

Background: Poxvirus encodes up to four TNF decoy receptors (vTNFRs) that mimic etanercept, an anti-TNF drug used in the clinic.
Results: vTNFRs display differences in ligand specificity and inhibitory potency.
Conclusion: Some vTNFRs are more specific and potent TNF inhibitors than etanercept.
Significance: This study may help to understand the role of vTNFRs in pathogenesis and improve the anti-TNF treatments.
Keywords: inflammation, poxvirus, receptor, tumor necrosis factor (TNF), virus, TNF ligand superfamily, etanercept, immune evasion, vTNFR
Abstract
The blockade of tumor necrosis factor (TNF) by etanercept, a soluble version of the human TNF receptor 2 (hTNFR2), is a well established strategy to inhibit adverse TNF-mediated inflammatory responses in the clinic. A similar strategy is employed by poxviruses, encoding four viral TNF decoy receptor homologues (vTNFRs) named cytokine response modifier B (CrmB), CrmC, CrmD, and CrmE. These vTNFRs are differentially expressed by poxviral species, suggesting distinct immunomodulatory properties. Whereas the human variola virus and mouse ectromelia virus encode one vTNFR, the broad host range cowpox virus encodes all vTNFRs. We report the first comprehensive study of the functional and binding properties of these four vTNFRs, providing an explanation for their expression profile among different poxviruses. In addition, the vTNFRs activities were compared with the hTNFR2 used in the clinic. Interestingly, CrmB from variola virus, the causative agent of smallpox, is the most potent TNFR of those tested here including hTNFR2. Furthermore, we demonstrate a new immunomodulatory activity of vTNFRs, showing that CrmB and CrmD also inhibit the activity of lymphotoxin β. Similarly, we report for the first time that the hTNFR2 blocks the biological activity of lymphotoxin β. The characterization of vTNFRs optimized during virus-host evolution to modulate the host immune response provides relevant information about their potential role in pathogenesis and may be used to improve anti-inflammatory therapies based on soluble decoy TNFRs.
Introduction
The human TNF ligand superfamily (TNFSF)3 comprises 19 cytokines that signal through 29 cellular receptors included in the TNF receptor superfamily (1). Among these, TNFα (TNF) and lymphotoxin α (LTα) exert important immune functions by their interaction with TNF receptor 1 and 2 (TNFR1 and TNFR2). Like other TNFSF members, TNF is produced after the enzymatic cleavage of a type II transmembrane TNF precursor that is processed by the TNF converting enzyme to release the soluble cytokine (2). By contrast, LTα is a secreted homotrimer of three α-subunits with no membrane precursor. A related cytokine, LTβ, is a non-secreted membrane cytokine that can appear in two different heterotrimers of α and β subunits, LTα1β2 and LTα2β1 (3). These two forms of LTβ signal through a distinct and specific receptor, LTβ receptor (LTβR) (4).
The importance of TNFSF signaling networks in antiviral defense is underpinned by the existence of numerous distinct viral proteins specifically dedicated to their modulation. Poxviruses, a family of complex DNA containing enveloped viruses that include important human and animal pathogens as well as broadly used vaccine vectors, have developed an array of proteins targeting TNF-induced intracellular pathways (5). A unique strategy developed by poxviruses for the control of TNF is the expression of secreted TNF binding proteins. The tanapox virus protein 2L displays a major histocompatibility complex I-like folding (6) and can bind TNF with high affinity and inhibit its activity (7). Orthologues are found in the other members of the Yatapoxvirus genus, Yaba-like disease virus and yaba monkey tumor virus, and in the genera Suipoxvirus and Cervidpoxvirus (7, 8). On the other hand, orthopoxviruses express secreted proteins with sequence similarity to the ligand binding region of cellular TNFRs (9), and these viral homologues have been included in a protein family termed viral TNFRs (vTNFRs). Five different vTNFRs have been identified: cytokine response modifier B (CrmB), CrmC, CrmD, CrmE, and a viral homologue of CD30 (10–15). The ectromelia virus (ECTV) CD30 interacts with CD30L to inhibit the CD30-CD30L interaction and to signal through membrane CD30L, causing down-regulation of the Th1 response (14). Nevertheless, the viral CD30 is not a major virulence factor in the classical mousepox model (16). The other four vTNFRs are able to bind TNF and inhibit its biological activity by mimicking the extracellular domain of the cellular TNFR1/2, as shown by the crystal structure of CrmE, the sole vTNFR structure available (17). A contribution of vTNFRs to poxvirus pathogenesis has been shown by using recombinant vaccinia viruses (VACVs) expressing CrmE, CrmB, or CrmC, which displayed increased virulence in an intranasal mouse infection (18). Additionally a CPXV lacking CrmB but not other vTNFRs displayed an increased LD50 in infected mice (19). Finally, the Leporipoxvirus myxoma virus expresses another vTNFR named M-T2, whose absence resulted in reduced clinical signs of illness in infected rabbits (20).
vTNFRs are differentially conserved among orthopoxviral species, but the reasons for this variability are not defined. Thus, variola virus (VARV), the causative agent of smallpox (21), encodes one copy of a single active vTNFR, CrmB that can efficiently inhibit human TNF and LTα (22). CrmB is also the only active vTNFR gene in monkeypox virus (MPXV) that harbors two copies of this vTNFR. On the other hand, some cowpox virus (CPXV) strains, with a broad host range and a current public health concern despite its generally low virulence in humans (23), encode all four vTNFRs, with the CrmB gene present in two copies in their genomes. In ECTV, a strict mouse pathogen where vTNFR gene conservation has been studied across isolates, the CrmD gene is present in two copies, whereas the remaining vTNFRs appear as pseudogenes (24–26). In most VACV strains vTNFRs genes are either deleted or truncated (27, 28), but strains Lister, Evans, and USSR encode both CrmC and CrmE (18, 29). This pattern (summarized in Table 1) suggests that vTNFRs have been gained or lost selectively across poxviral isolates and reflects the intrinsic unique evolutionary history of each isolate.
TABLE 1.
vTNFR gene conservation in selected Orthopoxvirus strains
na., not annotated in complete genome sequence; tr., truncated gene; -, gene absent.
| Virus | CrmB | CrmC | CrmD | CrmE |
|---|---|---|---|---|
| CPXV GRI-90 | D2L/I4Ra | A56R | K2R | K3R |
| CPXV BRb | CPXV005/CPXV226 | CPXV191 | CPXV221 | na./(tr.) |
| ECTV NAVAL | EVN002P (tr.)/EVN205P (tr.) | EVN175P (tr.) | EVN006/EVN201 | EVN005P (tr.)/EVN202P (tr.) |
| VARV BSH75 | G2R | - | - | - |
| VACV Lister | List002 (tr.)/List200 (tr.) | List172 | - | List195 |
| VACV WR | VACVWR004 (tr.)/VACVWR215 (tr.) | VACVWR179 (tr.) | - | - |
a The names of the genes are indicated.
b BR, Brighton Red; BSH75, Bangladesh 1975; WR, Western Reserve.
