylation process. Biochemistry 34, 14843–14851 [DOI] [PubMed] [Google Scholar]
- 23. Casnellie J. E. (1991) Assay of protein kinases using peptides with basic residues for phosphocellulose binding. Methods Enzymol. 200, 115–120 [DOI] [PubMed] [Google Scholar]
- 24. Lantz K. A., Hart S. G., Planey S. L., Roitman M. F., Ruiz-White I. A., Wolfe H. R., McLane M. P. (2010) Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 18, 1516–1523 [DOI] [PubMed] [Google Scholar]
- 25. Krishnan N., Koveal D., Miller D. H., Xue B., Akshinthala S. D., Kragelj J., Jensen M. R., Gauss C. M., Page R., Blackledge M., Muthuswamy S. K., Peti W., Tonks N. K. (2014) Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 10, 558–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goldstein B. J., Bittner-Kowalczyk A., White M. F., Harbeck M. (2000) Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J. Biol. Chem. 275, 4283–4289 [DOI] [PubMed] [Google Scholar]
- 27. Fan G., Lin G., Lucito R., Tonks N. K. (2013) Protein-tyrosine phosphatase 1B antagonized signaling by insulin-like growth factor-1 receptor and kinase BRK/PTK6 in ovarian cancer cells. J. Biol. Chem. 288, 24923–24934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu W., Harrison S. C., Eck M. J. (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595–602 [DOI] [PubMed] [Google Scholar]
- 29. Nada S., Okada M., MacAuley A., Cooper J. A., Nakagawa H. (1991) Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 351, 69–72 [DOI] [PubMed] [Google Scholar]
- 30. Park S. H., Lee K. H., Kim H., Lee S. T. (1997) Assignment of the human PTK6 gene encoding a non-receptor protein tyrosine kinase to 20q13.3 by fluorescence in situ hybridization. Cytogenet. Cell Genet. 77, 271–272 [DOI] [PubMed] [Google Scholar]
- 31. Kohmura N., Yagi T., Tomooka Y., Oyanagi M., Kominami R., Takeda N., Chiba J., Ikawa Y., Aizawa S. (1994) A novel nonreceptor tyrosine kinase, Srm: cloning and targeted disruption. Mol. Cell. Biol. 14, 6915–6925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pellicena P., Stowell K. R., Miller W. T. (1998) Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J. Biol. Chem. 273, 15325–15328 [DOI] [PubMed] [Google Scholar]
- 33. Sondhi D., Xu W., Songyang Z., Eck M. J., Cole P. A. (1998) Peptide and protein phosphorylation by protein tyrosine kinase Csk: insights into specificity and mechanism. Biochemistry 37, 165–172 [DOI] [PubMed] [Google Scholar]
- 34. Kawabuchi M., Satomi Y., Takao T., Shimonishi Y., Nada S., Nagai K., Tarakhovsky A., Okada M. (2000) Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature 404, 999–1003 [DOI] [PubMed] [Google Scholar]
- 35. Brdicka T., Pavlistová D., Leo A., Bruyns E., Korínek V., Angelisová P., Scherer J., Shevchenko A., Hilgert I., Cerný J., Drbal K., Kuramitsu Y., Kornacker B., Horejsí V., Schraven B. (2000) Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 191, 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blake R. A., Broome M. A., Liu X., Wu J., Gishizky M., Sun L., Courtneidge S. A. (2000) SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell. Biol. 20, 9018–9027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shima T., Nada S., Okada M. (2003) Transmembrane phosphoprotein Cbp senses cell adhesion signaling mediated by Src family kinase in lipid rafts. Proc. Natl. Acad. Sci. U.S.A. 100, 14897–14902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown D. A., London E. (1998) Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14, 111–136 [DOI] [PubMed] [Google Scholar]
- 39. Zhang S. Q., Yang W., Kontaridis M. I., Bivona T. G., Wen G., Araki T., Luo J., Thompson J. A., Schraven B. L., Philips M. R., Neel B. G. (2004) Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol. Cell 13, 341–355 [DOI] [PubMed] [Google Scholar]
- 40. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 41. Goel R. K., Miah S., Black K., Kalra N., Dai C., Lukong K. E. (2013) The unique N-terminal region of SRMS regulates enzymatic activity and phosphorylation of its novel substrate docking protein 1. FEBS J. 280, 4539–4559 [DOI] [PubMed] [Google Scholar]
- 42. Hrdinka M., Horejsi V. (2014) PAG–a multipurpose transmembrane adaptor protein. Oncogene 33, 4881–4892 [DOI] [PubMed] [Google Scholar]
- 43. Solheim S. A., Torgersen K. M., Taskén K., Berge T. (2008) Regulation of FynT function by dual domain docking on PAG/Cbp. J. Biol. Chem. 283, 2773–2783 [DOI] [PubMed] [Google Scholar]
- 44. Dubé N., Cheng A., Tremblay M. L. (2004) The role of protein tyrosine phosphatase 1B in Ras signaling. Proc. Natl. Acad. Sci. U.S.A. 101, 1834–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bentires-Alj M., Neel B. G. (2007) Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 67, 2420–2424 [DOI] [PubMed] [Google Scholar]
- 46. Julien S. G., Dubé N., Read M., Penney J., Paquet M., Han Y., Kennedy B. P., Muller W. J., Tremblay M. L. (2007) Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 39, 338–346 [DOI] [PubMed] [Google Scholar]