Importantly, vTNFRs are also differentially expressed during infection in terms of time of expression and abundance. Thus, CrmB orthologues appear to be expressed at early times of infection in all viral species (10, 11), whereas in ECTV and CPXV CrmD and CPXV CrmC are late genes (11). CPXV CrmC is expressed to higher levels than CrmD, whereas CrmD is produced in higher amounts by ECTV (12). Moreover, although vTNFRs were defined as soluble TNF inhibitors, CrmE from VACV encodes both soluble and membrane vTNFR activity (18). These differences in the expression kinetics and location together with the potential unique biochemical properties of each vTNFRs could explain their different expression pattern among poxvirus species. However, the latter has not been properly approached, and the characterization of the binding ligands and specificity of vTNFRs remain incomplete. Although previous studies on vTNFRs have been focused on a single or only a few proteins, a comprehensive analysis of the ligand host specificity of the vTNFRs and their orthologues to explore the implications of this variability in virus-host interaction is lacking. Furthermore, despite the structural similarity among TNFSF members, the binding of vTNFRs to other ligands distinct to TNF or LTα has been scarcely analyzed. Here we report the first comprehensive and comparative study of the affinity and the inhibitory activity of six different vTNFRs for TNF and LTα from mouse and human origin. We also demonstrate that CrmB and CrmD inhibit the biological activity of LTβ, adding a new immunomodulatory function to vTNFRs. Our study shows that there are significant specificity differences that may explain the vTNFR expression profiles among poxvirus species.
TNF inhibition with antagonists is an effective strategy to treat TNF-mediated chronic inflammatory diseases. In this study we have compared the biochemical properties of a soluble version of the human TNFR2, named etanercept (30), with those of the vTNFRs and found that some viral receptors are more potent and specific TNF inhibitors than etanercept.
Experimental Procedures
Cells and Reagents
L929 were grown in 10% FCS DMEM. Recombinant baculoviruses were grown in adherent Hi5 insect cells in 10% FCS TC-100 medium or suspension Hi5 cells maintained in Express Five (Life Technologies) medium for the generation of recombinant baculovirus or protein expression, respectively. Recombinant cytokines were obtained from R&D Systems and Peprotech Inc. The recombinant receptor mTNFR1-Fc was purchased from R&D Systems.
Construction of Recombinant Baculoviruses
The coding genes, lacking the signal peptide, were cloned into pFastBac1 (Life Technologies) for the generation of recombinant baculovirus. Genes were PCR-amplified from recombinant plasmids, except for CrmD from CPXV strain Brighton Red (CPXV221; GenBankTM accession number AAM13659), which was amplified from viral genomic DNA. CrmB (CPXV005; GenBankTM AAA60952) and CrmC (CPXV191; GenBankTM AAM13631) from CPXV strain Brighton Red were PCR-amplified from pAH21 and pRA112 (22), respectively. CrmE (K3R; GenBankTM AJ272008) from CPXV strain Elephantpox was amplified from pMS3 (13). The CrmB gene (G2R; GenBankTM AAA60933) from VARV strain Bangladesh 1975 was generated by site-directed mutagenesis of the camelpox virus orthologue. Permission from the World Health Organization was granted to hold VARV DNA, and its manipulation was performed in accordance to the established rules. A plasmid containing this sequence, pRA107, was used as template (22). pMS1 (13) was used to amplify the CrmD gene (E6; GenBankTM AJ567688) from ECTV strain Hampstead.
Viral genes were amplified by PCR using a pair of primers containing BamHI and XhoI restriction sites at the 5′- and the 3′-termini, respectively. Genes were ligated to BamHI-XhoI-digested pAL7 (31), a modified pFastBac1 vector bearing the honeybee melittin signal peptide at the 5′ termini and a C-terminal V5-His6 tag. The resulting constructions were named as pSP3 (ECTV CrmD), pSP8 (CPXV CrmE), pSP9 (CPXV CrmC), pSP10 (CPXV CrmB), pSP11 (CPXV CrmD), and pSP12 (VARV CrmB).
The ligand binding domain of human TNFR2 (amino acids 23–257; GenBankTM BC052977) was amplified from pBhTNFR2 (kindly provided by Daniela Maennel, University of Regensburg, Germany) and cloned into BamHI-NotI-digested pFastBac1 to generate pRM4. The human IgG1 Fc fragment was subcloned into pRM4 from pMS18 (14) using NotI and SphI restriction enzymes. The resulting construct coding for the extracellular domain of hTNFR2 fused to a human Fc was named pRM6.
Recombinant baculoviruses were generated using the Bac-to-Bac system (Life Technologies) (22). Viral stocks were amplified by infecting Hi5 cells at low viral multiplicity of infection (0.1–0.01 pfu/cell) to obtain high titer baculovirus stocks for protein production.
Protein Expression and Purification
Recombinant His-tagged proteins were purified from supernatants of Hi5 cells infected at high multiplicity using nickel-nitrilotriacetic acid columns (Qiagen) (22). hTNFR2-Fc was purified using protein A-coupled Sepharose (Sigma) followed by size exclusion chromatography. Protein stocks were dialyzed against PBS and quantified by both BCA assay (Pierce) and gel densitometry and stored at −80 °C. When both quantification methods did not provide comparable protein concentration values, the concentration obtained by gel densitometry was assumed for subsequent experiments.
Cytotoxicity Assays
The activity of recombinant vTNFRs and hTNFR2-Fc against hTNF, hLTα, mTNF, mLTα, mLTα1β2, and mLTα2β1 was tested by cytotoxicity assays on L929 cells (13). Briefly, 20 ng/ml hTNF, hLTα, and mTNF were incubated in the presence of increasing molar ratios of recombinant protein. Due to its low specific activity, mLTα, mLTα2β1, and mLTα1β2 were used at 780, 44, and 440 ng/ml, respectively. After 1 h of incubation at 37 °C, mixtures were added over L929 cells in 96-well plates, and actinomycin D (Sigma) at 4 μg/ml was supplemented. Cell viability was assessed after 18 h by using the Cell Titer Aqueous One Solution kit (Promega) and measuring the absorbance (A) at 492 nm with a Sunrise microplate reader (Tecan). Samples with cytokine but without inhibitor (0% viability) were used to normalize the background signal. After normalization, cell viability was calculated as the ratio SampleA492/ControlA492, where ControlA492 is the average absorbance from samples without cytokine (media, 100% viability).
Surface Plasmon Resonance (SPR) Assays
SPR experiments were performed using a Biacore X biosensor (GE Healthcare) and immobilizing recombinant vTNFRs and hTNFR2-Fc to CM4 chips (GE Healthcare) by amine coupling as described (22). For the determination of affinity constants, receptors were immobilized to a final response of 350–800 response units. At least 10 different concentrations (in the range 0.1–200 nm) of the cytokines were injected at 30 μl/min in HBS-EP and the collected sensorgrams were aligned and fitted to a 1:1 Langmuir model using the software Biaevaluation 3.2. The kinetic constants were determined from fittings containing no less than 6 concentration sensorgrams.
For SPR competition experiments, 2,000 response units of mTNFR1-Fc (R&D) were immobilized on a CM4 chip. Increasing concentrations of CrmE and CrmC were preincubated in HBS-EP buffer with 30 nm concentrations of each cytokine during 15 min on ice. The mixture was injected at 10 μl/min over the mTNFR1-Fc chip, and the response units at 10 s before the end of the injection was recorded.
For the binding screening of mouse TNFSF ligands to CrmD, the cytokines (TNFSF3, LTβ (LTα1β2); TNFSF4, OX40L; TNFSF5, CD40L; TNFSF6, FasL; TNFSF7, CD27L; TNFSF8, CD30L; TNFSF10, TRAIL; TNFSF11, RANKL; TNFSF12, TWEAK; TNFSF13, APRIL; TNFSF14, LIGHT; TNFSF15, TL1A) were injected at 100 nm in HBS-EP and a flow rate of 10 μl/min over ECTV CrmD immobilized onto a CM4 chip to high density (≈2000 response units).
Results
Expression and Purification of vTNFRs
In orthopoxviruses four distinct secreted TNF-binding proteins with similarity to the extracellular domain of cellular TNFRs have been described. These are named CrmB, CrmC, CrmD, and CrmE, and orthologues are conserved in different combinations among viral species (Table 1) (32). The N-terminal region of vTNFRs mimics the extracellular domain of TNFRs and corresponds to their TNF binding moiety (17). In both cellular and viral TNFRs, the N terminus contains up to four repetitions of cysteine-rich domains (CRDs). The cysteine residues, essential for the folding of the CRDs, are highly conserved in the first three CRDs, whereas the four canonical cysteines of the CRD4 of hTNFR2 are inconsistently conserved in vTNFRs (Fig. 1A). Nevertheless, the CRD4 is dispensable for ligand binding in hTNFR2 (33), and CrmE is known to be a three-CRD containing vTNFR (17). For the other vTNFRs, we assumed the previously proposed CRD configuration, where CrmB and CrmD have four and CrmC has three CRDs (10–12). Overall, significant differences can be found in the primary sequences of the TNF binding domain among vTNFRs (Fig. 1A). In fact, only CrmD and CrmB share >50% amino acid identity in this domain, whereas the identity of CrmE to any other vTNFR is ∼45% and down to 35% in the case of CrmC, the most divergent vTNFR (Fig. 1B). To address the possible implications of this diversity in terms of viral immunomodulation, we have characterized these proteins further on a side-by-side basis. In addition, as schematically depicted in Fig. 1C, CrmB and CrmD contain an additional C-terminal extension, termed the SECRET domain, that has been described as an independent folding domain with the capacity to block chemokine activity (22, 34), suggesting differential roles during infection as compared with the CrmC and CrmE proteins that lack this domain (Fig. 1C).
FIGURE 1.

Expression of recombinant vTNFRs. A, sequence alignment using Clustal Omega of the TNF binding domains of ECTV CrmD (gene E6, strain Hampstead, UniProt/TrEMBL accession no. O57300), CPXV CrmD (gene CPXV221, strain Brighton Red, UniProt/TrEMBL accession no. O57079), VARV CrmB (gene G2R, strain Bangladesh 1975, UniProt/TrEMBL accession no. P34015), CPXV CrmB (gene CPXV005, strain Brighton Red, UniProt/TrEMBL accession no. Q85308), CPXV CrmE (gene K3R, strain Elephantpox, UniProt/TrEMBL accession no. Q9DJL2), CPXV CrmC (gene CPXV191, strain Brighton Red, UniProt/TrEMBL accession no. Q9YP87), and human TNFR2 (UniProt/TrEMBL accession no. P20333). The sequences were aligned without the signal peptide and split into the different CRDs for clarity purposes. Cysteine residues are highlighted in each CRD. B, percentage of amino acid identity among the TNF binding domain of vTNFRs and hTNFR2 determined by using the web server SIAS and the substitution matrix BLOSUM62. C, schematic representation of the baculovirus expressed vTNFRs. The anti-TNFSF domain and the SECRET domain are indicated. Below, Coomassie Blue-stained gels showing 0.5–1 μg of each purified vTNFR and hTNFR2-Fc. Molecular mass is indicated in kDa.
We expressed and purified the four different vTNFRs encoded by CPXV as well as variants derived from VARV (CrmB) and ECTV (CrmD) (Fig. 1C). The vTNFRs were expressed in the baculovirus system fused to C-terminal V5 and His tags, and the secreted proteins were purified by affinity chromatography. Additionally, we purified the hTNFR2-Fc protein (hTNFR2 hereon), a molecule known in the clinic as etanercept and used for the treatment of TNF-mediated immune disorders. We aimed to compare the inhibitory and binding abilities of this anti-TNF drug with those of vTNFRs.
Anti-TNFSF Activity and Binding Affinity of vTNFRs
Overall, vTNFRs are known to bind either TNF alone or TNF as well as LTα. The proteins assessed here are derived from viruses infecting human and/or rodents. Therefore, to obtain comparative data for all purified vTNFRs, we performed SPR analysis to determine their binding affinities for both human and murine TNF and LTα (Table 2). Some examples of the sensorgram fittings performed to obtain the affinity constants are shown in Fig. 2. The results confirmed the differentiation in two groups, with the CrmB and CrmD proteins binding both TNF and LTα, whereas CrmC and CrmE were selective TNF binders. The affinity constants for TNF were globally 1 order of magnitude lower (100–2420 pm) than those calculated, when applicable, for LTα (3.11–50.40 nm). Remarkably, regarding LTα affinities, CrmB proteins showed a higher affinity for the human than for the mouse ligand, whereas the opposite scenario was found for CrmD proteins (Table 2). This observation hints at potential viral adaptations to different hosts.
TABLE 2.
Kinetic parameters, derived affinity constants, and half-life (t1/2) of the binding of the recombinant vTNFRs to mTNF, hTNF, mLTα, and hLTα
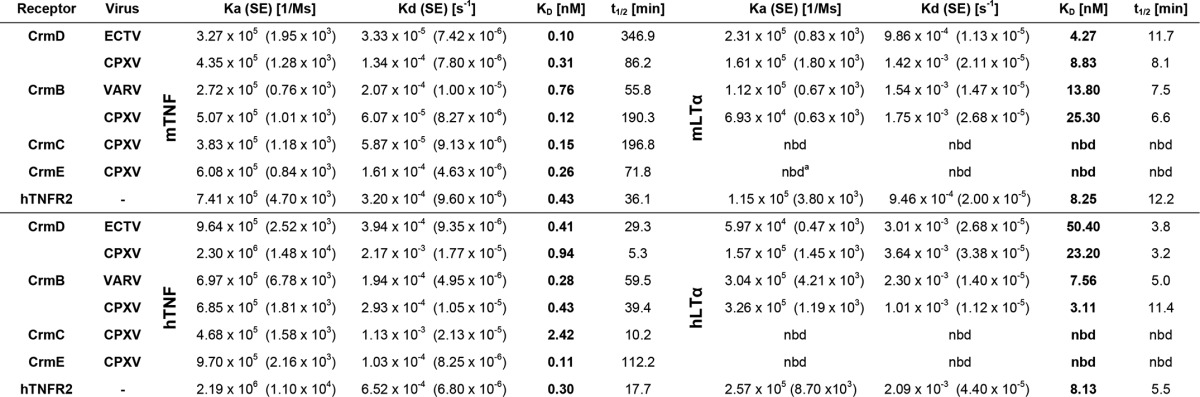
a nbd., no binding detected.
FIGURE 2.

Surface plasmon resonance analysis of the TNFSF members binding to vTNFRs. Four examples of the binding sensorgrams and fittings obtained for the determination of the kinetic constants of the TNFSF-vTNFR interactions: mTNF to CPXV CrmD (A), hTNF to CPXV CrmB (B), mLTα to ECTV CrmD (C), and hLTα to VARV CrmB (D). Binding and dissociation of several concentrations of TNFSF ligands at 30 μl/min were recorded and adjusted to a 1:1 Langmuir fitting (solid lines). The nanomolar concentration corresponding to each sensorgram is indicated. The arrowhead points the end of the injection.
Binding of the ligands can result in the blockade of their biologic activity, although this has been shown not always to be the case for vTNFRs. For instance, the CrmE protein binds TNF from different species while only inhibiting hTNF activity (13, 18). To obtain a complete understanding of the blocking capacity of the proteins under study, we performed biological activity assays. The addition of TNF or LTα to L929 cells induces cell death. In the presence of a blocking protein, cells will be protected from this effect, resulting in preservation of the cell monolayer. As shown in Fig. 3, the four cytokines efficiently induced cell death resulting in complete killing of the cells (0% viability).
FIGURE 3.

TNF and LTα inhibitory activity of vTNFRs and hTNFR2. Inhibition of the cytotoxicity induced by mTNF (A), hTNF (B), mLTα (C), and hLTα (D) in L929 cells in the presence of purified recombinant vTNFRs or hTNFR2 at the indicated cytokine:protein molar ratios. Cell viability was assessed as the absorbance at 492 nm using the Cell Titer Aqueous One Solution kit (Promega). Values were normalized with the absorbance recorded from samples containing only the corresponding cytokine, and these were set to zero. Data are represented as the percentage relative to the absorbance in the absence of cytokine (media). Means ± S.D. of triplicate samples of three representative experiments are shown.
As expected, CrmE and CrmC did not protect cells from hLTα- or mLTα-induced cytotoxicity (Fig. 3, C and D), in accordance with their lack of binding for these ligands. Although both CrmB and CrmD proteins effectively inhibited the activity of mLTα (Fig. 3C), only the CrmB proteins could protect cells from hLTα-induced cytotoxicity to some extent, achieving 40–50% cell survival when incubated with a 200-fold molar excess of vTNFR (Fig. 3D). This might reflect the higher binding affinity for this ligand of the CrmB proteins (KD 7.56 and 3.11 nm) as compared with that of the CrmD proteins (KD 50.40 and 23.20 nm).
Despite the similar affinity of CrmB and CrmD proteins for both hTNF and mTNF, interesting differences in their capacity to block these cytokines were detected. CrmB proteins protected from mTNF-induced cell death best, with 80% cell viability rates at a 1:5 molar ratio, whereas to achieve similar protection levels against hTNF, 1:50 ratios were required. Between CrmDs, however, although the ECTV orthologue effectively blocked both human and murine TNF (requiring 50-fold molar excess to reach full protection levels), the CPXV-derived protein was a surprisingly poor TNF inhibitor, protecting 70 and 30% of cells when incubated with mTNF and hTNF, respectively, at a 1:200 molar ratio. This phenomenon might be related to the reduced half-life (t½) of the CPXV CrmD-TNF complexes as compared with those of the ECTV CrmD-TNF complexes (Table 2).
As previously shown, CrmE was unable to protect from mTNF activity, whereas it protected from hTNF (Fig. 3, A and B). In contrast to what previous data suggested (13, 18), CrmE displayed high binding affinities for both mTNF (KD 0.26 nm) and hTNF (KD 0.11 nm) (Table 2), suggesting that, unlike for hTNF, the mTNF residues involved in the biological effect of this cytokine are not efficiently blocked by CrmE. CrmC showed the opposite specificity in terms of activity, inhibiting the mTNF cytotoxic effect but not that of hTNF (Fig. 3, A and B). This confirms previous findings obtained with CrmC-containing supernatants from VACV-infected cells (18). The activity of CrmC correlated with a relatively worse affinity for the human ligand, with KD values of 0.15 and 2.42 nm, respectively (Table 2), although differences in the binding mode might also contribute to this effect.
Importantly, although hTNFR2 bound all these ligands with an affinity in the same range as the vTNFRs (Table 2), it was found to be a much weaker inhibitor than vTNFRs (Fig. 3). The purified hTNFR2 expressed in the baculovirus system had a binding affinity for TNF similar to that of commercially available etanercept (not shown). Although the vTNFRs completely blocked the activity of murine ligands, these were only partially blocked (20 and 50% cell viability) by hTNFR2 at the highest molar ratios tested (Fig. 3, A and C). In the case of the human ligands, the degree of cell protection achieved with hTNFR2 was comparable to the vTNFRs activity at the highest dose (Fig. 3, B and D). However, CrmB, CrmD, and CrmE achieved complete protection from hTNF-induced cytotoxicity at lower doses than hTNFR2 (Fig. 3B). These results indicate that vTNFRs are more efficient anti-TNFSF proteins than one of the antagonists used in the clinic to block TNF activity.
Differences in the Blockage of the mTNF-TNFR1 and hTNF-TNFR1 Interaction Explain the Observed Specificity for CrmE and CrmC
To understand the reason for the lack of inhibition of hTNF activity by CrmC and mTNF by CrmE despite their high binding affinities (KD 2.42 and 0.26 nm, respectively), we next studied their ability to compete ligand-cell receptor interactions. Thus, we assayed by SPR the capacity of CrmC and CrmE to inhibit the interaction of mTNF and hTNF with mTNFR1, the cellular receptor that drives their cytotoxic effect on L929 cells (35). As shown in Fig. 4, increasing amounts of recombinant CrmE and CrmC were able to compete the binding of mTNF and hTNF to the mTNFR1. Although both CrmE and CrmC bind mTNF with similar high affinity, 40 nm CrmC was sufficient to reduce by 50% the mTNF-TNFR1 binding, whereas >160 nm CrmE was required to achieve a similar degree of inhibition (Fig. 4A). The opposite situation was found in the case of the hTNF-mTNFR1 interaction, where CrmE was able to inhibit binding of hTNF to the receptor almost completely, whereas CrmC only diminished it to 70% at the highest dose tested (Fig. 4B). These results showed that vTNFRs block the TNF binding to its cellular receptor and that CrmE and CrmC bind hTNF and mTNF using different molecular strategies in such a way that the residues involved in receptor interaction are only efficiently blocked in hTNF and mTNF, respectively, accounting for the observed differences in their biological activities.
FIGURE 4.
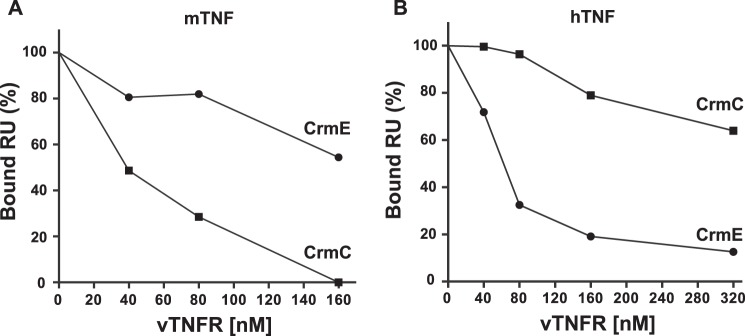
Inhibition of hTNF and mTNF binding to mTNFR1 by CrmE and CrmC. SPR competition experiment of 30 nm mTNF (A) and hTNF (B) binding to mTNFR1-coupled biosensor chips with increasing amounts of soluble CrmC (■) and CrmE (●). The percentage of binding refers to binding in the absence of soluble vTNFRs. RU, response units.
CrmD, CrmB, and hTNFR2 Bind mLTβ and Inhibit Its Cytotoxic Effect
Among the vTNFRs, CrmD and CrmB have been described as TNF and LTα inhibitors, whereas CrmC and CrmE are known to bind TNF only as confirmed in our study. However, vTNFRs may interact with other members of the TNFSF, which comprises up to 19 structurally related ligands (1). Specifically, CrmE was shown not to interact with any other TNFSF ligands besides TNF (13). Similarly, CD30L was proved to be the only ligand of vCD30 (16). However, a similar analysis has not been performed with the other vTNFRs yet. To search for possible further ligands, we performed a screening by SPR using CrmD from ECTV and all commercially available members of the mouse TNFSF. As shown in Fig. 5A we found that, in addition to mTNF, CrmD was able to bind mLTβ. This cytokine is a membrane ligand that can appear in two different heterotrimeric forms, LTα1β2 and LTα2β1 (3). We determined by SPR the affinity of CrmD and the other receptors for these two forms of mLTβ. As shown in Table 3, CrmB, CrmD, and hTNFR2 but not CrmE and CrmC bound both LTβ heterotrimers with affinities ranging from 14 to 100 nm. LTβ signals through a distinct cell receptor, LTβR (4). The interaction LTβ-LTβR is known to induce cell death in some adenocarcinoma cell lines (36). L929 cells express LTβR (37), and therefore, they are likely to be susceptible to LTβ-induced cytotoxicity. Thus, to study whether the observed vTNFR-mLTβ interactions could result in the blockade of the mLTβ biological activity, a cytotoxicity assay on L929 cells was set up. As shown in Fig. 5, B and C, both forms of mLTβ effectively induced cytotoxicity, which was differently inhibited by vTNFRs and hTNFR2. As expected, CrmE and CrmC did not prevent the killing effect of either mLTβ forms, whereas the CrmD and CrmB orthologues inhibited LTβ heterotrimers in a dose-dependent manner (Fig. 5, B and C). Interestingly, and in agreement with its higher affinity, CrmB proteins were more active than the CrmD orthologues against mLTα2β1 (Fig. 5C). In contrast, the CrmD orthologues were better mLTα1β2 inhibitors than the CrmB proteins (Fig. 5B). In summary, we show that CrmD and CrmB orthologues interfere efficiently not only with the activity of TNF and LTα but also with that of their newly identified ligand, LTβ.
FIGURE 5.

mLTβ inhibitory activity of vTNFRs. A, CrmD binding screening to mouse TNFSF by SPR. Cytokines were injected at 100 nm in HBS-EP over a ECTV CrmD coupled chip at 10 μl/min. The binding to CrmD of all commercially available mouse TNFSF members, from mTNFSF3 (mLTβ, from mLTα1β2) to mTNFSF15 (mTL1A) was recorded. As a control, mTNF was included in the screening. The arrowhead indicates the end of the injection. B and C, inhibition of the cytotoxicity induced by mLTα1β2 (B) and LTα2β1 (C) in L929 cells in the presence of purified recombinant vTNFRs and hTNFR2 at the indicated increasing cytokine:protein molar ratios. Values were normalized with the absorbance recorded from samples containing only the corresponding cytokine, and these were set to zero. Data are represented as the percentage relative to the absorbance in the absence of cytokine (media). Means ± S.D. of triplicate samples of three representative experiments are shown.
TABLE 3.
Kinetic parameters, derived affinity constants, and half-life (t1/2) of the binding of the two forms of mLTβ to ECTV CrmD, VARV CrmB, CrmC, CrmE, and hTNFR2
| Cytokine | Receptor | Ka (S.E.) | Kd (S.E.) | KD | t½ |
|---|---|---|---|---|---|
| 1/ms | s−1 | nm | min | ||
| mLTα1β2 | CrmD | 1.03 × 105 (7.62 × 102) | 1.72 × 10−3 (2.31 × 10−5) | 16.7 | 6.7 |
| CrmB | 1.29 × 105 (1.84 × 103) | 2.58 × 10−3 (4.85 × 10−5) | 20.0 | 4.5 | |
| CrmC | nbda | nbd | nbd | nbd | |
| CrmE | nbd | nbd | nbd | nbd | |
| hTNFR2 | 2.93 × 105 (1.00 × 104) | 5.24 × 10−3 (1.90 × 10−4) | 17.9 | 2.2 | |
| mLTα2β1 | CrmD | 4.25 × 104 (1.35 × 103) | 4.27 × 10−3 (4.89 × 10−5) | 100.0 | 2.7 |
| CrmB | 1.39 × 105 (1.59 × 103) | 1.94 × 10−3 (2.62 × 10−5) | 14.0 | 6.0 | |
| CrmC | nbd | nbd | nbd | nbd | |
| CrmE | nbd | nbd | nbd | nbd | |
| hTNFR2 | 2.96 × 105 (6.70 × 103) | 3.00 × 10−3 (3.70 × 10−5) | 10.1 | 3.8 |
a nbd, no binding detected.
The hTNFR2 blocked the activity of these two forms of mouse LTβ (Fig. 5, B and C), in accordance with its high affinities determined by SPR (KD 17.9 and 10.1 nm). This is, to our knowledge, the first evidence of a biological effect of TNFR2 on the activity of LTβ.
Discussion
The blockade of TNF activity is a potent strategy of immune modulation widely used in the clinic for the treatment of inflammatory diseases such as rheumatoid arthritis or ankylosing spondylitis. The finding that poxviruses have developed during their coevolution with their hosts, a similar strategy based on the secretion of different modified soluble versions of host TNFRs to modulate the inflammatory response suggests the importance of TNF during the pathogenesis of viral infections.
To better understand the differential contribution of vTNFRs to specific poxvirus-host interaction pairs and because most vTNFRs to date had been studied individually and under different conditions, we provide a direct comparison of the inhibitory capabilities and binding affinities of all vTNFRs produced, purified, and assayed on a side by side basis. This greatly expands our previous knowledge of the vTNFR family and allows an accurate interpretation of their potential roles. Up to date only the affinities of CrmB for hTNF, mTNF, and hLT (22, 29) and CrmC for hTNF and mTNF had been published (11, 29). In our analysis this has been expanded to these and new ligands as well as to all six analyzed vTNFRs. Of note, these preexisting affinity data agree with the affinities reported here for those same interactions. Moreover, we describe a new ligand for CrmB and CrmD, LTβ, and we report the vTNFRs activity against a ligand not studied before, the mouse LTα. Additionally, we compared the activities of the vTNFRs with those of a soluble cellular hTNFR2, or etanercept, a molecule used in the clinic as a TNF antagonist, to help understand potential determinants of specificity and activity differences.
CrmB and CrmD have been shown to interact with hLTα (10, 12), but their interaction with mLTα was not explored. We have shown that CrmB and CrmD interact with high affinity with both hLTα and mLTα. In contrast to previous observations (12), the CrmD orthologues failed to inhibit the cytotoxic effect of hLTα in our conditions. Loparev et al. (12) characterized CrmD as a hLTα inhibitor, but they used low hLTα concentrations at high CrmD:hLTα ratios (starting at 250:1 and reaching up to 4000:1). According to our data, CrmD orthologues, which bind mLTα with a KD in the nanomolar range, display a significant increase in the KD for hLTα. Interestingly, CrmB orthologues from VARV and CPXV bind hLTα with higher affinity than mLTα and are the only vTNFRs that inhibit to some extent the hLTα biological function. We conclude that CrmB is a much more potent hLTα inhibitor than CrmD, which may explain why a human virus such as VARV encodes CrmB rather than CrmD. Complementarily, because the affinity of CrmD-mLTα interaction is higher than that of CrmB-mLTα, CrmD could be a more efficient vTNFR for the mouse pathogen ECTV. Little is known about the role of LTα in defense against poxviruses, but the fact that the host-restricted VARV and ECTV encode vTNFRs that are active against LTα in a host-specific manner suggests an important function of this cytokine in the anti-viral response.
In contrast, CrmE and CrmC only interact with TNF. It is known that CrmE binds TNF from different species but only inhibits hTNF (13). Accordingly, we show that the binding affinities for both CrmE-hTNF and CrmE-mTNF interactions are similarly high, but only the interaction with hTNF results in an efficient cell protection. Surprisingly, despite this inability to inhibit mTNF, CrmE enhances the viral virulence in a mouse model when expressed from recombinant VACV (18). Interestingly, CrmC displays the reverse specificity. Both hTNF and mTNF are strongly bound by CrmC, whereas only the cytotoxicity of mTNF is prevented by this protein. In this regard the affinity constants did not anticipate the host specificity of CrmC and CrmE. Nevertheless, we show that CrmE is more efficient in blocking the hTNF-TNFR1 than the mTNF-TNFR1 interaction, suggesting that only in the case of hTNF the residues involved in the receptor binding are efficiently blocked by CrmE. In contrast, CrmC poorly affected the hTNF-TNFR1 interaction but disrupted the binding of mTNF. Therefore, CrmC and CrmE are specific mTNF and hTNF inhibitors, respectively, suggesting their expression pattern is related to the host tropism of the corresponding viral strains. This opposite specificity of CrmC and CrmE was previously described by Reading et al. (18) with protein-containing supernatants; here we confirm their observations with purified proteins and provide the affinity constants for the interaction of these vTNFRs with mTNF and hTNF.
The differences in the anti-TNFSF ligand activity among vTNFR orthologues have been vaguely explored. Similar to VARV, CrmB is the sole vTNFR encoded by MPXV. Gileva et al. (38) defined that, despite their high similarity, CrmB orthologues from VARV, CPXV, and MPXV show different anti-TNF properties, with the VARV orthologue being the most potent hTNF inhibitor. We have not detected significant differences between VARV CrmB and CPXV CrmB, probably because we have studied the CrmB orthologue from CPXV strain Brighton Red, whereas Gileva et al. (38) analyzed the orthologue from CPXV strain GRI-90. The seven-amino acid changes found between the TNF binding domain of the CrmB orthologues of these CPXV strains may confer distinct inhibitory activities. In fact, differing in only six residues, we have shown that CPXV CrmD is a much weaker TNF inhibitor than ECTV CrmD. This inefficient anti-TNF activity of the CrmD from CPXV is probably compensated by other vTNFRs expressed by this virus. In contrast, CrmD and CrmB are the only vTNFRs in ECTV and VARV, respectively, and may have been finely adapted during evolution to efficiently inhibit the TNFSF ligands found in their hosts (mouse or human). Thus, in the case of ECTV and VARV, a single vTNFRs seems to have co-opted all the immune modulatory activities provided by up to four different vTNFRs in CPXV.
We identify here a new ligand for vTNFRs, LTβ. Based on the LTα inhibitory activity of CrmB, Smith et al. (11) proposed that CrmB might bind LTβ. However, the observation that LTβ contains at least one α-subunit does not prove that a LTα binder can interact with LTβ, as underpinned by the fact that these cytokines signal through completely different receptors: TNFR1, TNFR2, and HVEM for LTα and LTβR for LTβ (1, 3). We found that CrmD and CrmB interact with high affinity with the two forms of mLTβ, mLTα1β2 and mLTα2β1, and demonstrated that they are able to block their activity. Although little is known about the role of LTβ in anti-viral responses, it has been shown to be essential in the defense against Theiler's murine encephalomyelitis virus (39). Our findings suggest that LTβ might play an important as yet undescribed role during the host response to poxviral infections.
The results presented here, summarized in Fig. 6, illustrate that every vTNFR possesses unique features of binding affinity and specificity as well as varying degrees of biological activity that are probably finely tuned in relationship with the infected host as well as the complement of immune-modulating proteins of each virus. As additional aspects when considering their roles in pathogenesis, it is important to note that vTNFRs are also differentially expressed during the time course of infection and that possible differences in the location of the vTNFRs such as the described expression of CrmE at the cell surface (18) may also influence their role in vivo.
FIGURE 6.

Summary of the inhibitory activity of vTNFRs against TNFSF members. A, vTNFR inhibitory activities diagram. The cytokines efficiently blocked by each vTNFR are indicated in the corresponding group. B, summary of the binding and inhibitory activities of all the studied vTNFRs. −, no inhibition detected; +, partial inhibition; ++, complete inhibition and low affinity (KD > 10 nm); +++, complete inhibition and high affinity (KD < 10 nm). Regarding the column of mLTβ, activities of CrmD and CrmB are referred to mLTα1β2 and mLTα2β1, respectively.
Etanercept, a soluble version of the cellular hTNFR2 used in the clinic as an anti-TNF drug, was included in our experiments. We demonstrate that hTNFR2 binds and inhibits both mouse forms of LTβ. Regarding the human LTβ, hTNFR2 is thought to interact only with the form LTα2β1 (40), but there are no data to confirm whether this interaction can actually induce any intracellular signaling. Therefore, we report here the first evidence of a biological effect of TNFR2 over LTβ that might have implications for the study of the molecular mechanisms of TNF-related inflammatory diseases treated clinically or experimentally with TNFR2. Etanercept is used for the treatment of diverse TNF-mediated inflammatory diseases to block the excess of soluble TNF that causes an exacerbated inflammatory response (41). However, as shown here, this molecule can inhibit not only TNF but also LTα and LTβ, two potentially unfavorable activities. Indeed, these cytokines are essential for the control of immune homeostasis and infections (42, 43), and an increased risk of infections, autoimmune disorders, and cancer are documented side effects of etanercept. Thus, molecular redesign of this medicine seems to be necessary to target TNF specifically and avoid undesirable effects. The required knowledge for this aim could be extracted from vTNFRs. For instance, a CrmE-like modification of etanercept would be particularly interesting to develop a more potent and specific anti-hTNF drug and reduce its side effects.
In summary, we provide the first comprehensive characterization of the different anti-TNFSF ligand activities of the vTNFR family. This analysis will help to better understand the role of these molecules in poxvirus pathogenesis and the relevance of TNFSF ligands in anti-viral defense. Additionally, the comparative study of the different vTNFRs provides a reference framework for the design of more powerful and specific anti-TNF treatments to be used in the clinic.
Acknowledgments
We thank Begoña Ruiz-Arguello for helpful discussions, Rocío Martín for excellent technical support, and Carolina Sánchez for the purification of hTNFR2-Fc.
This work was supported by Spanish Ministry of Economy and Competitiveness Grants SAF2009-07857 and SAF2012-38957. The authors declare that they have no conflicts of interest with the contents of this article.
- TNFSF
- human TNF ligand superfamily
- LT
- lymphotoxin
- TNFR
- TNF receptor
- vTNFR
- viral TNFR
- LTβR
- LTβ receptor
- Crm
- cytokine response modifier
- VACV
- vaccinia virus
- VARV
- variola virus
- MPXV
- monkeypox virus
- CPXV
- cowpox virus
- SPR
- surface plasmon resonance
- CRD
- cysteine-rich domain
- ECTV
- ectromelia virus
- h-
- human
- m-
- mouse.
References
- 1. Aggarwal B. B. (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3, 745–756 [DOI] [PubMed] [Google Scholar]
- 2. Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F., Castner B. J., Stocking K. L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K. A., Gerhart M., Davis R., Fitzner J. N., Johnson R. S., Paxton R. J., March C. J., Cerretti D. P. (1997) A metalloproteinase disintegrin that releases tumour necrosis factor-α from cells. Nature 385, 729–733 [DOI] [PubMed] [Google Scholar]
- 3. Browning J. L., Ngam-ek A., Lawton P., DeMarinis J., Tizard R., Chow E. P., Hession C., O'Brine-Greco B., Foley S. F., Ware C. F. (1993) Lymphotoxin β, a novel member of the TNF family that forms a heteromeric complex with lymphotoxin on the cell surface. Cell 72, 847–856 [DOI] [PubMed] [Google Scholar]
- 4. Crowe P. D., VanArsdale T. L., Walter B. N., Ware C. F., Hession C., Ehrenfels B., Browning J. L., Din W. S., Goodwin R. G., Smith C. A. (1994) A lymphotoxin-β-specific receptor. Science 264, 707–7108171323 [Google Scholar]
- 5. Rahman M. M., McFadden G. (2006) Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang Z., West A. P., Jr., Bjorkman P. J. (2009) Crystal structure of TNFα complexed with a poxvirus MHC-related TNF binding protein. Nat. Struct. Mol. Biol. 16, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brunetti C. R., Paulose-Murphy M., Singh R., Qin J., Barrett J. W., Tardivel A., Schneider P., Essani K., McFadden G. (2003) A secreted high affinity inhibitor of human TNF from tanapox virus. Proc. Natl. Acad. Sci. U.S.A. 100, 4831–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Afonso C. L., Delhon G., Tulman E. R., Lu Z., Zsak A., Becerra V. M., Zsak L., Kutish G. F., Rock D. L. (2005) Genome of deerpox virus. J. Virol. 79, 966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alejo A., Pontejo S. M., Alcami A. (2011) Poxviral TNFRs: properties and role in viral pathogenesis. Adv. Exp. Med. Biol. 691, 203–210 [DOI] [PubMed] [Google Scholar]
- 10. Hu F. Q., Smith C. A., Pickup D. J. (1994) Cowpox virus contains two copies of an early gene encoding a soluble secreted form of the type II TNF receptor. Virology 204, 343–356 [DOI] [PubMed] [Google Scholar]
- 11. Smith C. A., Hu F. Q., Smith T. D., Richards C. L., Smolak P., Goodwin R. G., Pickup D. J. (1996) Cowpox virus genome encodes a second soluble homologue of cellular TNF receptors, distinct from CrmB, that binds TNF but not LTα. Virology 223, 132–147 [DOI] [PubMed] [Google Scholar]
- 12. Loparev V. N., Parsons J. M., Knight J. C., Panus J. F., Ray C. A., Buller R. M., Pickup D. J., Esposito J. J. (1998) A third distinct tumor necrosis factor receptor of orthopoxviruses. Proc. Natl. Acad. Sci. U.S.A. 95, 3786–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saraiva M., Alcami A. (2001) CrmE, a novel soluble tumor necrosis factor receptor encoded by poxviruses. J. Virol. 75, 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saraiva M., Smith P., Fallon P. G., Alcami A. (2002) Inhibition of type 1 cytokine-mediated inflammation by a soluble CD30 homologue encoded by ectromelia (mousepox) virus. J. Exp. Med. 196, 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panus J. F., Smith C. A., Ray C. A., Smith T. D., Patel D. D., Pickup D. J. (2002) Cowpox virus encodes a fifth member of the tumor necrosis factor receptor family: a soluble, secreted CD30 homologue. Proc. Natl. Acad. Sci. U.S.A. 99, 8348–8353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alejo A., Saraiva M., Ruiz-Argüello M. B., Viejo-Borbolla A., de Marco M. F., Salguero F. J., Alcami A. (2009) A method for the generation of ectromelia virus (ECTV) recombinants: in vivo analysis of ECTV vCD30 deletion mutants. PLoS ONE 4, e5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graham S. C., Bahar M. W., Abrescia N. G., Smith G. L., Stuart D. I., Grimes J. M. (2007) Structure of CrmE, a virus-encoded tumour necrosis factor receptor. J. Mol. Biol. 372, 660–671 [DOI] [PubMed] [Google Scholar]
- 18. Reading P. C., Khanna A., Smith G. L. (2002) Vaccinia virus CrmE encodes a soluble and cell surface tumor necrosis factor receptor that contributes to virus virulence. Virology 292, 285–298 [DOI] [PubMed] [Google Scholar]
- 19. Palumbo G. J., Buller R. M., Glasgow W. C. (1994) Multigenic evasion of inflammation by poxviruses. J. Virol. 68, 1737–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Upton C., Stuart D., McFadden G. (1991) Identification and DNA sequence of the large subunit of the capping enzyme from Shope fibroma virus. Virology 183, 773–777 [DOI] [PubMed] [Google Scholar]
- 21. Fenner F. (1993) Smallpox: emergence, global spread, and eradication. Hist. Philos. Life Sci. 15, 397–420 [PubMed] [Google Scholar]
- 22. Alejo A., Ruiz-Argüello M. B., Ho Y., Smith V. P., Saraiva M., Alcami A. (2006) A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. U.S.A. 103, 5995–6000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vogel S., Sárdy M., Glos K., Korting H. C., Ruzicka T., Wollenberg A. (2012) The Munich outbreak of cutaneous cowpox infection: transmission by infected pet rats. Acta Derm. Venereol. 92, 126–131 [DOI] [PubMed] [Google Scholar]
- 24. Ribas G., Rivera J., Saraiva M., Campbell R. D., Alcami A. (2003) Genetic variability of immunomodulatory genes in ectromelia virus isolates detected by denaturing high-performance liquid chromatography. J. Virol. 77, 10139–10146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mavian C., López-Bueno A., Bryant N. A., Seeger K., Quail M. A., Harris D., Barrell B., Alcami A. (2014) The genome sequence of ectromelia virus Naval and Cornell isolates from outbreaks in North America. Virology 462, 218–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen N., Danila M. I., Feng Z., Buller R. M., Wang C., Han X., Lefkowitz E. J., Upton C. (2003) The genomic sequence of ectromelia virus, the causative agent of mousepox. Virology 317, 165–186 [DOI] [PubMed] [Google Scholar]
- 27. Goebel S. J., Johnson G. P., Perkus M. E., Davis S. W., Winslow J. P., Paoletti E. (1990) The complete DNA sequence of vaccinia virus. Virology 179, 247–266, 517–563 [DOI] [PubMed] [Google Scholar]
- 28. Howard S. T., Chan Y. S., Smith G. L. (1991) Vaccinia virus homologues of the Shope fibroma virus inverted terminal repeat proteins and a discontinuous ORF related to the tumor necrosis factor receptor family. Virology 180, 633–647 [DOI] [PubMed] [Google Scholar]
- 29. Alcamí A., Khanna A., Paul N. L., Smith G. L. (1999) Vaccinia virus strains Lister, USSR, and Evans express soluble and cell-surface tumour necrosis factor receptors. J. Gen. Virol. 80, 949–959 [DOI] [PubMed] [Google Scholar]
- 30. Tracey D., Klareskog L., Sasso E. H., Salfeld J. G., Tak P. P. (2008) Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol. Ther. 117, 244–279 [DOI] [PubMed] [Google Scholar]
- 31. Montanuy I., Alejo A., Alcami A. (2011) Glycosaminoglycans mediate retention of the poxvirus type I interferon binding protein at the cell surface to locally block interferon antiviral responses. FASEB J. 25, 1960–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruiz-Arguello M. B., Alejo A., Alcami A. (2005) Secreted tumour necrosis factor inhibitors encoded by poxviruses. in Society for General Microbiology Symposium 64: Molecular Pathogenesis of Virus Infections (Digard P. E., Nash A. A., Randall R. E. eds.), pp. 269–289, Cambridge University Press, Cambridge, UK [Google Scholar]
- 33. Mukai Y., Nakamura T., Yoshikawa M., Yoshioka Y., Tsunoda S., Nakagawa S., Yamagata Y., Tsutsumi Y. (2010) Solution of the structure of the TNF-TNFR2 complex. Sci. Signal. 3, ra83. [DOI] [PubMed] [Google Scholar]
- 34. Xue X., Lu Q., Wei H., Wang D., Chen D., He G., Huang L., Wang H., Wang X. (2011) Structural basis of chemokine sequestration by CrmD, a poxvirus-encoded tumor necrosis factor receptor. PLoS Pathog. 7, e1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanlangenakker N., Bertrand M. J., Bogaert P., Vandenabeele P., Vanden Berghe T. (2011) TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2, e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Browning J. L., Miatkowski K., Sizing I., Griffiths D., Zafari M., Benjamin C. D., Meier W., Mackay F. (1996) Signaling through the lymphotoxin β receptor induces the death of some adenocarcinoma tumor lines. J. Exp. Med. 183, 867–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hehlgans T., Müller P., Stopfer P., Männel D. N. (2003) Activation of the lymphotoxin-β receptor induces NFkappaB-dependent interleukin-6 and MIP-2 secretion in mouse fibrosarcoma cells. Eur. Cytokine Netw. 14, 103–107 [PubMed] [Google Scholar]
- 38. Gileva I. P., Nepomnyashchikh T. S., Antonets D. V., Lebedev L. R., Kochneva G. V., Grazhdantseva A. V., Shchelkunov S. N. (2006) Properties of the recombinant TNF-binding proteins from variola, monkeypox, and cowpox viruses are different. Biochim. Biophys. Acta 1764, 1710–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin X., Ma X., Rodriguez M., Feng X., Zoecklein L., Fu Y. X., Roos R. P. (2003) Membrane lymphotoxin is required for resistance to Theiler's virus infection. Int. Immunol. 15, 955–962 [DOI] [PubMed] [Google Scholar]
- 40. Schneider K., Potter K. G., Ware C. F. (2004) Lymphotoxin and LIGHT signaling pathways and target genes. Immunol. Rev. 202, 49–66 [DOI] [PubMed] [Google Scholar]
- 41. Kerensky T. A., Gottlieb A. B., Yaniv S., Au S. C. (2012) Etanercept: efficacy and safety for approved indications. Expert. Opin. Drug Saf. 11, 121–139 [DOI] [PubMed] [Google Scholar]
- 42. Kruglov A. A., Grivennikov S. I., Kuprash D. V., Winsauer C., Prepens S., Seleznik G. M., Eberl G., Littman D. R., Heikenwalder M., Tumanov A. V., Nedospasov S. A. (2013) Nonredundant function of soluble LTα3 produced by innate lymphoid cells in intestinal homeostasis. Science 342, 1243–1246 [DOI] [PubMed] [Google Scholar]
- 43. Upadhyay V., Fu Y. X. (2013) Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat. Rev. Immunol. 13, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]